Introduction
Multiple myeloma (MM) is a malignant cancer of
plasma cells, which represent ~13% of all hematological
malignancies (1), with >80% of
patients with MM developing myeloma bone disease (MBD). MBD is
characterized by severe bone pain, vertebral compression fractures
and pathological fractures, which are caused by osteoclastic bone
resorption and impaired osteoblastic bone formation induced by the
interaction between myeloma cells and the bone marrow
microenvironment. However, the molecular mechanism by which MM
leads to the development of MBD remains to be elucidated.
Genes that are expressed at high levels in the bone
marrow of patients with MM have been identified (2), one of which is macrophage inhibitory
cytokine-1 (MIC-1). MIC-1 belongs to the human transforming growth
factor β superfamily. Also known as growth differentiation
factor-15, placental bone morphogenetic protein, prostate-derived
factor and NSAID-activated gene-1 (3–6),
MIC-1 has multiple functions. In a previous in vitro study,
MIC-1 was shown to inhibit the activation of macrophages (7). MIC-1 may be an important factor in
the development of several types of tumor, including MM (8). In patients with MM, MIC-1 is
expressed at high levels in the mesenchymal stem cells of the bone
marrow, and high levels of MIC-1 in the patients' serum predicts a
poor prognosis (2). MIC-1 may be
involved in osteoclastogenesis; in patients with prostate cancer
with bone metastases, MIC-1 has been found to induce the maturation
of osteoclasts (9). However,
whether MIC-1 is involved in the development of MBD in patients
with MM remains to be elucidated. In present study, in order to
evaluate the effect of target inhibition of MIC-1 in RPMI-8226
cells on osteoclastic differentiation of peripheral blood
mononuclear cells (PBMNCs) and the mechanisms by which MIC-1
contributes to the maturation of osteoclasts, a lentiviral RNAi
system directed toward the MIC-1 gene was designed and constructed
in RPMI-8226 cells. A co-culture system was used in the present
study to determine the role of MIC-1 on osteoclastic
differentiation. The results from the present study offered a
potential strategy for the treatment of MBD in patients with
MM.
Materials and methods
Preparation of the lentiviral vector
bearing MIC-1 short hairpin (sh)RNA
To understand the potential effect of MIC-1 in
RPMI-8226 cells, an MIC-1 shRNA was designed to specifically knock
down the gene expression of MIC-1. The MIC-1 shRNA was designed by
Genechem (Shanghai, China), based on the MIC-1 cDNA sequence
(GenBank accession no. AF019770.1; http://www.ncbi.nlm.nih.gov/nuccore/AF019770.1). A
control shRNA encoding a nonspecific shRNA was used as a negative
control. The MIC-1 shRNA and negative control shRNA were
synthesized by Genechem (Shanghai, China). The synthesized
oligonucleotides were designed, synthesized and inserted into a
GV115 vector (GeneChem) to construct a lentiviral vector, according
to the manufacturer's protocol, to generate lentiviral transfer
plasmids: Lv-shRNA, bearing MIC-1 shRNA, and Lv-NC, bearing control
shRNA as a negative control. The control shRNA had no significant
homology to any human gene sequence. The lentiviral vectors were
prepared by transfecting the Lv-shRNA or Lv-NC with packaging
plasmids into 293T cells, as described previously (10).
Cell culture and lentiviral
transduction
The RPMI-8226 cells (Wuhan University, Wuhan, China)
were cultured in RPMI-1640 medium (Gibco; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) supplemented with 10% fetal calf serum
(Gibco; Thermo Fisher Scientific, Inc.) at 37°C in a humidified
atmosphere containing 5% CO2. After 3 days, the RPMI-8226 cells
were transduced with the lentiviral vectors at a multiplicity of
infection of 15 in the presence of 2 µg/ml polybrene
(Sigma-Aldrich, St. Louis, MO, USA). Following incubation at 37°C
for 8 h, the cells were washed twice with phosphate-buffered saline
(PBS), and the mRNA and protein levels of MIC-1 in the cells were
measured using reverse transcription-quantitative polymerase chain
reaction (RT-qPCR) and Western blot analyses, respectively, to
evaluate the viral infection efficiency.
