Introduction
Acute lung injury (ALI) and acute respiratory
distress syndrome are well-defined and readily recognized clinical
disorders caused by numerous clinical insults to the lung or due to
predispositions to lung injury (1–3). ALI
is a frequent complication following sepsis in critically ill
patients and lipopolysaccharide (LPS) is thought to be the most
important pathogen that leads to the development of ALI in sepsis
(4,5).
Mitochondrial damage is associated with numerous
human diseases, including ALI (6–9).
Mitochondria are involved and serve a central role in the
integration and circulation of death signals initiating inside the
cells, including oxidative stress and DNA damage, and in regulating
cell death/apoptosis pathways (10–12).
Mitochondrial regulation of apoptosis is predominantly mediated via
the release of cytochrome c, apoptosis-inducing factor
(AIF), and second mitochondria-derived activator of caspases (Smac)
and ultimately caspase activation (13–15).
Dexmedetomidine (DEX), a highly selective and potent
α2-adrenoreceptor agonist, provides excellent sedation and
analgesia with minimal cardiovascular effects. Previous studies
have shown that DEX attenuates LPS, ischemia-reperfusion and
ventilator-induced lung injury in animal models; however, the
mechanism remains unclear (16–19).
Additionally, the anti-inflammatory and antiapoptotic effects of
DEX have been demonstrated in previous studies (20–23).
DEX has also been reported to have an effect on mitochondrial
permeability transition pore (mPTP) in neutrophil and isolated rat
hearts following ischemia/reperfusion injury (24–26).
The present study hypothesized that DEX may provide protective
effect against LPS-induced ALI by alleviating oxidative stress,
mitochondrial damage and mitochondria-dependent apoptosis.
Materials and methods
Animals
All experimental protocols were approved by the
Animal Care and Use Committee of the Southern Medical University
(Guangzhou, China). The care of animals was in accordance with the
National Institute of Health guidelines and with those of the
Chinese National guidelines. A total of 24 Male Sprague-Dawley rats
(weight, 180–220 g) were purchased from the Experimental Animal
Center at South Medical University (Guangzhou, China) and were
allowed to acclimate for 1 week prior to experiments. Animals were
housed in individual cages in a temperature-controlled (25±2°C)
room, under a 12 h light/dark cycle, with ad libitum access
to food and water.
Animal model
As previously described (27), ALI was induced by intratracheal
administration of LPS. Briefly, the animals were intramuscularly
anesthetized with an injection of sodium pentobarbital (30 mg/kg).
The rats were placed in a supine position on a warming device and
the trachea was surgically exposed by a cervical middle line
incision in the skin. The rats were subsequently challenged
intratracheally with either 0.5 ml sterile normal saline (NS) alone
or 0.5 ml NS with LPS (5 mg/kg body weight; Escherichia coli
0111:B4; Sigma-Aldrich; Merck Millipore, Darmstadt, Germany)
stabbing the trachea with a microsyringe. Prior to ALI induction,
the rats were pretreated with DEX (10 or 50 µg/kg) for 30 min. NS
was used as the vehicle control.
Cell culture and stimulation
Rat type I alveolar epithelial cells (AECs) were
obtained from ScienCell (San Diego, CA, USA) and grown at 37°C in
5% CO2 in Dulbecco's modified Eagle medium (DMEM,
Sigma-Aldrich; Merck Millipore), containing low glucose, penicillin
(100 U/ml, Sigma-Aldrich; Merck Millipore), streptomycin (100
units, Sigma-Aldrich; Merck Millipore) and 10% bovine serum
(Sigma-Aldrich; Merck Millipore). The AECs were respectively
pretreated with 10 and 50 µg/l DEX for 30 min, followed by
stimulation of 5 µg/ml LPS for 12 h. DMEM was used as a vehicle
control.
Experimental design
In vivo, rats were intramuscularly
anesthetized with an injection of sodium pentobarbital (30 mg/kg),
and were then sacrificed by decollation at 24 h post-LPS treatment.
