Introduction
Traumatic brain injury (TBI) is normally caused by
an external mechanical force that traumatically injures the brain.
It can be classified based on severity, nature of the impact or
other features (1–3). Currently, TBI is one of the major
causes of death and disability worldwide, especially in children
and young adults. The number of young cases that suffer from brain
injuries has been drastically increasing over the years (1,2).
Although the number of drugs that are capable of
ameliorating cerebral injuries remain scarce, a significant
progress has been made in understanding the pathological mechanism
of TBI and in developing a possible treatment. It is known that
following the occurrence of TBI, other events such as inflammatory
reactions, oxidative stress responses, and cytotoxicity injuries
develop, and together they elicit a secondary craniocerebral
injury, mainly driven by the inflammatory component (4,5). The
inflammatory reaction in the brain usually involves the necrosis or
death of numerous neurons as well as the activation of microglial
cells (6–8). Peripheral blood lymphocytes,
neutrophil granulocytes and thrombin invade the central nervous
system (CNS), passing through the injured blood-brain barrier
(9–12). Consequently, the expression of many
cytokines is up- or downregulated at the same time that resident
microglia cells are activated. Previous studies suggested that TBI,
of either moderate or severe levels, induces chronic microglial
activation that may negatively affect prognosis. It was also
suggested that the activation of these cells may be the result of
delayed cell death and tissue loss (5). Overall, all these changes in the
immune system further exacerbate the apoptosis and necrosis of
neurons, aggravating the injury and further impairing the
neurological function (13–15).
On the other hand, recent studies have demonstrated
that regulatory T cells (Tregs) are involved in immunomodulation,
alleviating the inflammatory reaction and tissue damage (6–12).
CD4 T cells that constitute Tregs are usually associated with CD25
and FoxP3 markers. Sakaguchi et al (13) confirmed that CD25 is a surface
phenotypic marker for suppressive CD4 cells in mice. On the other
hand, FoxP3 belongs to the forkhead/winged-helix family of
transcription factors, acting as the ‘master regulator’ in the
establishment of Tregs as a terminally differentiated and lineage
committed subset of CD4 T cells (13).
Tregs can reduce cerebral infarction and improve
behavioral function, and are positively associated with
neuro-regeneration and alleviation of local inflammation. They have
also shown to regulate immunity and inflammation in several
diseases, including brain ischemia, viral myocarditis, inflammatory
bowel disease, and myasthenia gravis (16,17).
However, their underlying pathological and physiological mechanisms
continue unknown. In certain studies in atherosclerosis, it was
found that Tregs stimulate the secretion of anti-inflammatory
cytokines of interleukin (IL)-10 and transforming growth factor
(TGF)-β (18–22).
Since few studies have examined the function of
Tregs in TBI patients, little is known about the physiological
functions of Tregs in this condition. However, clinical trials have
corroborated that the upregulation of Tregs in TBI patients
significantly improves their clinical condition (12). Therefore, more studies are required
to confirm their neuro-protective effects in TBI, since a better
understanding can remarkably improve diagnosis, treatment and
prognosis of the condition, as well as provide theoretical basis
for novel treatment of TBI.
By analyzing the expression of Tregs and
corresponding cytokines in the peripheral and CNSs, the present
study aimed to elucidate the association between the changes of the
immune system and in presence of TBI, by upregulating and
downregulating the Tregs concentration in a mouse model of TBI.
Co-cultures of Tregs and microglia cells were
performed to determine the immunological effect of Tregs. Our data
showed that Tregs considerably suppressed microglia activation,
while in the group of Treg depletion, microglia activation
exhibited the highest level.
Moreover, JNK and p38 MAPK signaling pathways have
been reported as critical factors during neuro-inflammation. In the
current study, the expression patterns of JNK1/2/3, p38 MAPK,
ERK1/2 and NF-κB proteins were identified among all the studied
groups. However, our findings suggested that in the group with
exogenous Treg injection, the expression levels of JNK1/2/3, p38
MAPK, ERK1/2 and NF-κB proteins were significantly reduced,
compared to their levels in other groups. The data indicated that
Tregs attenuate the inflammatory response by suppressing the JNK
and NF-κB signaling pathways.
Materials and methods
Animals
C57BL/6 mice, 6–8 weeks of age, were used in the
current study. Animals were randomly separated into two groups,
sham and TBI group. In both groups, food and tap water were
provided without restriction and the housing temperature was
strictly controlled. All the animal protocols were approved by the
Committee of Ethics and performed at Animal Experiments of the
Experimental Animal Research Institute. Every effort was made to
minimize animal suffering.
Mouse controlled cortical impact (CCI)
model of moderate TBI
The CCI model was used to induce traumatic brain
injuries in mice. TBI group consisted of 8 mice, which were
anesthetized via IP injection of a Ketamine/xylazine mixture (87.7
mg/ml Ketamine and 12.3 mg/ml Xylazine) (1 ml/kg). The heads of
mice were shaved between the ears and sterilized with 10% iodine,
followed by 70% ethanol. Subsequently, a midline incision was
performed to expose the skull, and the anatomical landmarks Lambda
(caudal aspect) and Bregma (frontal aspect) were identified, by
drawing a circle in the center with a 4 mm diameter and 0.5 mm away
from the midline. The marked circle was cut along and the bone dust
was gently blown away, in case the dura mater was damaged.
Afterwards, the injury was achieved by introducing the impactor tip
into the brain with a 1.0-mm depth of impact. The diameter of the
impactor tip was 3 mm. Parameters to produce moderate TBI were set
as follows: the velocity of the actuator was 3 m/sec, and impact
duration was 100 msec. After injury, the area was carefully cleaned
from any residual blood with a cotton-tipped applicator. Once
bleeding stopped, the wound was sutured. The mice then returned a
the clean cage and recovered from the surgery. The sham group
consisted of 7 mice, which did not receive the injury.
The comparison of Treg levels between TBI group and
Sham group were conducted at days 1, 3 and 7 post-injury. Brain
tissues were harvested, fixed and embedded with paraffin for
further examination of the number of Treg cells present at these
time points. Formalin-fixed hematoxylin-eosin stained sections were
also examined by a pathologist, who was blinded to the status of
the mice, by light microscopy.
Splenocyte isolation
All the steps were performed under a sterile laminar
flow hood to assure sterility of the procedure. Animals were
euthanized by cervical dislocation. After sterilization of the
abdomen area with 70% ethanol, a first transversal cut, without
opening the peritoneal cavity, was performed. The peritoneal sac
was identified and excised in order to collect the spleen. The
spleen was cleared from residual debris and fat tissue, by washing
it twice with 1X phosphate-buffered saline (PBS). Subsequently, the
spleen was cut in two halves, by using microscope glass slides
previously sterilized with 70% ethanol and fire. Spleen contents
were removed by squeezing the two halves between the slides to
extract the cellular matrix contained within the spleen capsule.
The remaining fibrous tissue was discarded and cell aggregates
obtained from this procedure were resuspended in complete RPMI-1640
medium.
Lysis of erythrocytes
Red blood cells were lysed by resuspending the
obtained cell pellet in 5 ml of ACK buffer for 90 sec. Immediately
afterwards, the cell suspension was washed with 45 ml of 1X PBS to
remove any residual of the ACK buffer. Cells were centrifuged at
400 × g for 10 min and subsequently resuspended in 10 ml of 1X PBS.
Successful lysis of red cells was confirmed by the presence of a
white cellular pellet. Number of cells were determined.
Magnetic sorting of Tregs
CD4+CD25+ Tregs were isolated
from 1×108 splenocytes, and according to the
manufacturer's guideline (cat. no. 130-091-041; Miltenyi Biotec
GmbH, Bergisch Gladbach, Germany). Specifically, splenocytes were
labelled with 100 µl of biotinylated antibody cocktail specific for
non-CD4+ T cells and diluted with 400 µl of RPMI-1640
10% FBS for 15 min, then 200 µl of the antibody specific for biotin
and conjugated with microbeads was added and further diluted with
300 µl of RPMI-1640 10% FBS and the mix was incubated for 10 min.
After labeling, cells were washed once with RPMI-1640 10% FBS and
re-suspended in 6 ml of RPMI-1640 containing 10% FBS. Indirect
separation of CD4+ cells was obtained by loading 2 ml of
the labeled cells onto an LD separation column (Miltenyi Biotec
GmbH). During this procedure, CD4-negative cells remained in the
column, since the magnetic beads were attached to their surfaces,
whereas, CD4-positive cells, flew through the column and were
finally collected in a new tube. The final number of
CD4+ cells was determined.
The cell population was further enriched in Tregs
through a positive selection of CD25+ cell surface
marker. Briefly, the obtained cells were labeled with an antibody
against CD25-PE, and incubated for 15 min. The cells were incubated
for another 15 min, but this time in the presence of an anti-PE
antibody conjugated with microbeads (Miltenyi Biotec GmbH). The
cells were washed once with 10 ml of RPMI 10% FBS and then
re-suspended in 6 ml of complete medium.
CD4+CD25− cells were separated from
CD4+CD25+ cells by loading the labelled mix
into the LS positive selection column (Miltenyi Biotec GmbH), 1 ml
at a time. Finally, CD4+CD25+ cells were
collected and quantified.
Flow cytometry analysis
The detection of CD4+CD25+
cells with CD4-FITC and anti-CD25-PE antibodies was performed as
follows: 2.5×105 CD4+CD25+ cells
were resuspended in 100 µl of PBS 0.2% BSA. 1 µl of each antibody:
CD4-FITC and anti-CD25-PE was added to the sample (1:100 dilution).