RT-qPCR
Total RNA was extracted from the cells using TRIzol
reagent (Invitrogen; Thermo Fisher Scientific, Inc.). The RNA (4
µg) was reverse transcribed into cDNA using Thermoscript RT-PCR
System reagent (Gibco; Thermo Fisher Scientific, Inc.), according
to the manufacturer's protocol, prior for use in qPCR. The qPCR was
performed on an Applied Biosystems PRISM 7300 sequence detection
system (Applied Biosystems; Thermo Fisher Scientific, Inc.) using a
SYBR Green PCR master mix (DRR041A; Takara Bio, Inc., Tokyo,
Japan). A total volume of 20μl, containing 2μl cDNA, 10μl SYBR
Premix Ex Taq, 0.4μl Forward Primer, 0.4μl Reverse Primer, 0.4μl
ROX Reference Dye II and 6.8μl dH2O, was used in the q-PCR
reaction. Each sample was analyzed in triplicate. The
quantification cycle (Cq) value of each sample was normalized to an
endogenous control, GAPDH. The 2−ΔΔCq method was used to
relatively quantify the mRNA levels of MIC-1 (11). The primer (Genechem) sequences for
human MIC-1 were: Sense 5′-GTTGCGGAAACGCTACGA−3′ and antisense
5′-AACAGAGCCCGGTGAAGG-3′. Primers for the control (GAPDH) were:
Sense 5′-TGACTTCAACAGCGACACCCA-3′ and antisense
5′-CACCCTGTTGCTGTAGCCAAA-3′. The qPCR program was as follows: 45
cycles of 95°C for 15 sec, 60°C for 30 sec and 72°C for 45 sec,
followed by 72°C for 10 min.
Isolation and culture of PBMNCs
The PBMNCs were isolated, as described previously
(10) from the peripheral blood of
healthy donors, who had provided written informed consent. The
present study was approved by the Ethics Committee of Fujian
Medical University. A total of 10 ml human blood was obtaubed from
peripheral blood samples, and was mixed with 10 ml lymphocyte
separation medium (Hao Yang Biological Manufacture Co. Ltd.,
Tianjin, China) in a centrifuge tube. The mixture was centrifuged
at 2,000 × g at room temperature for 20 mins. Following washing
with PBS once and α-minimal essential medium (α-MEM) twice, the
nucleated cells were grown in α-MEM supplemented with 10% (v/v)
fetal bovine serum (FBS), 50 ng/ml macrophage-colony-stimulating
factor (M-CSF; PeproTech, Rocky Hill, NJ, USA) and 100 ng/ml
receptor activator of nuclear factor-кB ligand (RANKL; Peprotech)
at 37°C in 5% CO2 humidified air. The medium was replaced every 3
days.
Co-culture system
To investigate the potential effect of MIC-1 on the
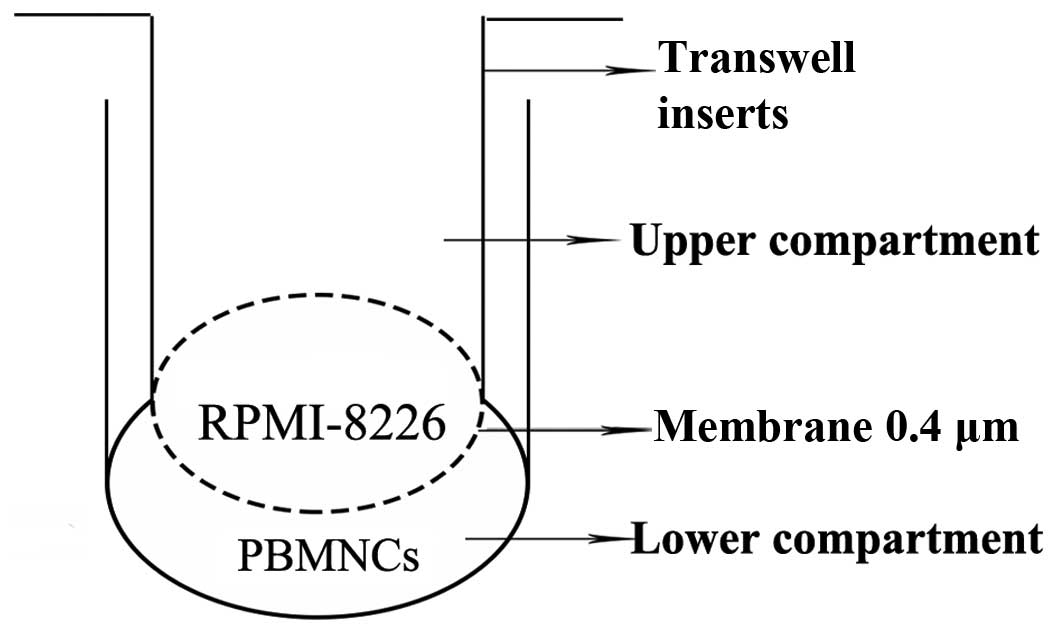
differentiation capacity of the PBMNCs, a Transwell insert system
(Corning, Inc., New York, NY, USA) was used (Fig. 1) to co-culture the PBMNCs
(1×106 cells/well) and the RPMI-8226 cells
(2×103 cells/well) transduced with Lv-shRNA or Lv-NC.