The lung injury oxygenation index
(PaO2/FIO2), histopathological changes, lung
microvascular permeability and wet-to-dry (W/D) weight ratio were
measured. To investigate the underlying mechanisms of DEX treatment
in LPS-induced ALI, terminal deoxynucleotidyl transferase dUTP nick
end labeling (TUNEL) staining, serum lipid peroxidation, caspase 3
activity, and the expression levels of Bax, Bcl-2 and cleaved
caspase 3 were investigated.
In vitro, following 12 h stimulation,
oxidative stress, as determined by reactive oxygen species (ROS)
production, mitochondrial function, as determined by mitochondrial
membrane potential (MMP) and cellular ATP, and
mitochondria-dependent apoptosis, as determined by cell apoptotic
rate, cytochrome c release and caspase-3 activity were
detected.
Histological examination
The lungs were harvested 24 h following LPS
injection. The right middle lobes of the lungs were fixed with 10%
formalin, embedded in paraffin and sectioned (4 µm thickness).
Following deparaffinization and rehydration, the tissue sections
were stained with hematoxylin and eosin. The pathological sections
were observed and assessed in a blinded manner.
Lung microvascular permeability
assay
The permeability assay was performed, as previously
described (4). Briefly, Evans blue
(EB; 20 mg/kg; Sigma-Aldrich) was injected intravenously through
the femoral vein. After 30 min, the animals were intramuscularly
anesthetized with an injection of sodium pentobarbital (30 mg/kg),
and were then sacrificed by decollation, and a midline thoracotomy
was performed. The superior and inferior vena cava were
subsequently ligated, the aorta was transected, and 20 ml normal
saline solution was injected into the right ventricle at a pressure
of 20 cm H2O to wash out the pulmonary intravascular
content. A sample of lung tissue was weighed, homogenized and
immersed in N,N-dimethylformamide (Sigma-Aldrich). The
homogenate was incubated at room temperature for 48 h. Eluted EB
was measured at 620 nm using an automatic microplate reader
(SpectraMax M5; Molecular Devices, Sunnyvale, CA, USA), and the
quantity was expressed as µg/100 mg dry tissue.
Measurement of lung W/D weight
ratio
The harvested wet lung was weighed and subsequently
placed in an oven for 48 h at 80°C, followed by weighing when it
was dried. The lung W/D weight ratio was calculated.
Oxygenation index
(PaO2/FiO2)
analysis
At 24 h following ALI (or sham), animals were
anesthetized and administered endotracheal intubation with a
20-gauge catheter. The animals were mechanically ventilated with
pure oxygen at 7 ml/kg (120 breaths/min). Following 20 min
ventilation, the arterial blood was obtained from the carotid
artery and measured using a commercial blood gas analyzer (ABL8000;
Radiometer Copenhagen, Copenhagen, Denmark).
Measurement of mitochondrial membrane
potential (MMP) in vitro
The MMP was determined using the potential-sensitive
fluorescent dye, JC-1. This dual-emission probe changes color from
red-orange to green as the mitochondrial membrane depolarizes. The
JC-1 (5 µmol/l) was loaded onto AECs for 15 min at 37°C. The
results were visualized using a confocal microscope (×650; LSM 780
NLO; Carl Zeiss, Jena, Germany). Alternatively, the fluorescence
intensity was monitored using flow cytometry (BD Immunocytometry
Systems, Franklin Lakes, NJ, USA) at an excitation/emission of
525/590 nm.
Measurement of cellular ATP in
vitro
Intracellular ATP was determined using a
luciferase-based assay (CellTiter-Glo, Madison, WI, USA), according
to the manufacturer's protocol. Following the addition of 100 µl
CellTiter-Glo reagent to 100 µl cell suspension containing 10,000
cells in each well of a standard opaque-walled 96-well plate, the
plates were allowed to incubate at room temperature for 10 min, and
the luminescence was recorded in an automatic microplate reader
(SpectraMax M5; Molecular Devices).