After 10 min incubation, the cells were washed and analyzed using
flow cytometry to determine the obtained cell purity. Generally, an
80% purity of CD4+CD25+ cells can be achieved
with this protocol, according to the manufacturer's
instructions.
In vitro expansion of Tregs
In the present study, we used mouse Treg expansion
kit (cat. no. 130-095-925; Miltenyi Biotec GmbH) to conduct in
vitro expansion of Treg cells. All procedures were carried out
according to the manufacturer's instructions. Treg cells were
resuspended at a concentration of 2×106 cells/ml in
complete culture medium containing 2,000 U/ml rIL-2. Tregs (50 µl)
were incubated with 50 µl of CD3/CD28 MACSiBead Particles in a well
of a 96-well flat bottom plate (day 0). At day 1, 100 µl media
including 2,000 U/ml IL-2 was added, and at day 3, cells were split
and then 100 µl medium including 2,000 U/ml rIL-2 was added. After
7 days of culture, CD3/CD28 MACSiBead Particles were removed
together with the CD3/CD28 cells bound to them by magnetic cell
separation. Treg population was collected for further analysis.
Immunohistochemistry. The paraffin-embedded tissue
from each group was cut into 6 µm sections and repaired with high
voltage. Sections were cooled at RT and then rinsed twice in
deionized water and threse times in PBS (5 min/wash). The sections
were incubated with FOXP3 primary antibody (1:400, ab20034; Abcam,
Cambridge, UK) diluted in 2% BSA at 4°C overnight. Not bound FoxP3
antibodies were removed by rinsing the tissue twice with PBS (2
min/wash). Finally, the non-biotinylated goat anti-rabbit IgG
secondary antibody was incubated with the primary antibody for 20
min at room temperature (RT), followed by three rinses with PBS (5
min/wash). The colored reaction product was developed using Simple
Stain DAB solution.
Isolation and culture of microglia
cells
Procedures were conducted under sterile conditions.
The base of a 75 cm2 culture flask was coated with 5 ml
poly-D-lysine (10 µg/ml) and allowed to set overnight at RT. Excess
of poly-D-lysine was removed and the flask was washed once with
PBS. The cleaned culture cabinet was exposed to additional 30 min
of UV light to maximize the sterilization process. Brains from five
fetal BALB/c mice (1–3 days old) were harvested and subjected to
disruption by incubating them with 2 ml of trypsin/EDTA
(0.05/0.02%) solution, in the incubator at 37°C for 10 min to allow
for enzymatic disaggregation. Following neutralization of the
trypsin with media, large pieces of tissues were further
disaggregated by passing the cell suspension through a 70 µm nylon
cell strainer. Finally, the suspended cells were added to a
poly-D-lysine coated flask and were allowed to grow for 10–14 days,
replacing the media every 3 days. Once the cells were 80–90%
confluent, they were transferred to a poly-D-lysine pre-coated
24-well tissue culture plate (1×105 cells/well) and
incubated at 37°C in a humidified 5% CO2 atmosphere.
Within three days of culture, the obtained cells were assayed for
their purity by flow cytometry using the anti-CD11b monoclonal
antibody.
Western blotting analysis
Protein extracts from mouse brain tissues were
obtained by grinding the tissue into powder in liquid nitrogen with
RIPA buffer. The extracted proteins were quantified using BCA assay
kit (Thermo Fisher Scientific, Waltham, MA, USA). Subsequently, 40
µg of protein extracts were separated by SDS-PAGE, then transferred
onto a PVDF membrane (Millipore Corp., Billerica, MA, USA) for
detection of specific proteins. The membranes were blocked for 30
min at RT with 5% non-fat dry milk and incubated for 2 h with
primary antibodies. All the primary antibodies: ERK1/2 (cat. no.
ab17942, diluted 1:1,000), JNK1+JNK2+JNK3 antibody (EPR18841-95)
(cat. no. ab208035, diluted 1:1,000), JNK1+JNK2+JNK3 (phospho
T183+T183+T221) (cat. no. ab124956, diluted 1:1,000), p38 MAPK
(cat. no. ab197348, diluted 1:1,000), p38 (phospho Y182) antibody
(cat. no. ab47363, diluted 1:1,000) were purchased from Abcam.
Western blot analysis were conducted by incubating the membrane
with the above-mentioned antibodies overnight in a humidified
container at 4°C. Following binding of primary antibodies to the
specific proteins in the extract, the membrane was washed with PBST
three times and finally incubated with HRP-conjugated secondary
antibody for 2 h. The specific proteins were detected and
quantified using ECL (Millipore, Corp.) and Quantity One software
(Bio-Rad, Berkeley, CA, USA).
RNA extraction and qPCR analysis
Total RNAs from the tissues of TBI and sham groups
were isolated from harvested cells and mouse brain tissues with
TRIzol (Invitrogen Life Technologies, Carlsbad, CA, USA), according
to the manufacturer's instructions. RNAs were reverse transcribed
using the PrimeScript™ RT-PCR kit (Takara Bio, Dalian, China).
Real-time PCR reactions were performed using SYBR Premix
DimerEraser system (Takara Bio). The PCR primers were designed as
follows: IL-10 forward, 5′-ACAACATACTGCTAACCGACTCCT-3′ and reverse,
5′-TGCTCCACTGCCTTGCTCTTAT-3′; and TGF-β forward,
5′-TGTCGTGGCAGTCCTTCTCAA-3′ and reverse,
5′-GCAGGTCAATGTCGGTGTAGC-3′. The PCR primers for tumor necrosis
factor (TNF)-α were: forward, 5′-AGCCAGGAGGGAGAACAGAAAC-3′ and
reverse, 5′-GCCACAAGCAGGAATGAGAAGAG-3′; The PCR primers for IL-1β
were: forward, 5′-ATCTCGCAGCAGCACATCAAC-3′ and reverse,
5′-TAGAGCGTCGTCGTGTAGTTG-3′; The PCR primers for IL-6 were:
forward, 5′-CCACCAAGAACGATAGTCAATTCCA-3′ and reverse,
5′-GGTATCCTCTGTGAAGTCTCCTCTC-3′. PCR cycles were carried out by
initial denaturation at 95°C for 5 min, then running 40 cycles of
95°C for 10 sec and 60°C for 1 min. Duplicate experiments were
conducted to calculate the mean ΔΔCq, mean RQ (fold-change) and
standard deviation.
ELISA
The level of each protein, TGF-β, IL-1β, IL-10, IL-6
and TNF-α, was determined with ELISA, using the following
commercial kits: TGF-β Quantikine ELISA kit (cat. nο. MB100B;
R&D Systems, Inc., Minneapolis, MN, USA), IL-1β Quantikine
ELISA kit (cat. nο. MLB00C; R&D Systems, Inc.), IL-10
Quantikine ELISA kit (cat. nο. M1000B; R&D Systems, Inc.), IL-6
Quantikine ELISA kit (cat. nο. M6000B; R&D Systems, Inc.) and
TNF-α Quantikine ELISA kit (cat. nο. MB100B; R&D Systems, Inc.)
Briefly, 100 µl of each standard and sample were incubated with
pre-coated antibody plates for 2.5 h at RT with gentle agitation.
Following an extensive wash with the provided buffer, 100 µl of
biotinylated antibody were added to the wells for 1 h at RT with
gentle shaking. The amount of cytokine present in the sample or
standard solution present in each well was then determined by
incubating the previous mix with 100 µl of streptavidin solution
for 45 min at RT and then with 100 µl of TMB One-Step substrate
reagent. The colorimetric reaction was stopped after 30 min of
incubation at RT by adding 50 µl of stop solution to each well. The
absorbance at 450 nm was immediately measured using a microplate
reader (Bio-Rad, Hercules, CA, USA).
Statistical analysis
Results were presented as means ± standard error of
the mean, and all the analysis were performed using GraphPad Prism
software (GraphPad Software, Inc., San Diego, CA, USA) and SPSS
17.0 (SPSS, Inc., Chicago, IL, USA). Categorical variables were
compared using Pearson Chi-square test, continuous variables
between groups were compared using Student's t-test or ANOVA. The
numbers of circulating Tregs at different time points were compared
using ANOVA. P<0.05 was considered to indicate a statistically
significant difference.