The PBMNCs were seeded into the lower compartment following
placement of a cover glass and dentine slides on the bottom. The
cells were cultured in α-MEM (Gibco; Thermo Fisher Scientific,
Inc.) containing 10% FBS, 50 ng/ml M-CSF and 100 ng/ml RANKL. After
12 h of culture at 37°C in 5% CO2, the non-adherent
cells in the culture were removed by gently washing with fresh
medium; the RPMI-8226 cells transduced with Lv-shRNA or Lv-NC were
cultured in the upper compartment. RPMI-8226 cells without viral
transduction were also used as a control.
Tartrate-resistant acid phosphatase
(TRAP) staining and resorption pit formation assay
The PBMNCs (1×106 cells/well), which were
cultured on cover glasses and dentine slides for 14 or 21 days,
were stained using a leukocyte acid phosphatase kit
(Sigma-Aldrich), and the cells were then counterstained with
hematoxylin (Sigma-Aldrich), as previously described (10). A mature osteoclast was identified
as a cell with at least three TRAP-positive nuclei. To determine
the number of mature osteoclasts in each group, 10 randomly
selected fields from the three cultures in each group were examined
under a microscope (BX43; Olympus, Tokyo, Japan). For the
resorption pit assay, the cells on dentine slices were stained with
toluidine blue and then scanned under a microscope (BX43; Olympus).
Osteoclastic bone resorption was measured, based on the area of
resorption pits per field from three cultures in each group.
Western blot analysis
The PBMNCs were harvested after co-culture for 14
days for protein analysis. The protein extracts from these cells
were separated by 6–15% SDS-PAGE gel electrophoresis and then
transferred onto polyvinylidene fluoride (PVDF; Beyotime Institute
of Biotechnology, Shanghai, China) membranes. Following blocking
with Tris-buffered saline with 0.2% Tween (TBST) containing 5%
non-fat milk at 4°C overnight, the membranes were incubated with
the following antibodies diluted in TBST for 2 h at 4°C: Rabbit
anti-RANKL (cat no. sc-9073; 1:2,000), rabbit anti–GAPDH (cat no.
sc-25778; 1:2,000), rabbit anti-c-fos (cat no. sc-52, 1:2,000),
rabbit anti-c-Jun N-terminal kinase (c-JNK; cat no. sc-572;
1:2,000), rabbit anti-p-c-Jun N-terminal kinase (p-c-JNK; cat no.
sc-135642; 1:2,000), rabbit anti-P-38 (cat no. sc-535; 1:2,000),
rabbit anti-p-P-38 (cat no. sc-101759; 1:2,000), rabbit anti-c-jun
(cat no. sc-44; 1:2,000), rabbit anti-p-c-jun (cat no. sc-7980-R;
1:2,000), all purchased from Santa Cruz Biotechnology, Inc. (Santa
Cruz, CA, USA), and rabbit anti-extracellular signal-regulated
kinase 1/2 (Erk1/2; cat no. 4695; 1:2,000; Cell Signaling
Technology, Inc., Danvers, MA, USA) or rabbit anti- p-Erk1/2 (cat
no. 4376; 1:2,000; Cell Signaling Technology, Danvers, MA, USA).
Following washing three times with TBST (pH 7.5), the membranes
were incubated with horseradish peroxidase-conjugated goat
anti-rabbit secondary antibody (cat no. bs-0295G; 1:1,000; Beijing
Biosynthesis Biotechnology, Beijing, China) for 1 h at room
temperature. The membranes were washed three times with TBST (pH
7.5). The resulting protein bands were visualized using an enhanced
chemiluminescence reagent kit (Beyotime Institute of
Biotechnology).
Statistical analysis
Statistical analysis was performed using SPSS 16.0
software (SPSS, Inc., Chicago, IL, USA). The data are expressed as
the mean ± standard deviation. One-way analysis of variance and
Student-Newman-Keuls analysis were used to evaluate the statistical
significance of differences among the groups. P<0.05 was
considered to indicate a statistically significant difference.