TUNEL staining
Lung histopathological slides were dewaxed and
incubated with proteinase K. The slides were stained using a TUNEL
kit (Biovision, Mountain View, CA, USA), according to the
manufacturer's instructions. Subsequently, the cells were
counterstained with Hoechst 33258 (Sigma-Aldrich) and examined
under a fluorescence microscope (ECLIPSE FNl, Nikon).
Measurement of cell apoptosis in
vitro
Cell apoptosis was detected using an annexin V-FITC
apoptosis detection kit (BD Biosciences, Franklin Lakes, NJ, USA).
Following induction with H2O2, the cells
(~1×105 cells/ml) were washed twice with PBS and
suspended in 1X binding buffer. A total of 5 µl annexin V-FITC and
10 µl propidium iodide (PI; 50 µg/ml; Sigma-Aldrich) was added to
the cell suspension. Following incubation at room temperature for
20 min in the dark, the fluorescence of the cells was determined
immediately using a flow cytometer (BD Immunocytometry
Systems).
Measurement of cytosolic cytochrome c
in vitro
Cytosolic cytochrome c content was estimated
using a cytochrome c ELISA kit (Biovision), as described
previously (28). The cell
homogenates were centrifuged (10,000 × g for 60 min at 4°C)
and the supernatant (cytosolic fraction) was collected and
subjected to protein estimation using the bicinchoninic acid method
(BCA). The samples were treated with a conjugate reagent,
transferred to a cytochrome c antibody-coated microwell
plate and incubated at room temperature for 60 min. The wells were
washed and treated with a substrate and incubated for 30 min at
room temperature, followed by the addition of a stop solution. The
optical density was measured at 450 nm using an automatic
microplate reader (SpectraMax M5). A serial dilution of cytochrome
c calibrator was subjected to the assay, along with the
samples. The values were plotted and the concentration of
cytochrome c was calibrated from the standard curve.
Measurement of caspase 3 activity in
vitro
Caspase 3 activity was determined using a caspase 3
activity assay kit, according to the manufacturer's instructions
(Biovision). The cells were lysed in caspase 3 sample lysis buffer.
The homogenates were centrifuged at 10,000 × g and the
supernatant was collected for protein estimation using BCA and for
the caspase 3 assay. The cell lysates were exposed to the DEVD
substrate conjugate provided in the kit. The sample was measured in
an automatic microplate reader (SpectraMax M5) at excitation of 400
nm and emission of 505 nm.
Western blot analysis for Bax, Bcl-2
and cleaved caspase 3 in vivo
The lung tissues were homogenized and analyzed by
western blotting. Protein concentrations were determined using the
BCA method. An equal quantity of protein was loaded onto 10% sodium
dodecyl sulphate-polyacrylamide gels for electrophoresis. Following
electrophoresis, the proteins were electroblotted onto
polyvinylidene difluoride membranes. Membranes were blocked with
blocking solution (5% skimmed milk diluted with PBS) at room
temperature for 2 h, followed by incubation with primary antibodies
against β-actin (1:5000 dilution; cat. no. ab8227; Abcam,
Cambridge, UK), Bax (1:1,000 dilution; cat. no. ab32503; Abcam),
Bcl-2 (1:1,000 dilution; cat. no. ab59348; Abcam) and cleaved
caspase 3 (1:1,000 dilution; cat. no. ab13847; Abcam) overnight at
4°C. The membranes were subsequently incubated with horseradish
peroxidase-conjugated secondary antibody (1:5,000, cat. no. ab6721,
Abcam), and the protein expression was detected using an enhanced
chemiluminescence reagent [cat. no. abs920B-500, Absin
Biotechnology Co., Ltd., Shanghai, China].
Measurement of ROS levels in
vitro
Intracellular ROS levels were assessed using a
DCFH-DA probe (Sigma-Aldrich). The cells were treated with DCFH-DA
(10 µM) after LPS stimulation for 20 min at 37°C. Following
incubation, the cells were washed and analyzed using an automatic
microplate reader (SpectraMax M5). The relative intensity of DCF
fluorescence was determined at a wavelength of 535 nm, as compared
with the sham group cells.