Results
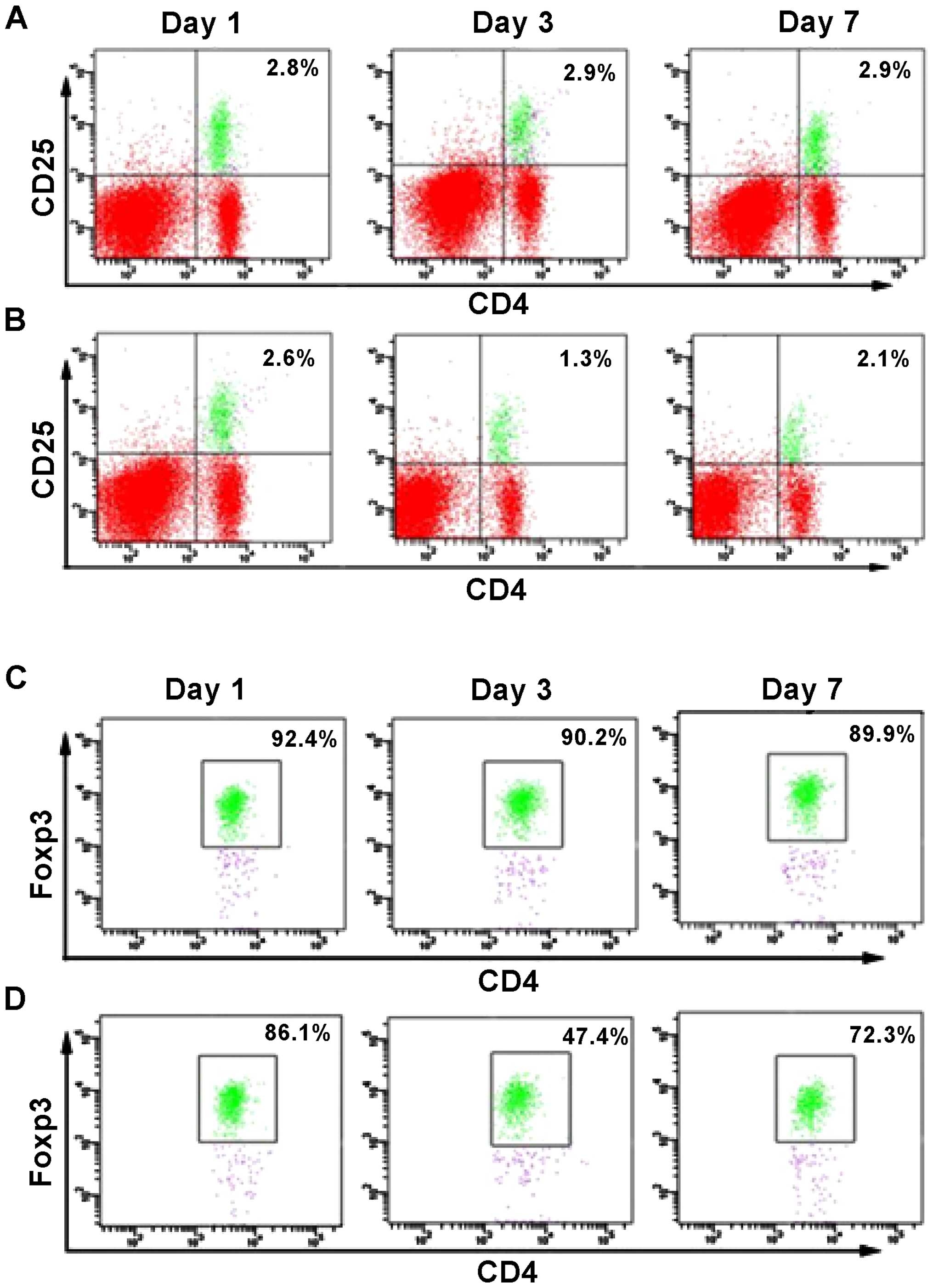
A previous report (12) evidenced a reduced number of
circulating Treg cells when patients suffered from TBI, compared to
healthy individuals. However, it is unknown at which time point the
number of Tregs is significantly affected by the presence of TBI.
Therefore, we conducted flow cytometry analysis to determine the
percentage of CD4+CD25+FoxP3+
cells at different days after the brain injury (Fig. 1B and D), and compared these values
with the number of cells present in the sham control group
(Fig. 1A and C). No significant
change in the percentage of CD4+FoxP3+ and
CD4+CD25+ cells was observed in the sham
group. However, at day 1 after TIB induction, the percentage of
FoxP3+ cells was reduced to 1.3% compared to 2.6%, which
corresponded to the sham group. Similarly, the percentage of
CD4+CD25+ cells was reduced to 47.4%,
compared to 86.1% (sham group). However, the percentage of the two
cell populations increased at day 7 post-induction of TBI, as a
difference with the observed values at day 1. Thus, the results
suggested that between 1 and 7 days following TBI, the number of
FoxP3+ cells was significantly reduced in the mouse
brain, with a difference from the control group. Notably, this
decrease was more pronounced at day 3 post-induction of TBI