Results
Expression of MIC-1 in RPMI-8226
cells
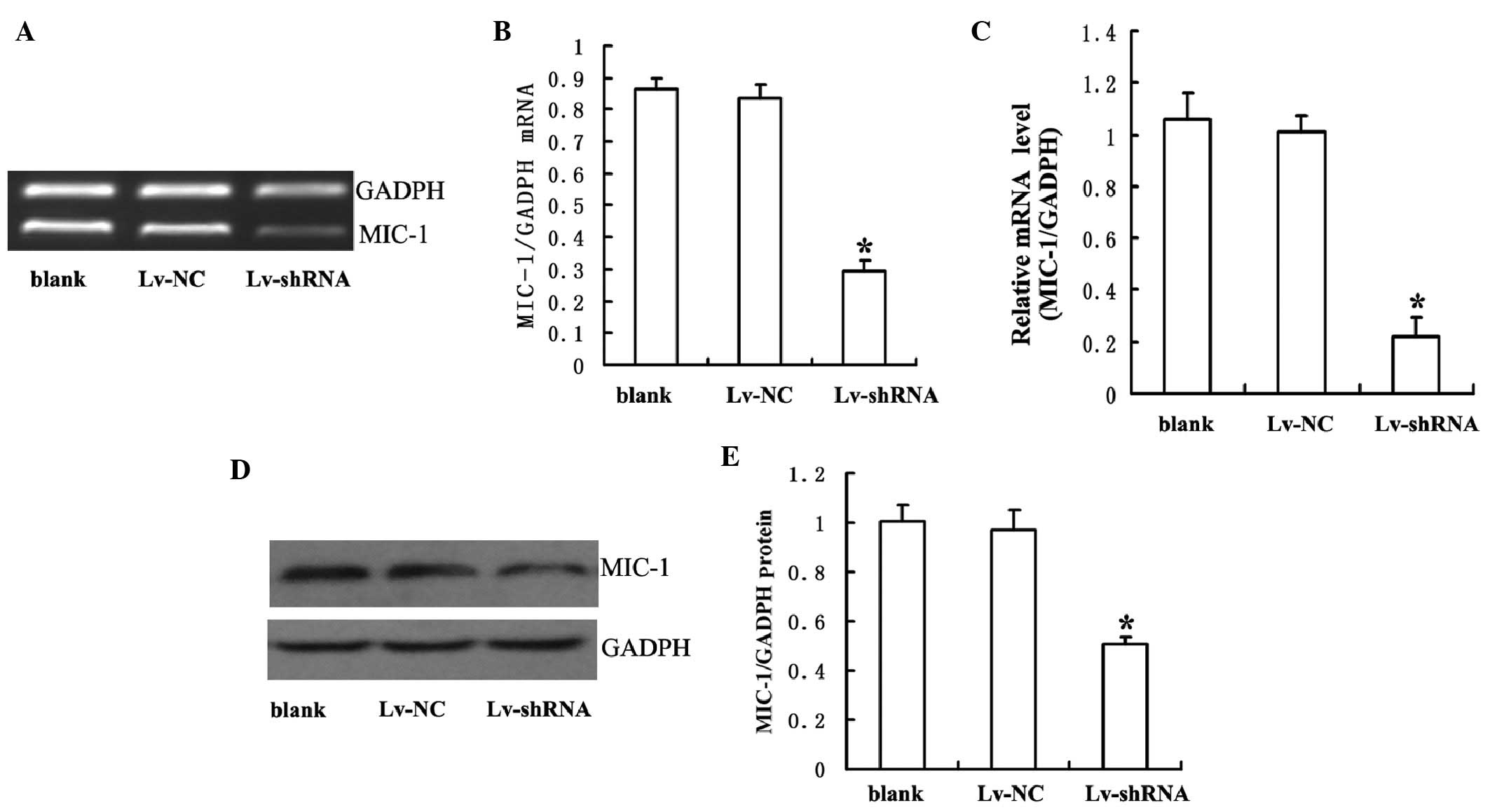
To determine whether MIC-1 shRNA successfully
knocked down the gene expression of MIC-1, the present study
prepared lentiviral vectors bearing either MIC-1 shRNA (Lv-shRNA)
or non-specific sequence (Lv-NC). Following transduction with
Lv-shRNA or Lv-NC for 14 days, the RPMI-8226 cells were harvested
and the expression levels of MIC-1 in these cells were analyzed
using RT-qPCR and Western blot analyses. As shown in Fig. 2, the mRNA and protein levels of
MIC-1 were significantly lower in the cells transduced with
Lv-shRNA, compared with those in the cells transduced with Lv-NC or
those without viral tranduction.