Measurement of lipid peroxides in
vivo
The serum was obtained as described above. Lipid
peroxides (LPOs) were measured using a commercially available kit
(Cayman Chemical Co., Ann Arbor, MI, USA), according to the
manufacturer's protocol. The LPO content was measured at 500 nm
using an automatic microplate reader (SpectraMax M5).
Statistical analysis
All variables are presented as the mean ± standard
deviation. Differences between the groups were determined using
one-way analysis of variance with the least significant difference
multiple-comparison test and Student's t-test when appropriate.
P<0.05 was considered to indicate a statistically significant
difference.
Results
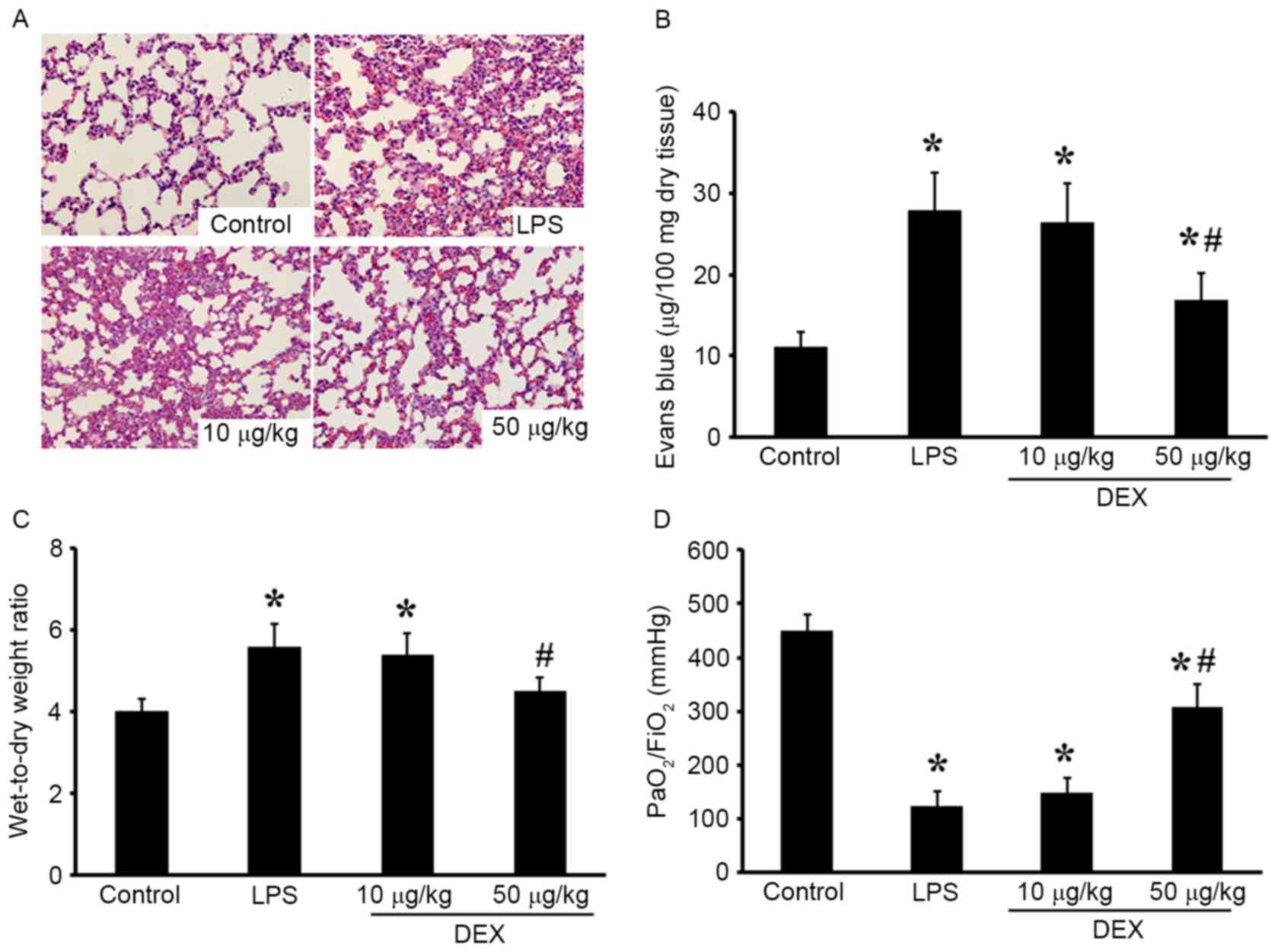
DEX attenuates LPS-induced ALI in
rats
The present study examined the effects of DEX on
LPS-induced ALI in rats. Lung histological sections of
LPS-challenged rats exhibited accumulation of a large number of
neutrophils in the intra and interalveolar space, a thickened
alveolar wall, less alveolar space and interstitial congestion, and
these alterations were markedly attenuated by pretreatment with DEX
(50 µg/kg) (Fig. 1A).