(Fig. 1). These results also
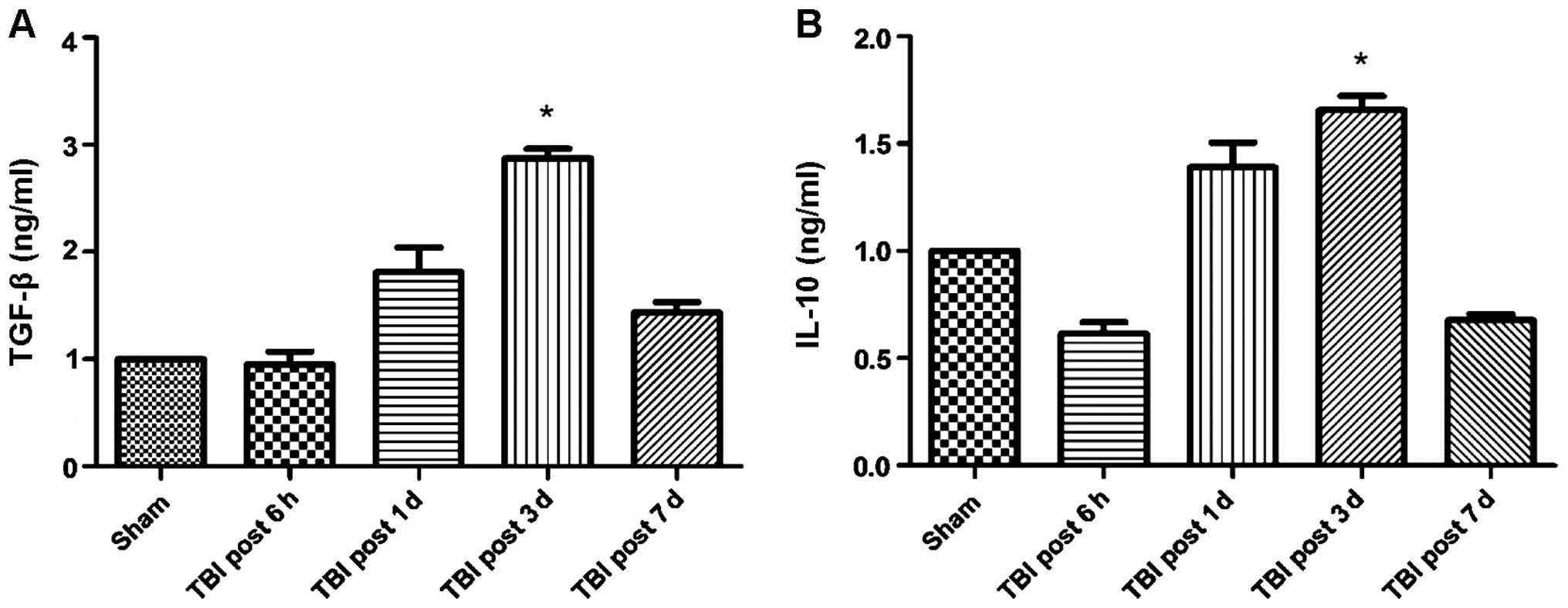
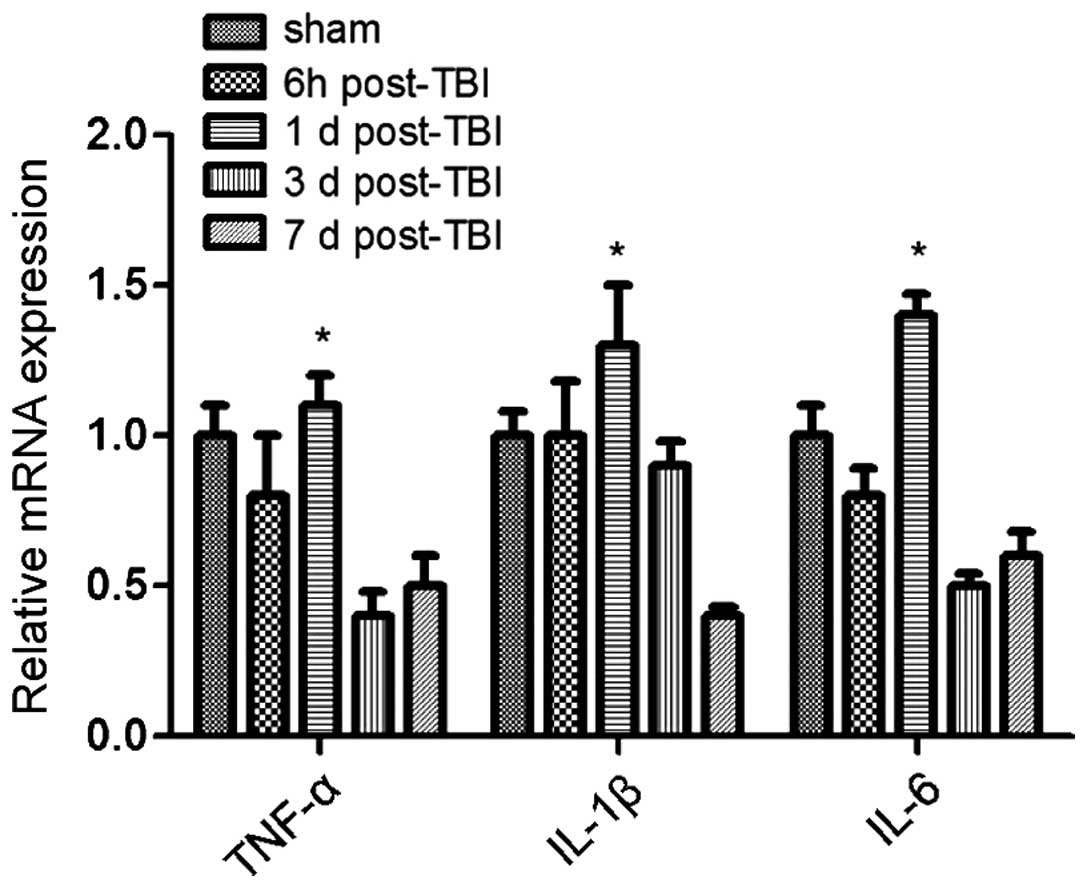
correlated with changes in cytokine levels. Specifically, the
expression of the anti-inflammatory cytokines TGF-β and IL-10
increased over time following TBI induction, whereas the expression
of pro-inflammatory cytokines TNF-α, IL-1β and IL-6 was
significantly reduced (Figs. 2 and
3). Of note, this effect was more
pronounced at 3 days post-induction of TBI, compared to 7 days. In
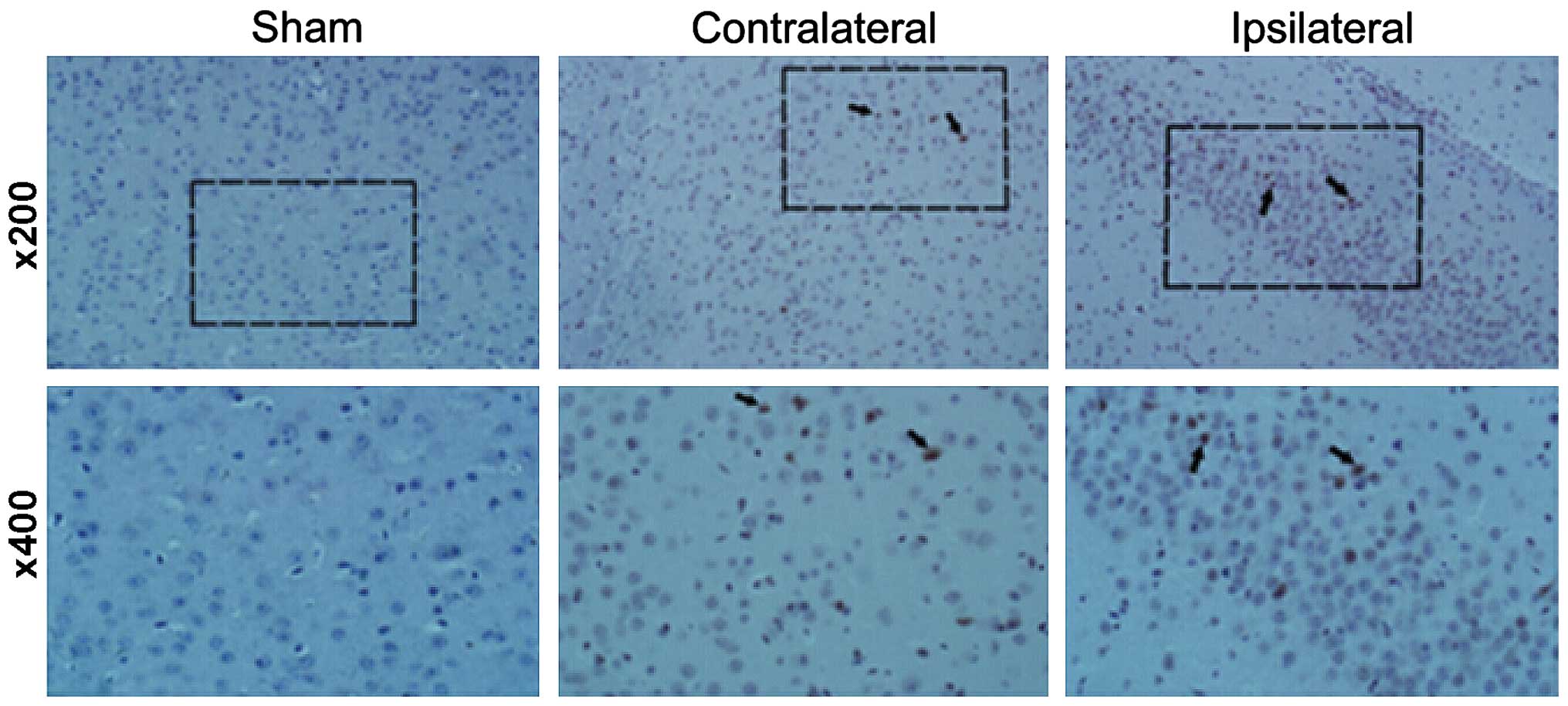
addition, IHC staining for FoxP3 revealed that the expression of
FoxP3 in the two TBI groups, contralateral and ipsilateral
(Fig. 4), were significantly
increased compared with the sham group.
Pre-clinical and clinical studies have reported that
TBI can induce chronic, as well as acute neuro-degeneration. Among
classical chronic neurodegenerative disorders, microglial
activation has been identified to contribute to delayed cell death
and tissue loss. Therefore, we examined the microglia cell
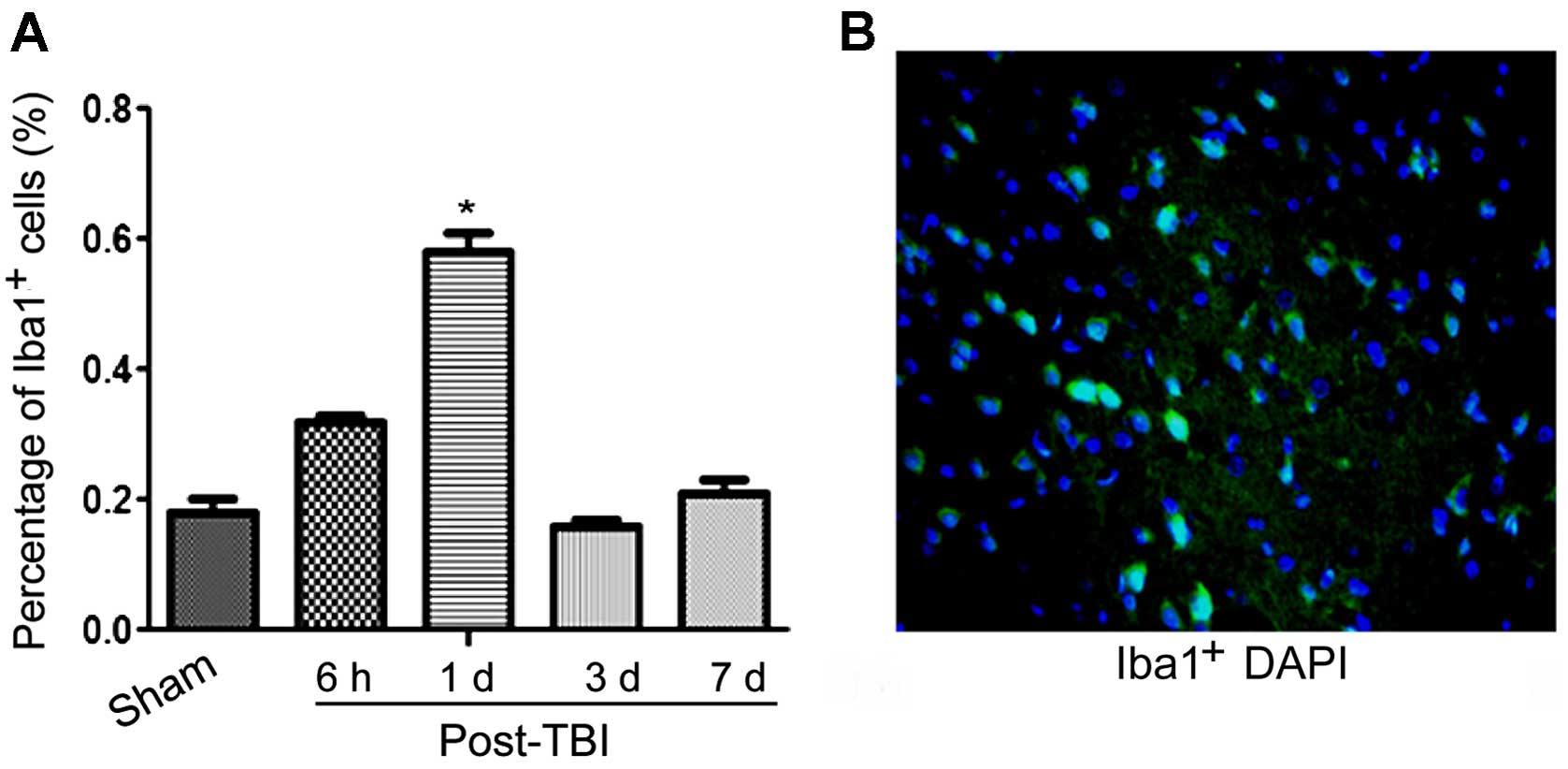
involvement in this condition. As shown in Fig. 5, our data suggested that the
activation of microglia cells was significantly higher at 1 day
post-TBI, compared to the levels observed in the sham group.