MIC-1 is essential for the
osteoclastic differentiation of PBMNCs
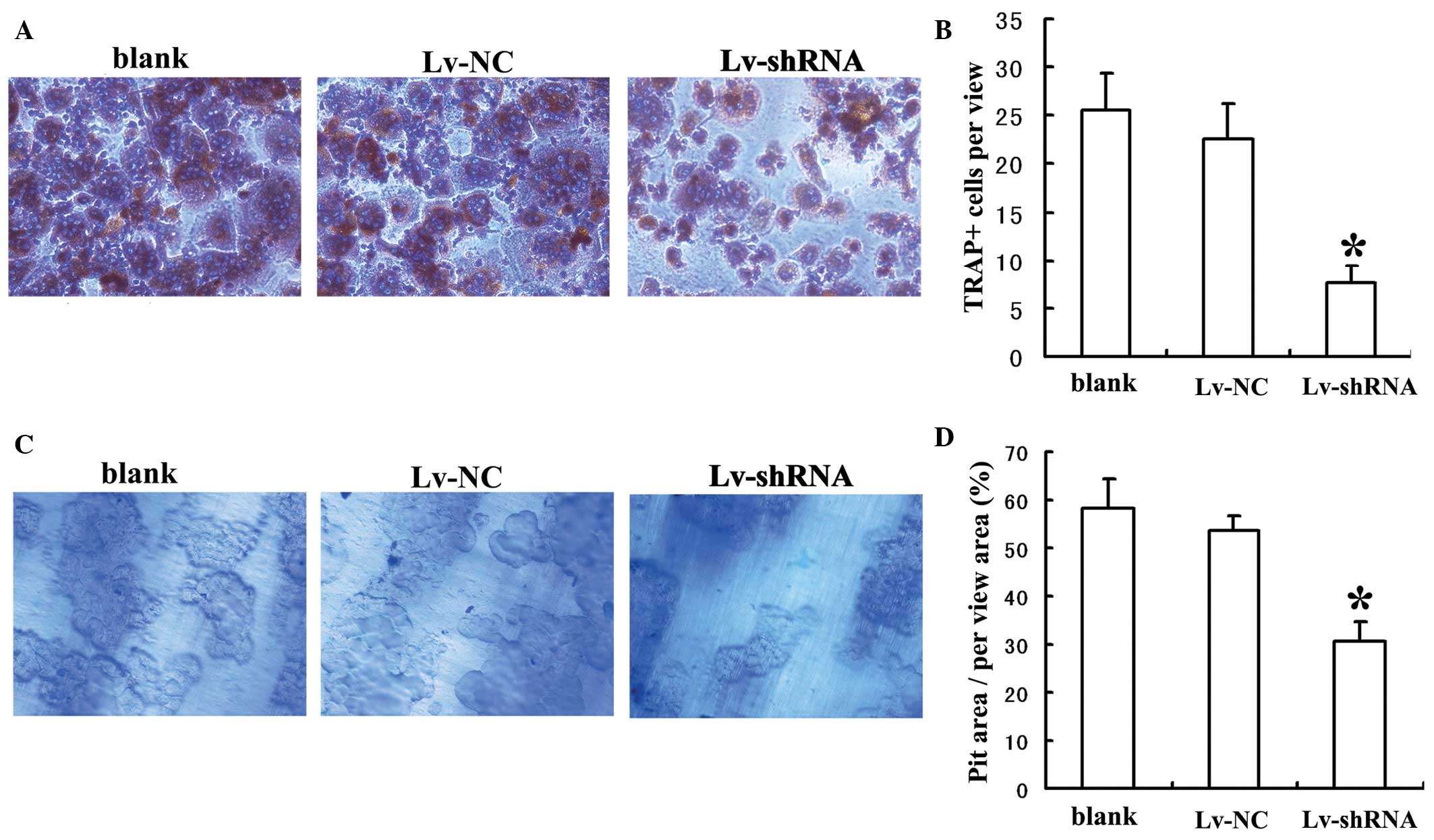
To investigate the role of MIC-1 in the osteoclastic
differentiation of PBMNCs, the PBMNCs were stained for TRAP
following co-culture with RPMI-8226 cells for 14 days. The number
of TRAP(+) cells was significantly lower when the PBMNCs were
co-cultured with the RPMI-8226 cells transduced with Lv-shRNA,
compared with when they were co-cultured with RPMI-8226 cells
transduced with Lv-NC or without viral transduction (Fig. 3A and B). To investigate the role of
MIC-1 in bone resorption by differentiated PBMNCs, the PBMNCs were
stained with toluidine blue following co-culture with RPMI-8226
cells for 14 days. The percentage of pit area within a randomly
selected area was significantly lower when the PBMNCs were
co-cultured with the RPMI-8226 cells transduced with Lv-shRNA,
compared with those co-cultured with the RPMI-8226 cells transduced
with Lv-NC or without viral transduction (Fig. 3C and D). Taken together, these
results suggested that MIC-1 was required for the osteoclastic
differentiation and bone resorption activities of PBMNCs.
To confirm the role of MIC-1 in the osteoclastic
differentiation of PBMNCs, the present study examined whether the
expression of the osteoclast-specific gene, RANKL, was altered by
the decreased expression of MIC-1. As shown in Fig. 4A and B, the protein level of RANKL
in the PBMNCs was significantly lower when the cells were
co-cultured with the RPMI-8226 cells transduced with Lv-shRNA,
compared with those co-cultured with the RPMI-8226 cells transduced
with Lv-NC or without viral transduction.