Additionally, the lung microvascular permeability, reflected by EB
content in lung tissue and W/D weight ratio of the lung were
increased in LPS-challenged rats compared with the control animals.
DEX pretreatment reduced the EB content in lung and the W/D weight
ratio of lung compared with the LPS group (P<0.05 compared with
the LPS group for 50 µg/kg DEX group; P>0.05 compared with the
LPS group for 10 µg/kg DEX group; Fig.
1B-D). In addition, the
PaO2/FiO2 was
significantly decreased in LPS-challenged rats, which was improved
by DEX treatment (P<0.05 compared with the LPS group for 50
µg/kg DEX group; P>0.05 compared with the LPS group for 10 µg/kg
DEX group; Fig. 1D). These results
indicated that DEX pretreatment attenuated LPS-induced ALI in
rats.
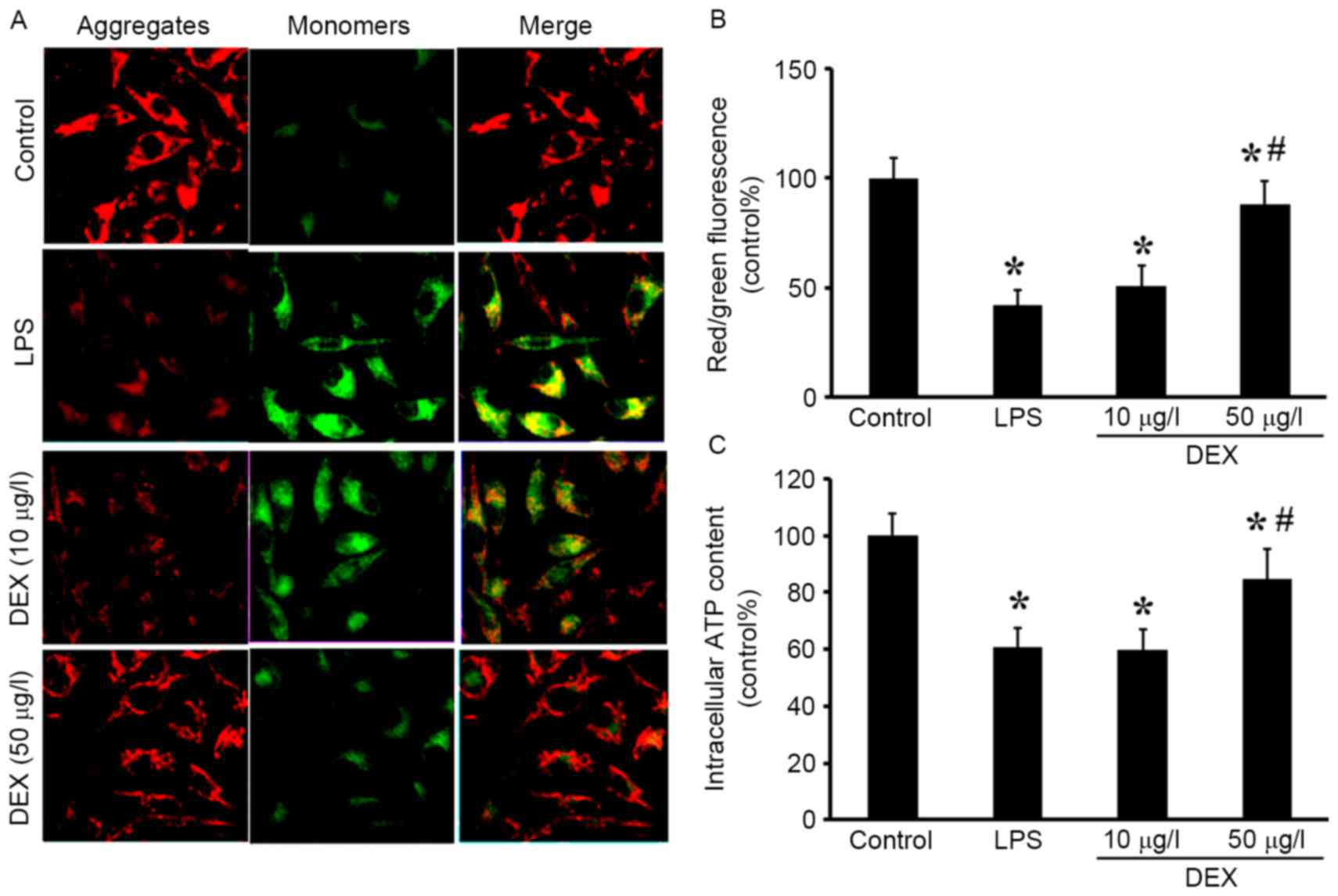
DEX prevents LPS-induced mitochondrial
dysfunction
To establish whether DEX attenuated LPS-induced
mitochondrial dysfunction, the level of MMP and cellular ATP levels
were evaluated. JC-1, the potential-sensitive fluorescent dye,
forms aggregates in normally polarized mitochondria and monomers in
damaged and depolarized mitochondria. The color of this
dual-emission probe changed from red-orange to green as the
mitochondrial membrane turned depolarized. As shown in Fig. 2A and B, the control cells were
clearly red. However, LPS exposure rapidly caused MMP dissipation,
as shown by the increase in green fluorescence and the concomitant
disappearance of red fluorescence. Pretreatment with DEX
significantly attenuated the changes in MMP, as indicated by the
repression of green fluorescence and restoration of red
fluorescence (P<0.05 compared with the LPS group for 50 µg/l DEX
group; P>0.05 compared with the LPS group for 10 µg/l DEX
group). In addition, the intracellular ATP level was decreased by
LPS stimulation compared with the control group, indicating
mitochondrial dysfunction in the AECs. Following pretreatment with
the DEX, the ATP levels were increased compared with the LPS group
(P<0.05 compared with the LPS group for 50 µg/l DEX group;
P>0.05 compared with the LPS group for 10 µg/l DEX group;
Fig. 2C).