In the animal model of cerebral hemorrhage,
investigators found that the proliferation of Tregs after active
immunization can stimulate the secretion of large amount of
anti-inflammatory cytokines, thereby suppressing inflammatory
reactions and mitigating tissue damage. Subsequently, the
compromised neuro-functions were safeguarded effectively. In order
to determine whether Tregs render a neuro-protective effect in the
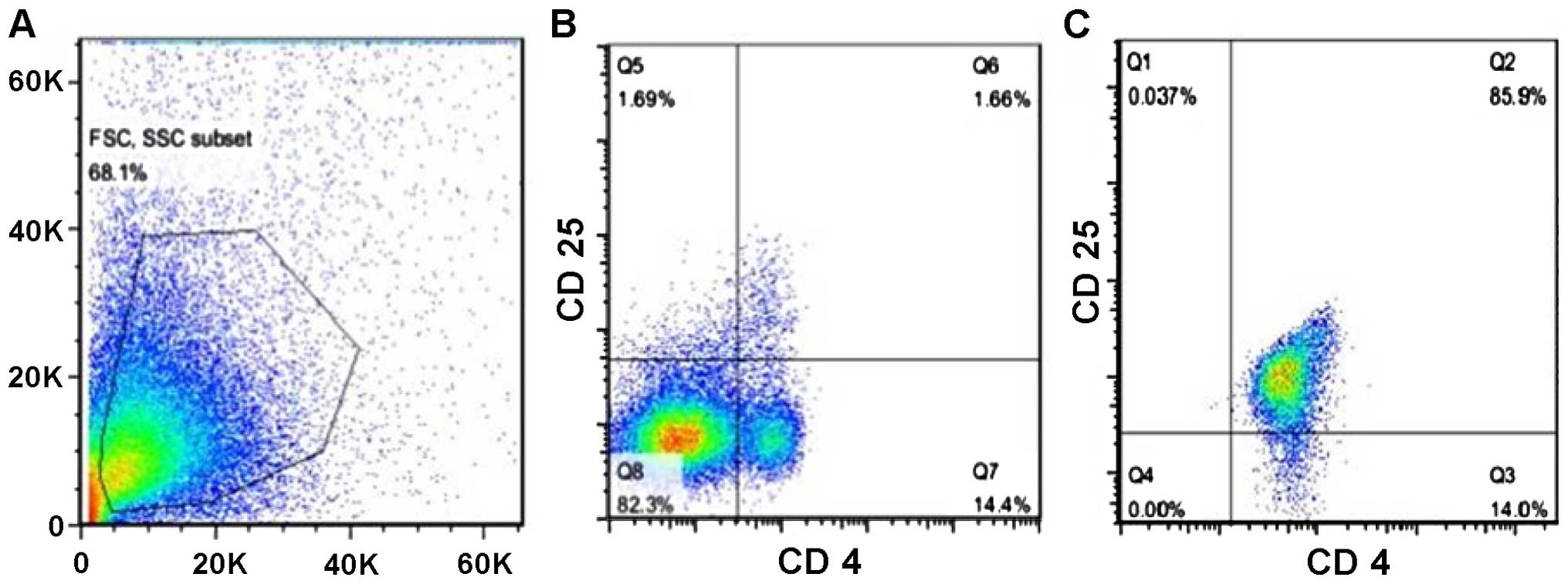
presence of TBI, we isolated Tregs from mouse spleens and enriched
them by magnetic sorting and finally, we injected them back into
the brain of mice from the TBI group. After the isolation and
enrichment, the percentage of Tregs reached 85.9% purity (Fig. 6).
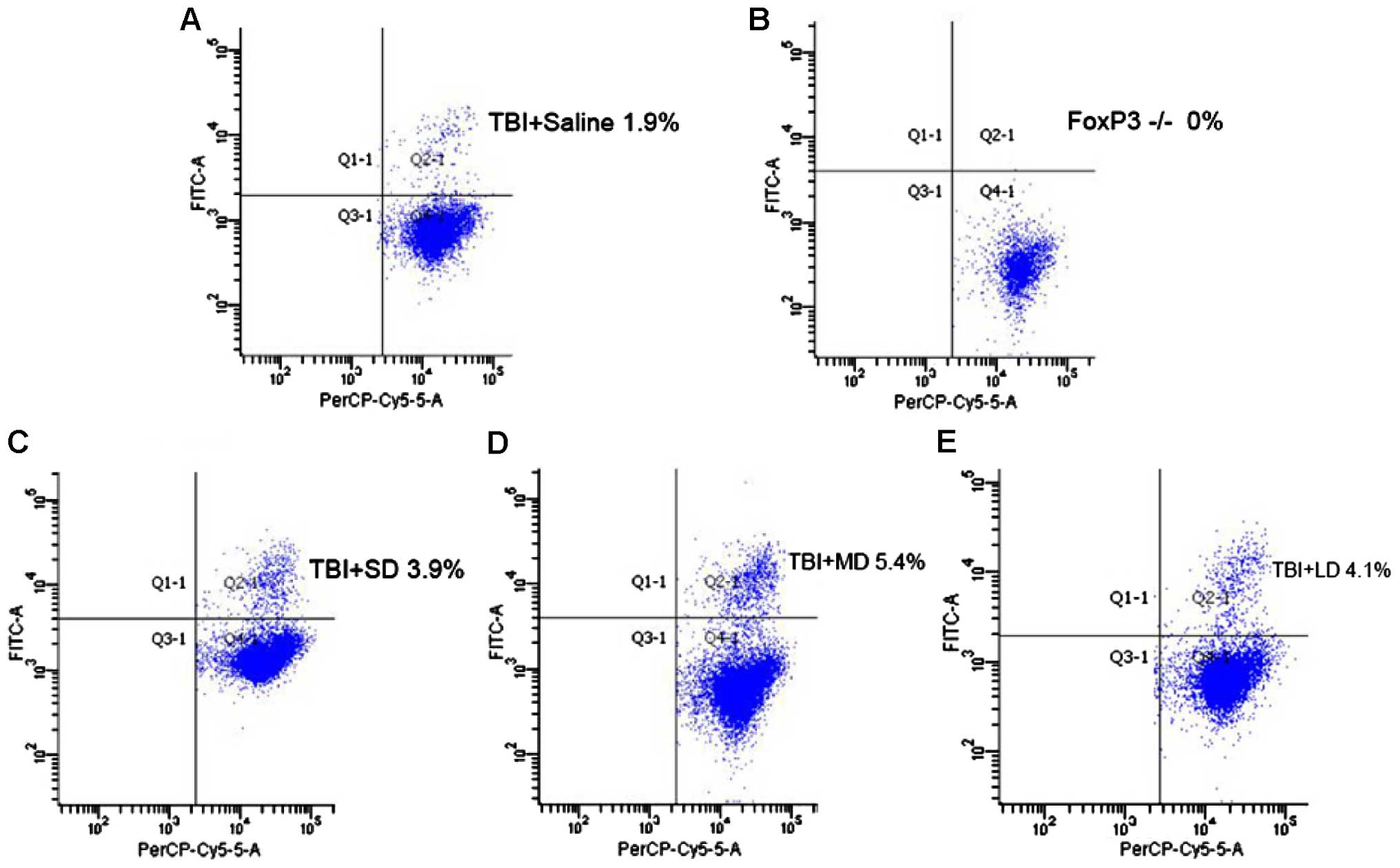
To investigate the role that Tregs play in
immune-modulation, we subdivided mice that received the brain
injury into five groups: i) Saline group; ii) Tregs depletion
group; iii) small-dose (SD) group, treatment with
1.25×105 Tregs; iv) moderate-dose (MD) group, treatment
with 2.5×105 Tregs; and v) large-dose (LD) group,
treatment with 5×105 Tregs. After TBI development and,
following euthanasia, the percentage of Tregs was measured for each
group with flow cytometry. According to Fig. 7, the result showed a significant
increase of Tregs in all the mouse groups, which received Tregs:
3.9% (LD), 5.4% (MD), 4.1% (HD), compared with the saline (1.9%)
and Treg depleted (0%) groups. However, the mouse group that
received intermediate dose of Tregs showed a higher amount of Treg,
rather than the group that received the higher dose.
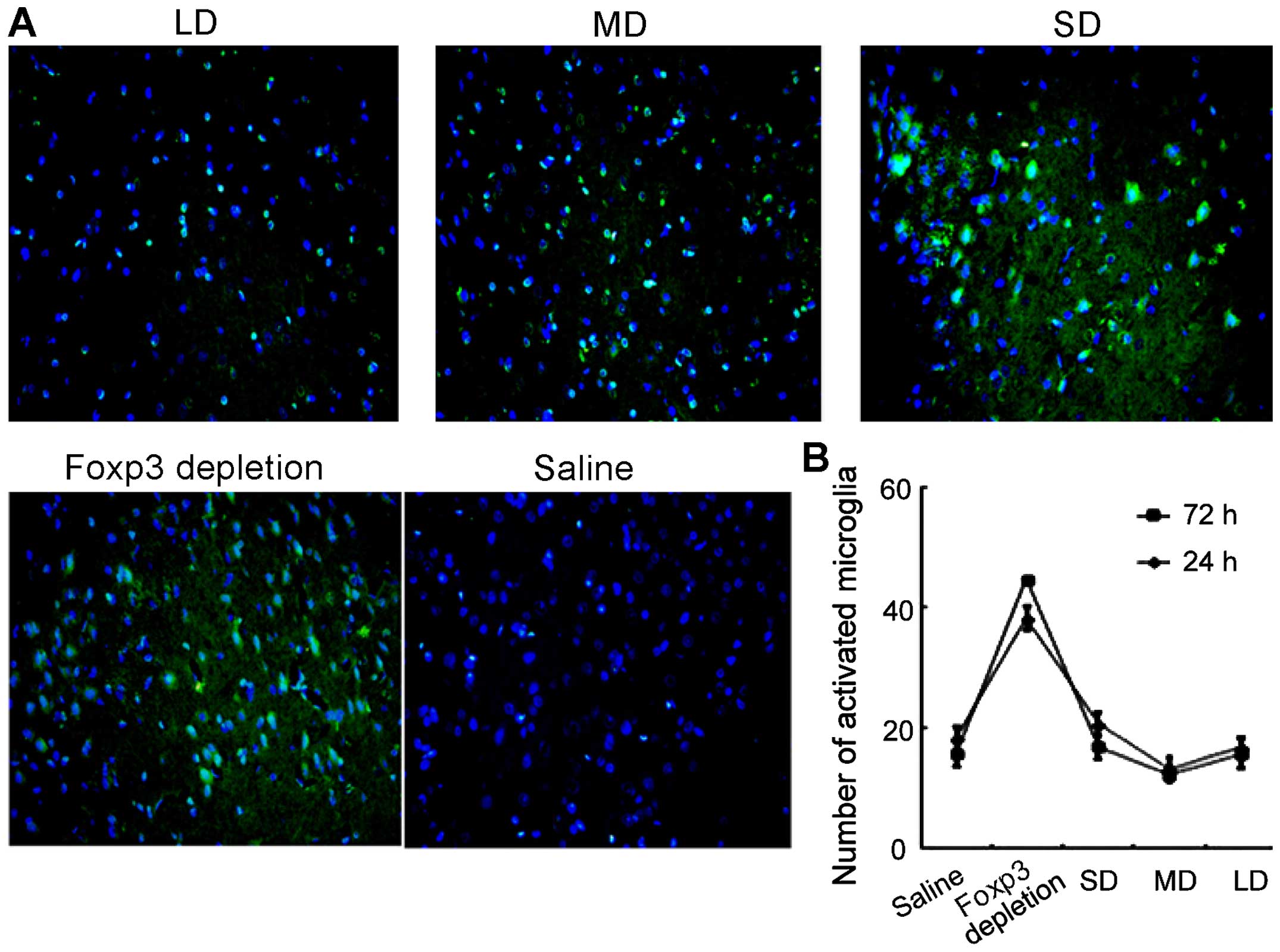
Furthermore, microglia activation was also evaluated
using immunofluorescence. As shown in Fig. 8, levels of microglia activation
were correlated with the expression of Tregs. In FoxP3 depletion
group, the microglia activation level was the highest. However, as
the dose of exogenous injection of Tregs continued to increase, the
activation of microglia cells was suppressed. In the MD group, the
microglia activation level was the lowest.