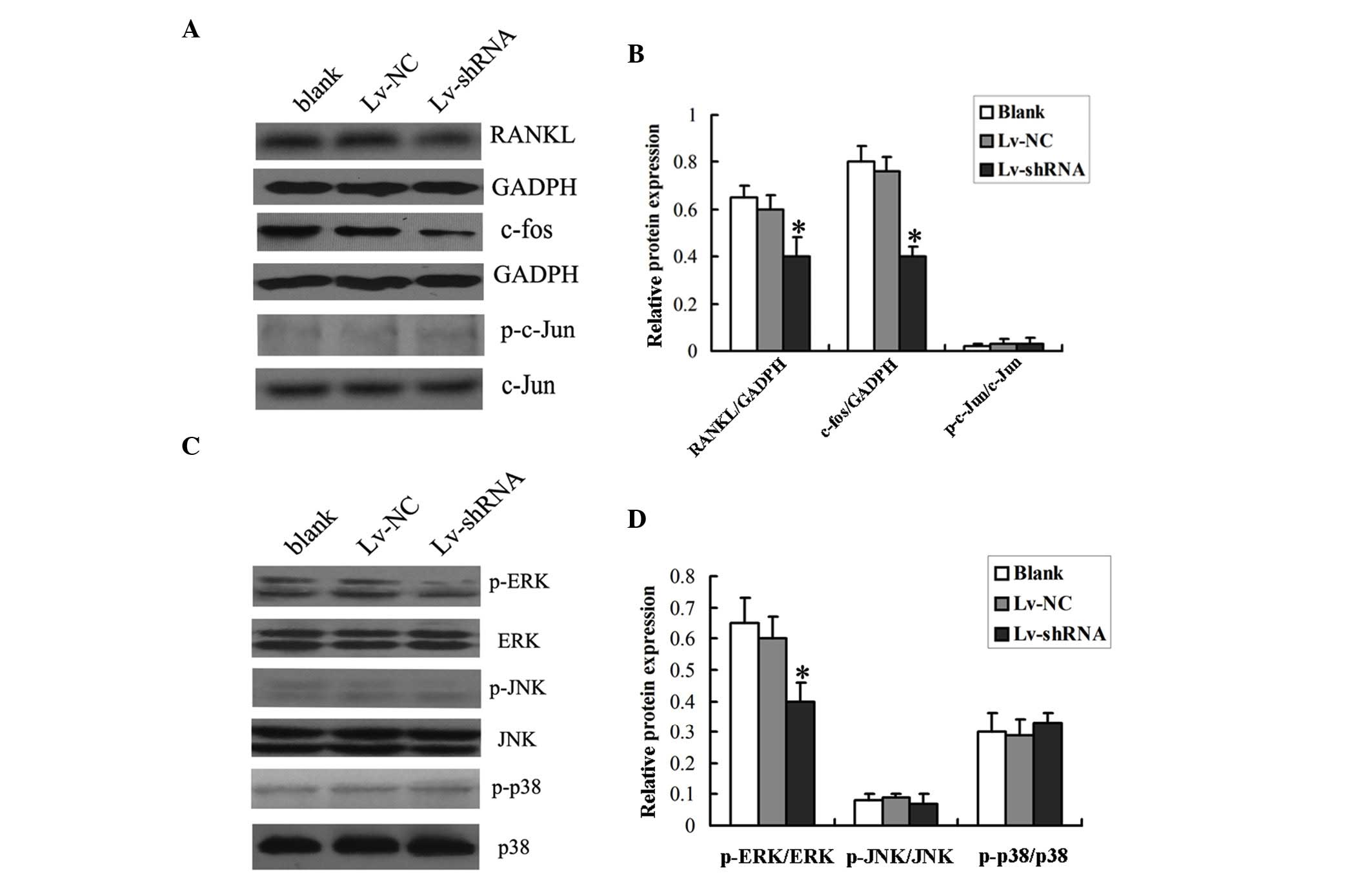 | Figure 4.Expression of RANKL and the
phosphorylation status of the Erk1/2 signaling pathway in PBMNCs.
PBMNCs were co-cultured with RPMI-8226 cells infected with Lv-shRNA
or Lv-NC for 14 days prior to harvesting for the analysis of the
expression levels of (A) RANKL and c-fos, and the phosphorylation
status of c-jun. GADPH was used as internal control. (B)
Densitometric analysis of the western blot was performed. (C) The
phosphorylation status of Erk1/2, p-38 and c-JNK was determined by
western blotting. (D) Densitometric analysis of the western blot
was performed. The data are expressed as the mean ± standard
deviation (*P<0.05 vs. Lv-NC group or Blank group). Blank, cells
without viral transduction; PBMNCs, peripheral blood mononuclear
cells; shRNA, short hairpin RNA; NC, negative control; RANKL,
receptor activator of nuclear factor-κB ligand; p-ERK,
phosphorylated-extracellular signal-regulated kinase; t-ERK, total
ERK; JNK, c-Jun N-terminal kinase. |
Subsequently, the present study investigated which
signaling pathway was activated by RANKL by analyzing the
phosphorylation status of the ERK1/2, c-JNK and p38
mitogen-activated protein kinase (MAPK) proteins using Western blot
analysis. It was found that the protein levels of phosphorylated
Erk1/2, but not those of c-JNK or p38, were significantly lower in
the PBMNCs co-cultured with the RPMI-8226 cells transduced with
Lv-shRNA, compared with those co-cultured with RPMI-8226 cells
transduced with Lv-NC or without viral transduction (Fig. 4C and D), indicating that the
MCI-1-induced increase in RANKL may activate the Erk1/2
pathway.