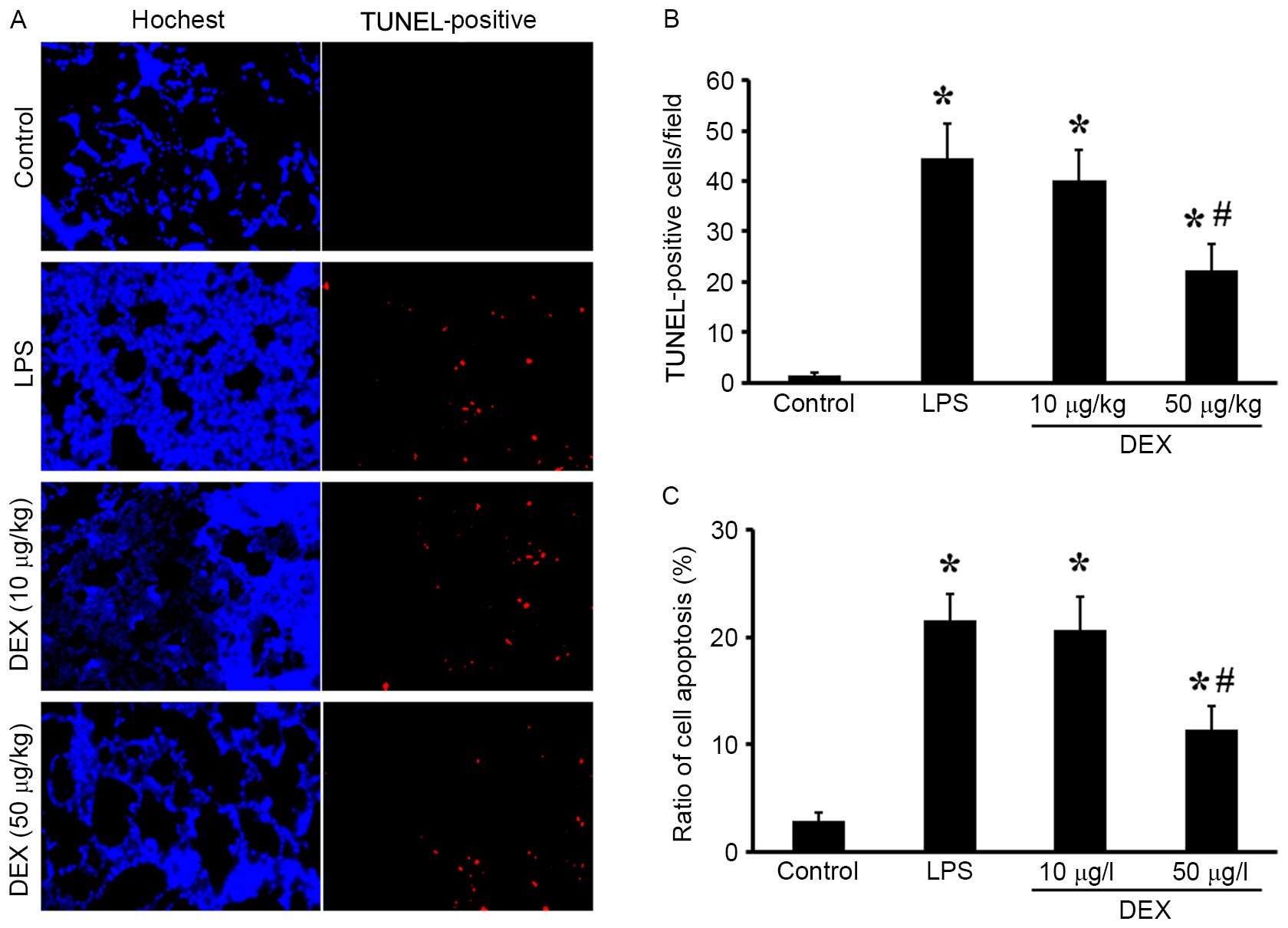
DEX inhibits the LPS-induced
mitochondrial-dependent apoptosis
To determine the protective effects of DEX against
LPS-induced apoptosis, TUNEL and annexin-V/PI double staining
assays were performed. In vivo, LPS-challenged animals
exhibited a significant increase in red marked apoptotic cells,
which was reduced by DEX (50 µg/kg) pretreatment (Fig. 3A and B). In vitro, the rates
of cell apoptosis were markedly increased following LPS exposure,
compared with the control group, and were subsequently reduced by
DEX pretreatment (P<0.05 compared with the LPS group for 50 µg/l
DEX group; P>0.05 compared with the LPS group for 10 µg/l DEX
group; Fig. 3C).