The degree and progression of inflammatory reactions
were evaluated by measuring the expression levels of pro- and
anti-inflammatory cytokines, using ELISA and PCR. Our results
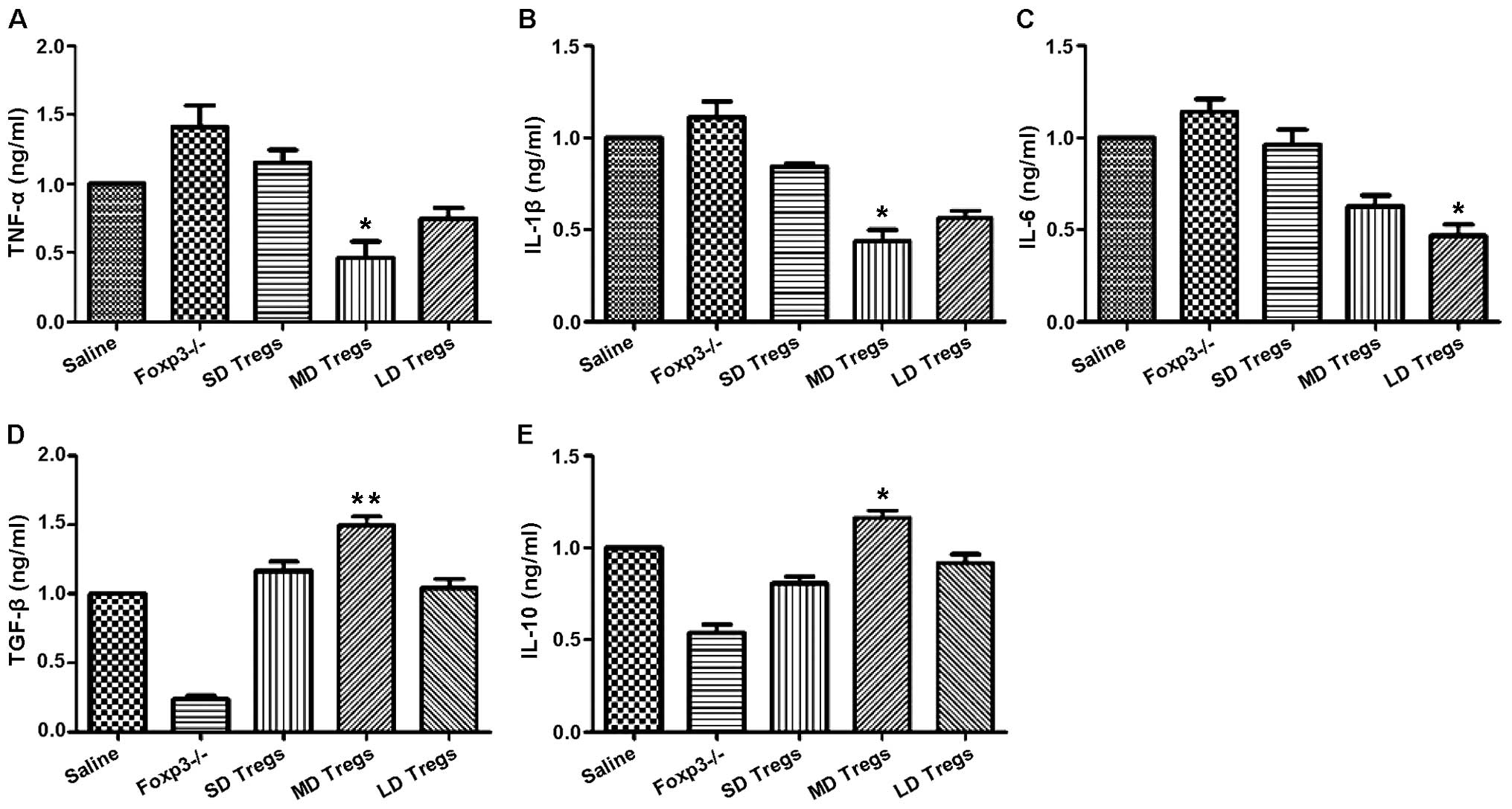
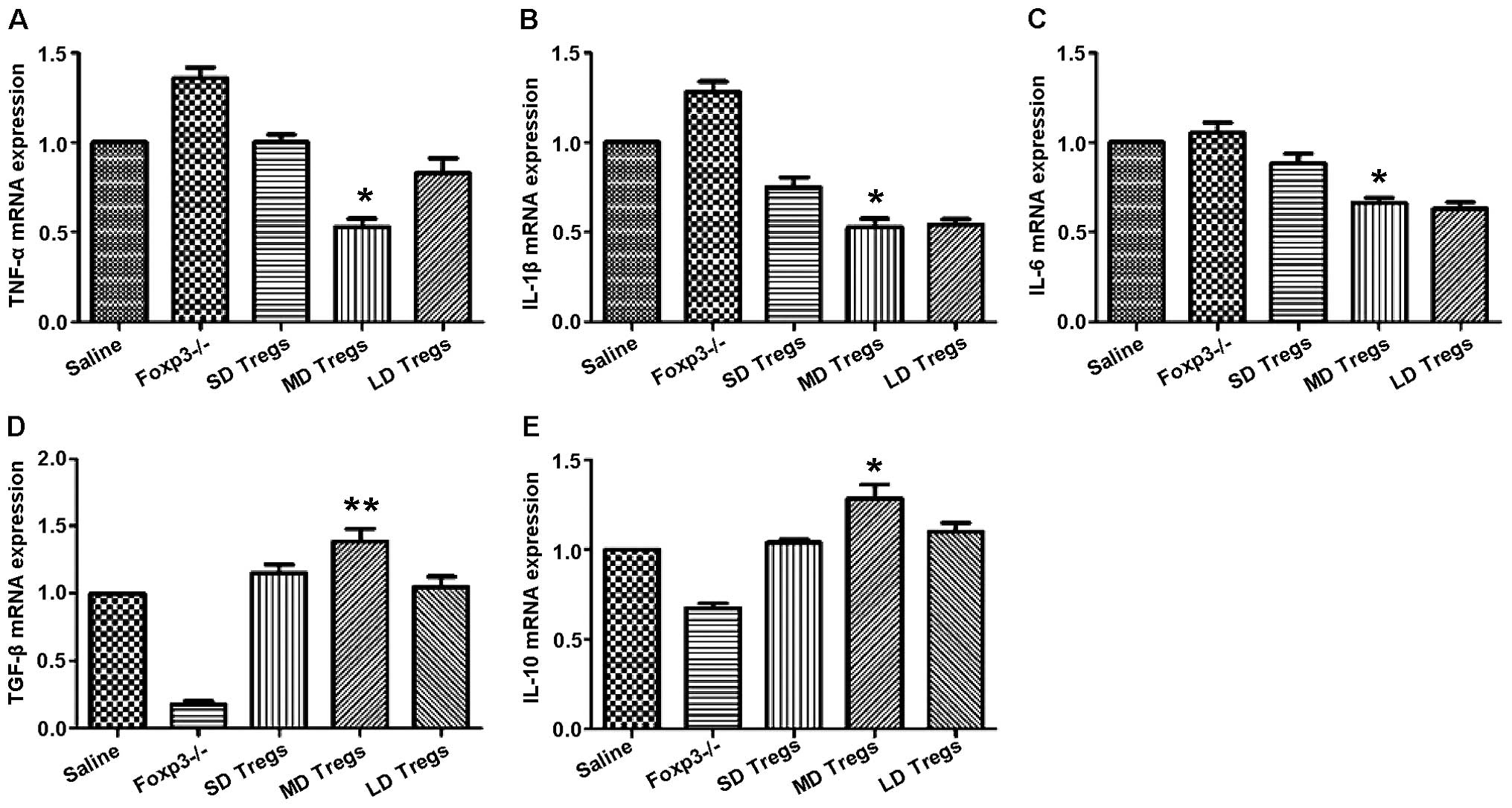
indicated that the expressions of anti-inflammatory cytokines
(TGF-β, IL-10) were significantly higher in MD group compared to
those of other groups (Fig. 9);
the levels of pro-inflammatory cytokines, including IL-6, IL-1β,
TNF-α, were significantly enhanced in FoxP3 depletion group
(Fig. 9).