The present study then investigated whether c-fos
and c-jun, important downstream molecules of ERK1/2, were involved
in the differentiation of osteoclasts by analyzing the
phosphorylation status of c-fos and c-jun using Western blot
analysis. It was found that the protein levels of total c-fos were
markedly reduced in the PBMNCs co-cultured with the RPMI-8226 cells
transduced with Lv-shRNA, compared with those co-cultured with the
RPMI-8226 cells transduced with Lv-NC or without viral
transduction. However, no changes were observed in the protein
levels of total c-jun or phosphorylated c-jun in the PBMNCs
co-cultured with the RPMI-8226 cells transduced with either
Lv-shRNA or Lv-NC (Fig. 4C and D).
Taken together, these results suggested that MIC-1 regulated the
osteoclastic differentiation and bone resorption activities of the
PBMNCs via the Erk1/2 signaling pathway.
Discussion
In the present study, the role of MIC-1 in the
osteoclastic differentiation of PBMNCs was examined. A co-culture
system was used, in which PBMNCs co-cultured with RPMI-8226 cells
were induced to differentiate into osteoclasts and to resorb bone.
To knock down the expression of MIC-1 in the RPMI-8226 cells, a
lentiviral vector was used to deliver the MIC-1 shRNA to the
RPMI-8226 cells. It was found that MIC-1 shRNA efficiently
decreased the expression of MIC-1 in the RPMI-8226 cells (Fig. 2). The present study then
demonstrated that reduced expression of MIC-1 inhibited the
osteoclastic differentiation of PBMNCs (Fig. 3). Finally, it was shown that the
expression of the osteoclast-specific gene, RANKL, and the levels
of dephosphorylated Erk1/2 were decreased in those PBMNCs, which
were co-cultured with the RPMI-8226 cells transduced with Lv-shRNA
(Fig. 4A and C). These results led
to the conclusion that MIC-1 promoted osteoclastic differentiation
of PBMNCs through inhibition of the RANKL-Erk1/2 signaling
pathway.
Patients with MM often develop MBD with osteoclastic
differentiation, induced by cytokines and other soluble factors in
the bone marrow (12). In the bone
marrow of patients with MM, MIC-1 is expressed at high levels
(2). It has been suggested that
MIC-1 may be involved in the regulation of osteoclastogenesis
(13,14). The results of the study
demonstrated that MIC-1 was required in order for RPMI-8226 to
induce osteoclastic differentiation of the PBMNCs (Figs. 3 and 4), supporting the role of MCI-1 in the
development of MBD in patients with MM.
The osteoclast-specific marker gene, RANKL, is
involved in MCI-1 stimulated osteoclastic differentiation. Binding
to its receptor RANK, RANKL stimulates the osteoclastic
differentiation of monocyte macrophages and the maturation of
osteoclasts (15). In
RANKL-deficient mice, severe osteopetrosis and a defect in tooth
eruption have been observed (16).
In patients with MM, RANKL has been shown to be involved
osteoclastic differentiation (17–19).
In the present study, it was shown that the expression of RANKL in
the PBMNCs was reduced when the expression of MIC-1 in the
co-cultured RPMI-8226 cells was knocked down (Fig. 4). RANKL binds to RANK and activates
several MAPK signaling pathways (20), including the Erk1/2, JNK and P-38
pathways (21). The present study
demonstrated that reduced expression of MIC-1 in the RPMI-8226
cells appeared to decrease the phosphorylation of Erk1/2, but not
JNK or P-38, indicating the involvement of the Erk1/2 pathway in
MIC-1-induced osteoclastic differentiation of PBMNCs (22,23).
Consistent with this observation, the expression levels of c-fos
and c-jun, downstream molecules of the Erk1/2 signaling pathway in
PBMNCs, were also decreased when the expression of MCI-1 in the
co-cultured RPMI-8226 cells was reduced.
In conclusion, the present study found that MIC-1
was involved in the development of MBD in patients with MM by
promoting the osteoclastic differentiation of PBMNCs by activating
the RANKL-Erk1/2 signaling pathway. Thus, MIC-1 may offer potential
as a target gene in the development of strategies to treat MM.
References
|
1
|
Kyle RA and Rajkumar SV: Criteria for
diagnosis, staging, risk stratification and response assessment of
multiple myeloma. Leukemia. 23:3–9. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Corre J, Mahtouk K, Attal M, Gadelorge M,
Huynh A, Fleury-Cappellesso S, Danho C, Laharrague P, Klein B, Rème
T and Bourin P: Bone marrow mesenchymal stem cells are abnormal in
multiple myeloma. Leukemia. 21:1079–1088. 2007.PubMed/NCBI
|
|
3
|
Hromas R, Hufford M, Sutton J, Xu D, Li Y
and Lu L: PLAB, a novel placental bone morphogenetic protein.