Previous studies reported that TBI induces a
neuroinflammatory response that involves the infiltration of white
blood cells into the CNS and activation of resident microglia. To
investigate the correlation between microglia activation and TBI,
IHC analysis was conducted in the following group: Saline, Treg
depleted and mice injected with different concentrations of Tregs,
using FoxP3 antibody. The results showed that in the group that
received Treg cells, the activation of microglia was enormously
hampered, compared to the activation in the csaline group and the
Treg depleted group (Fig. 10).
Our data revealed that Tregs inhibited the inflammatory response by
suppressing the microglia activation.
To elucidate the underlying mechanism of the
Treg-mediated immune suppression, we conducted western blot
analysis. Specifically, we investigated the JNK-NF-κB signaling
pathway. Detections of Erk1/2, p38 MAPK, NF-κB and JNK1 at the
protein level were performed. Our results indicated that the
expression levels of p-Erk1/2, p-p38 MAPK, p-NF-κB and p-JNK were
significantly reduced in the groups treated with Tregs, especially
in the MD group (Fig. 11), while
the expression of total Erk1/2, p38 MAPK, NF-κB and JNK1 remained
constant. These results indicated that Tregs suppressed
inflammatory reactions by inhibiting the JNK-NF-κB pathway.
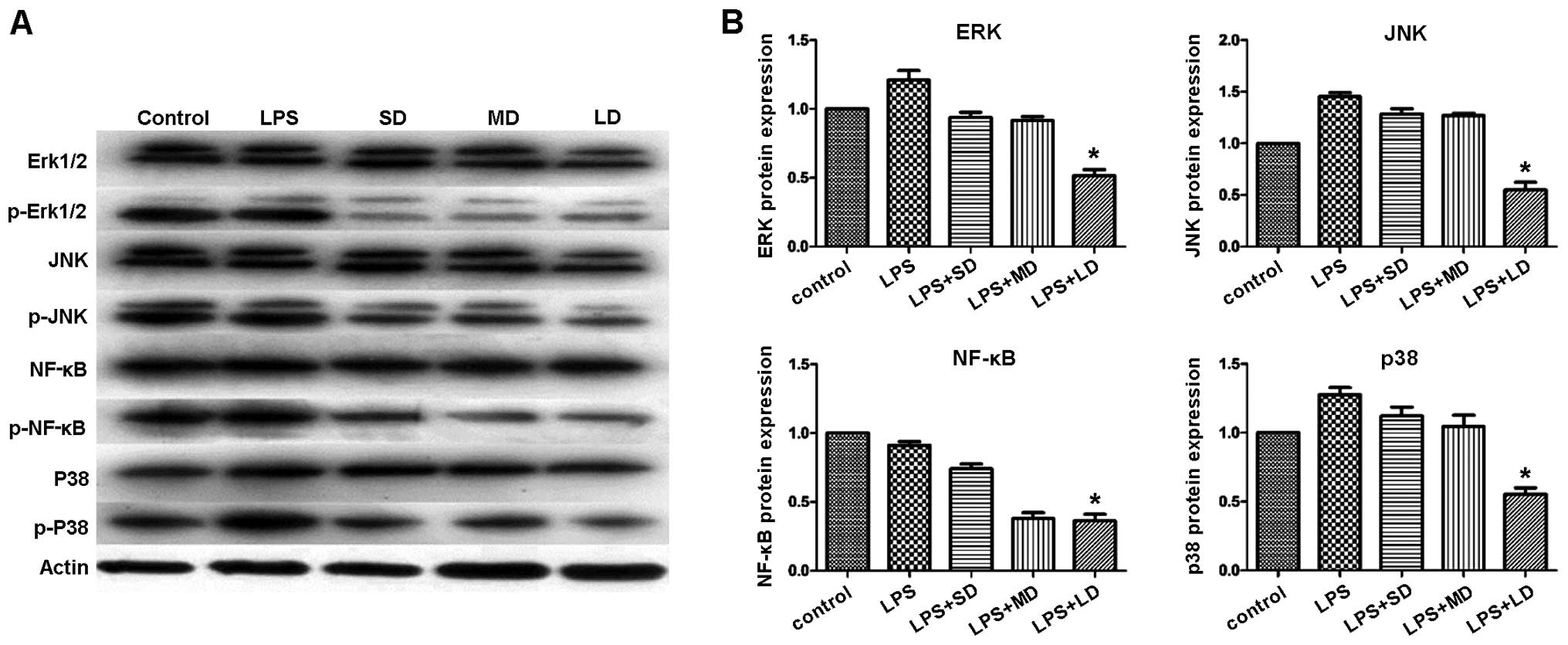 | Figure 11.p-erk1/2, p-p38 MAPK, p-NF-κB and
p-JNK protein expression, respectively, significantly reduced in
the mouse groups treated with Tregs. (A) Western blot analysis
showing the protein expression of p-Erk1/2, p-JNK, p-NF-κB, and
p-p38 MAPK. (B) Quantification of the protein levels for p-Erk1/2,
p-JNK, p-NF-κB, and p-p38 MAPK in the different mouse groups.
*P<0.05. SD, small-dose; MD, moderate-dose; LD, large-dose. |
Discussion
Regulatory pathways mediated by Tregs constitute an
essential homeostatic mechanism of the immune system (7–12).
Distinct Treg subsets were found to coexist in the intestinal
mucosa and mesenteric lymph nodes, including the ‘natural’ and
‘adaptive’ CD4+ forkhead box p3 (FoxP3+)
Tregs, as well as Th1 and Th3 cells. Tregs that develop in the
thymus are commonly known as natural Tregs (nTregs). Previous
studies have reported that Tregs play a role in immunomodulation,
and in the regulation of numerous cytokine expressions and several
signaling pathways (23,24).
Inflammation is characterized by an interplay
modulation of pro- and anti-inflammatory cytokines. Cytokines are
commonly classified as pro-inflammatory: IL-1, TNF, interferon
(IFN)-γ, IL-12, IL-18, whereas, IL-4, IL-10, IL-13, IFN-α and TGF-β
are considered anti-inflammatory cytokines (25–31).
In the present study, the inflammatory response was evaluated by
comparing the expression levels of anti-inflammatory cytokines
(IL-10, TGF-β) and pro-inflammatory cytokines (TNF-α, IL-1β, IL-6)
between TBI and Sham groups, using qPCR and ELISA analysis. Our
data suggested that the expression of IL-10 and TGF-β was
significantly lower in TBI group compared with those in the sham
group, while the expression of TNF-α, IL-1β, IL-6 was significantly
higher in the injured group compared with their expression in the
sham group. However, after increasing the concentration of Tregs in
the TBI group, the expression of IL-10 and TGF-β was enhanced,
while the expression of TNF-α, IL-1β, and IL-6 was decreased,
compared to the sham group (32–36).
Therefore, we concluded that the development of TBI can elicit an
inflammatory reaction, albeit its effect can be neutralized and may
even be reversed by the presence of Tregs.
The CNS is considered an immune-privileged site
because the entry of lymphocytes to this area is tightly controlled
by the endothelial blood-brain barrier (BBB) and blood-spinal cord
barrier (BSCB) (27–29). Under normal conditions, the number
of leukocytes travelling into the CNS is small, however, in the
presence of TBI and its associated secondary inflammatory reaction,
a large number of circulating immunocompetent cells readily gain
access into the CNS. This enhanced vascular permeability and
increased leukocyte cell infiltration into the CNS during TBI is
due to severe BBB and BSCB alterations.
Besides the leukocytes infiltration into the CNS,
inducible Tregs (iTregs) are also developed from naïve CD4 T cells
(nTregs) in the lymphoid tissues (37). This induction takes place in
response to an adequate antigen stimulation and in the presence of
anti-inflammatory cytokines, such as TGF-β and IL-10, while in the
absence of pro-inflammatory cytokines such as IL-6, IL-12 and
IFN-γ. By contrast, after TBI, Treg levels are significantly
increased, possibly due to the trafficking of nTregs and iTregs
into the CNS (22).
Microglia, a common immune cell that resides in the
CNS, has been also involved in the TBI pathophysiology. Microglia
are considered a source of highly cytotoxic substances. By
delivering deriving oxygen-free radicals, subsequent reaction
products, hydrogen peroxide and peroxynitrite, microglia are
potentially able to injure cells, produce oxidative damage and
induce neurodegeneration. Increased immunoreactivity in the brain
was detected after TBI, followed by the accumulation and activation
of microglia cells (25,30).
To investigate the interplay between Tregs and
microglia cells, we conducted an experiment consisting of five
groups: i) Saline (control), ii) TBI, iii) TBI+SD Treg, iv) TBI+MD
Tregs, and v) TBI+LD Tregs. In the TBI group, the maximized effect
of accumulation and activation of microglia cells was observed,
while, in the TBI+SD Treg group, microglia activation induced by
TBI was suppressed, compared to the control group. Similar trend
was detected using the IHC method. Accordingly, qPCR and ELISA
analysis confirmed that the expression of pro-inflammatory
cytokines (TNF-α, IL-1β, and IL-6) in the TBI group were
significantly higher than that in any other group, while the
expression of TNF-α, IL-1β, and IL-6 was significantly decreased in
the TBI+MD Tregs group. The results were consistent with the
results of Sakaguchi (31). Our
experiments showed that Tregs suppress the inflammatory response,
thereby exhibiting neuroprotective effects and improving prognosis
of TBI in the mouse model.
The current study focused on identifying the
immunomodulatory role of Tregs in the mouse model of TBI,
confirming the immune suppressive and neuroprotective functions of
Tregs. The findings of the present study provide theoretically
basis for the development of novel methods for TBI management and
treatment.
Acknowledgements
The present study was supported by GuiZhou Province
Scientific Award for Society Development SY (2012) 3118.
References
|
1
|
Brasure M, Lamberty GJ, Sayer NA, Nelson
NW, Macdonald R, Ouellette J and Wilt TJ: Participation after
multidisciplinary rehabilitation for moderate to severe traumatic
brain injury in adults: a systematic review. Arch Phys Med Rehabil.
94:1398–1420. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Faden AI: Microglial activation and
traumatic brain injury. Ann Neurol. 70:345–346. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Whalen MJ, Carlos TM, Wisniewski SR, Clark
RS, Mellick JA, Marion DW and Kochanek PM: Effect of neutropenia
and granulocyte colony stimulating factor-induced neutrophilia on
blood-brain barrier permeability and brain edema after traumatic
brain injury in rats. Crit Care Med. 28:3710–3717. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ahn MJ, Sherwood ER, Prough DS, Lin CY and
DeWitt DS: The effects of traumatic brain injury on cerebral blood
flow and brain tissue nitric oxide levels and cytokine expression.
J Neurotrauma. 21:1431–1442. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Readnower RD, Chavko M, Adeeb S, Conroy
MD, Pauly JR, McCarron RM and Sullivan PG: Increase in blood-brain
barrier permeability, oxidative stress, and activated microglia in
a rat model of blast-induced traumatic brain injury. J Neurosci
Res. 88:3530–3539. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Liesz A, Suri-Payer E, Veltkamp C, Doerr
H, Sommer C, Rivest S, Giese T and Veltkamp R: Regulatory T cells
are key cerebroprotective immunomodulators in acute experimental
stroke. Nat Med. 15:192–199. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ishibashi S, Maric D, Mou Y, Ohtani R,
Ruetzler C and Hallenbeck JM: Mucosal tolerance to E-selectin
promotes the survival of newly generated neuroblasts via regulatory
T-cell induction after stroke in spontaneously hypertensive rats. J
Cereb Blood Flow Metab. 29:606–620. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shi Y, Fukuoka M, Li G, Liu Y, Chen M,
Konviser M, Chen X, Opavsky MA and Liu PP: Regulatory T cells
protect mice against coxsackievirus-induced myocarditis through the
transforming growth factor β-coxsackie-adenovirus receptor pathway.
Circulation. 121:2624–2634. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Strisciuglio C and van Deventer S:
Regulatory T cells as potential targets for immunotherapy in
inflammatory bowel disease. Immunotherapy. 2:749–752. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liu R, Zhou Q, La Cava A, Campagnolo DI,
Van Kaer L and Shi FD: Expansion of regulatory T cells via
IL-2/anti-IL-2 mAb complexes suppresses experimental myasthenia.
Eur J Immunol. 40:1577–1589. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Meng X, Zhang K, Li J, Dong M, Yang J, An
G, Qin W, Gao F, Zhang C and Zhang Y: Statins induce the
accumulation of regulatory T cells in atherosclerotic plaque. Mol
Med. 18:598–605. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li M, Lin YP, Chen JL, Li H, Jiang RC and
Zhang JN: Role of regulatory T cell in clinical outcome of
traumatic brain injury. Chin Med J (Engl). 128:1072–1078. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sakaguchi S, Sakaguchi N, Asano M, Itoh M
and Toda M: Immunologic self-tolerance maintained by activated T
cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a
single mechanism of self-tolerance causes various autoimmune
diseases. J Immunol. 155:1151–1164. 1995.PubMed/NCBI
|
|
14
|
Sakaguchi S, Miyara M, Costantino CM and
Hafler DA: FOXP3+ regulatory T cells in the human immune
system. Nat Rev Immunol. 10:490–500. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Vignali DA, Collison LW and Workman CJ:
How regulatory T cells work. Nat Rev Immunol. 8:523–532. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yuan R, Maeda Y, Li W, Lu W, Cook S and
Dowling P: Erythropoietin: A potent inducer of peripheral
immuno/inflammatory modulation in autoimmune EAE. PLoS One.
3:e19242008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Dénes A, Humphreys N, Lane TE, Grencis R
and Rothwell N: Chronic systemic infection exacerbates ischemic
brain damage via a CCL5 (regulated on activation, normal T-cell
expressed and secreted)-mediated proinflammatory response in mice.
J Neurosci. 30:10086–10095. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bassil R, Zhu B, Lahoud Y, Riella LV,
Yagita H, Elyaman W and Khoury SJ: Notch ligand delta-like 4
blockade alleviates experimental autoimmune encephalomyelitis by
promoting regulatory T cell development. J Immunol. 187:2322–2328.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Louveau A, Smirnov I, Keyes TJ, Eccles JD,
Rouhani SJ, Peske JD, Derecki NC, Castle D, Mandell JW, Lee KS, et
al: Structural and functional features of central nervous system
lymphatic vessels. Nature. 523:337–341. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sakaguchi S, Ono M, Setoguchi R, Yagi H,
Hori S, Fehervari Z, Shimizu J, Takahashi T and Nomura T:
Foxp3+ CD25+ CD4+ natural
regulatory T cells in dominant self-tolerance and autoimmune
disease. Immunol Rev. 212:8–27. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ghajar J: Traumatic brain injury. Lancet.
356:923–929. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ziebell JM and Morganti-Kossmann MC:
Involvement of pro- and anti-inflammatory cytokines and chemokines
in the pathophysiology of traumatic brain injury.
Neurotherapeutics. 7:22–30. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Scheffold A, Murphy KM and Hofer T:
Competition for cytokines: T(reg) cells take all. Nat Immunol.
8:1285–1287. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang Q, Cui F, Fang L, Hong J, Zheng B
and Zhang JZ: TNF-alpha impairs differentiation and function of
TGF-beta-induced Treg cells in autoimmune diseases through Akt and
Smad3 signaling pathway. J Mol Cell Biol. 5:85–98. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Nagamoto-Combs K, McNeal DW, Morecraft RJ
and Combs CK: Prolonged microgliosis in the rhesus monkey central
nervous system after traumatic brain injury. J Neurotrauma.
24:1719–1742. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Abbott NJ: Inflammatory mediators and
modulation of blood-brain barrier permeability. Cell Mol Neurobiol.
20:131–147. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Amiry-Moghaddam M and Ottersen OP: The
molecular basis of water transport in the brain. Nat Rev Neurosci.
4:991–1001. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Balda MS, Whitney JA, Flores C, González
S, Cereijido M and Matter K: Functional dissociation of
paracellular permeability and transepithelial electrical resistance
and disruption of the apical-basolateral intramembrane diffusion
barrier by expression of a mutant tight junction membrane protein.
J Cell Biol. 134:1031–1049. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Di Giovanni S, Movsesyan V, Ahmed F,
Cernak I, Schinelli S, Stoica B and Faden AI: Cell cycle inhibition
provides neuroprotection and reduces glial proliferation and scar
formation after traumatic brain injury. Proc Natl Acad Sci USA.
102:8333–8338. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ramlackhansingh AF, Brooks DJ, Greenwood
RJ, Bose SK, Turkheimer FE, Kinnunen KM, Gentleman S, Heckemann RA,
Gunanayagam K, Gelosa G, et al: Inflammation after trauma:
Microglial activation and traumatic brain injury. Ann Neurol.
70:374–383. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sakaguchi S: Naturally arising
Foxp3-expressing CD25+CD4+ regulatory T cells
in immunological tolerance to self and non-self. Nat Immunol.
6:345–352. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
32
|
Schwartz RH: Natural regulatory T cells
and self-tolerance. Nat Immunol. 6:327–330. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
La Cava A, Van Kaer L and Fu-Dong-Shi:
CD4+CD25+ Tregs and NKT cells: Regulators
regulating regulators. Trends Immunol. 27:322–327. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Balandina A, Lécart S, Dartevelle P,
Saoudi A and Berrih-Aknin S: Functional defect of regulatory
CD4(+)CD25(+) T cells in the thymus of patients with autoimmune
myasthenia gravis. Blood. 105:735–741. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Fattorossi A, Battaglia A, Buzzonetti A,
Ciaraffa F, Scambia G and Evoli A: Circulating and thymic CD4 CD25
T regulatory cells in myasthenia gravis: Effect of
immunosuppressive treatment. Immunology. 116:134–141. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Liu R, La Cava A, Bai XF, Jee Y, Price M,
Campagnolo DI, Christadoss P, Vollmer TL, Van Kaer L and Shi FD:
Cooperation of invariant NKT cells and
CD4+CD25+ T regulatory cells in the
prevention of autoimmune myasthenia. J Immunol. 175:7898–7904.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ji J and Cloyd MW: HIV-1 binding to CD4 on
CD4+CD25+ regulatory T cells enhances their suppressive function
and induces them to home to, and accumulate in, peripheral and
mucosal lymphoid tissues: An additional mechanism of
immunosuppression. Int Immunol. 21:283–294. 2009. View Article : Google Scholar : PubMed/NCBI
|