Biochim Biophys Acta. 1354:40–44. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Paralkar VM, Vail AL, Grasser WA, Brown
TA, Xu H, Vukicevic S, Ke HZ, Qi H, Owen TA and Thompson DD:
Cloning and characterization of a novel member of the transforming
growth factor-beta/bone morphogenetic protein family. J Biol Chem.
273:13760–13767. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Böttner M, Suter-Crazzolara C, Schober A
and Unsicker K: Expression of a novel member of the TGF-beta
superfamily, growth/differentiation factor-15/macrophage-inhibiting
cytokine-1 (GDF-15/MIC-1) in adult rat tissues. Cell Tissue Res.
297:103–110. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Baek SJ, Horowitz JM and Eling TE:
Molecular cloning and characterization of human nonsteroidal
anti-inflammatory drug-activated gene promoter. Basal transcription
is mediated by Sp1 and Sp3. J Biol Chem. 276:33384–33392. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bootcov MR, Bauskin AR, Valenzuela SM,
Moore AG, Bansal M, He XY, Zhang HP, Donnellan M, Mahler S, Pryor
K, et al: Mic-1, a novel macrophage inhibitory cytokine, is a
divergent member of the TGF-beta superfamily. Proc Natl Acad Sci
USA. 94:11514–11519. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tarkun P, Atesoglu E Birtas, Mehtap O,
Musul MM and Hacihanefioglu A: Serum growth differentiation factor
15 levels in newly diagnosed multiple myeloma patients. Acta
Haematol. 131:173–178. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wakchoure S, Swain TM, Hentunen TA,
Bauskin AR, Brown DA, Breit SN, Vuopala KS, Harris KW and Selander
KS: Expression of macrophage inhibitory cytokine-1 in prostate
cancer bone metastases induces osteoclast activation and weight
loss. Prostate. 69:652–661. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zeng Z, Zhang C and Chen J:
Lentivirus-mediated RNA interference of DC-STAMP expression
inhibits the fusion and resorptive activity of human osteoclasts. J
Bone Miner Metab. 31:409–416. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Roodman GD: Pathogenesis of myeloma bone
disease. Leukemia. 23:435–441. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Khosla S: Minireview: The OPG/RANKL/RANK
system. Endocrinology. 142:5050–5055. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sung B, Prasad S, Yadav VR, Gupta SC,
Reuter S, Yamamoto N, Murakami A and Aggarwal BB: RANKL signaling
and osteoclastogenesis is negatively regulated by cardamonin. PLoS
One. 8:e641182013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lacey DL, Timms E, Tan HL, Kelley MJ,
Dunstan CR, Burgess T, Elliott R, Colombero A, Elliott G, Scully S,
et al: Osteoprotegerin ligand is a cytokine that regulates
osteoclast differentiation and activation. Cell. 93:165–176. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kong YY, Yoshida H, Sarosi I, Tan HL,
Timms E, Capparelli C, Morony S, Oliveira-dos-Santos AJ, Van G,
Itie A, et al: OPGL is a key regulator of osteoclastogenesis,
lymphocyte development and lymph-node organogenesis. Nature.
397:315–323. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Schramek D, Leibbrandt A, Sigl V, Kenner
L, Pospisilik JA, Lee HJ, Hanada R, Joshi PA, Aliprantis A,
Glimcher L, et al: Osteoclast differentiation factor RANKL controls
development of progestin-driven mammary cancer. Nature. 468:98–102.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sung B, Oyajobi B and Aggarwal BB:
Plumbagin inhibits osteoclastogenesis and reduces human breast
cancer-induced osteolytic bone metastasis in mice through
suppression of RANKL signaling. Mol Cancer Ther. 11:350–359. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ai LS, Sun CY, Zhang L, Zhou SC, Chu ZB,
Qin Y, Wang YD, Zeng W, Yan H, Guo T, et al: Inhibition of BDNF in
multiple myeloma blocks osteoclastogenesis via down-regulated
stroma-derived RANKL expression both in vitro and in vivo. PLoS
One. 7:e462872012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wada T, Nakashima T, Hiroshi N and
Penninger JM: RANKL-RANK signaling in osteoclastogenesis and bone
disease. Trends Mol Med. 12:17–25. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Huang P, Han J and Hui L: MAPK signaling
in inflammation-associated cancer development. Protein Cell.
1:218–226. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang W and Liu HT: MAPK signal pathways
in the regulation of cell proliferation in mammalian cells. Cell
Res. 12:9–18. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Feng X: RANKing intracellular signaling in
osteoclasts. IUBMB Life. 57:389–395. 2005. View Article : Google Scholar : PubMed/NCBI
|