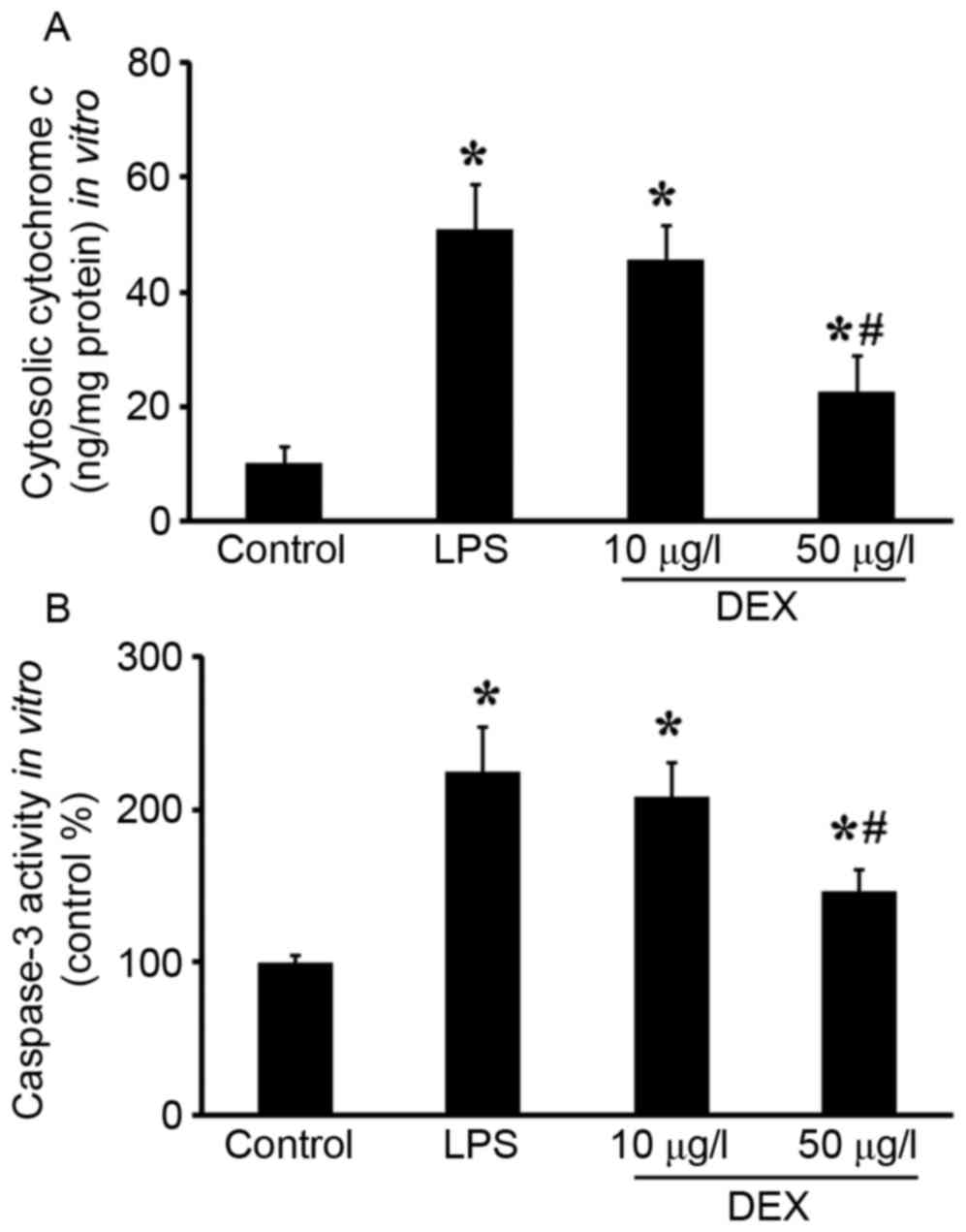
Mitochondrial regulation of apoptosis is mediated
through the release of cytochrome c, AIF and Smac, and
ultimately caspase activation. In present study, cytochrome
c release, caspase 3 activity and cleaved caspase 3
expression were detected. As shown in Fig. 4, increased cytosolic cytochrome
c and caspase 3 activities were observed in vitro,
which was reduced by DEX pretreatment (P<0.05 compared with the
LPS group for 50 µg/l DEX group; P>0.05 compared with the LPS
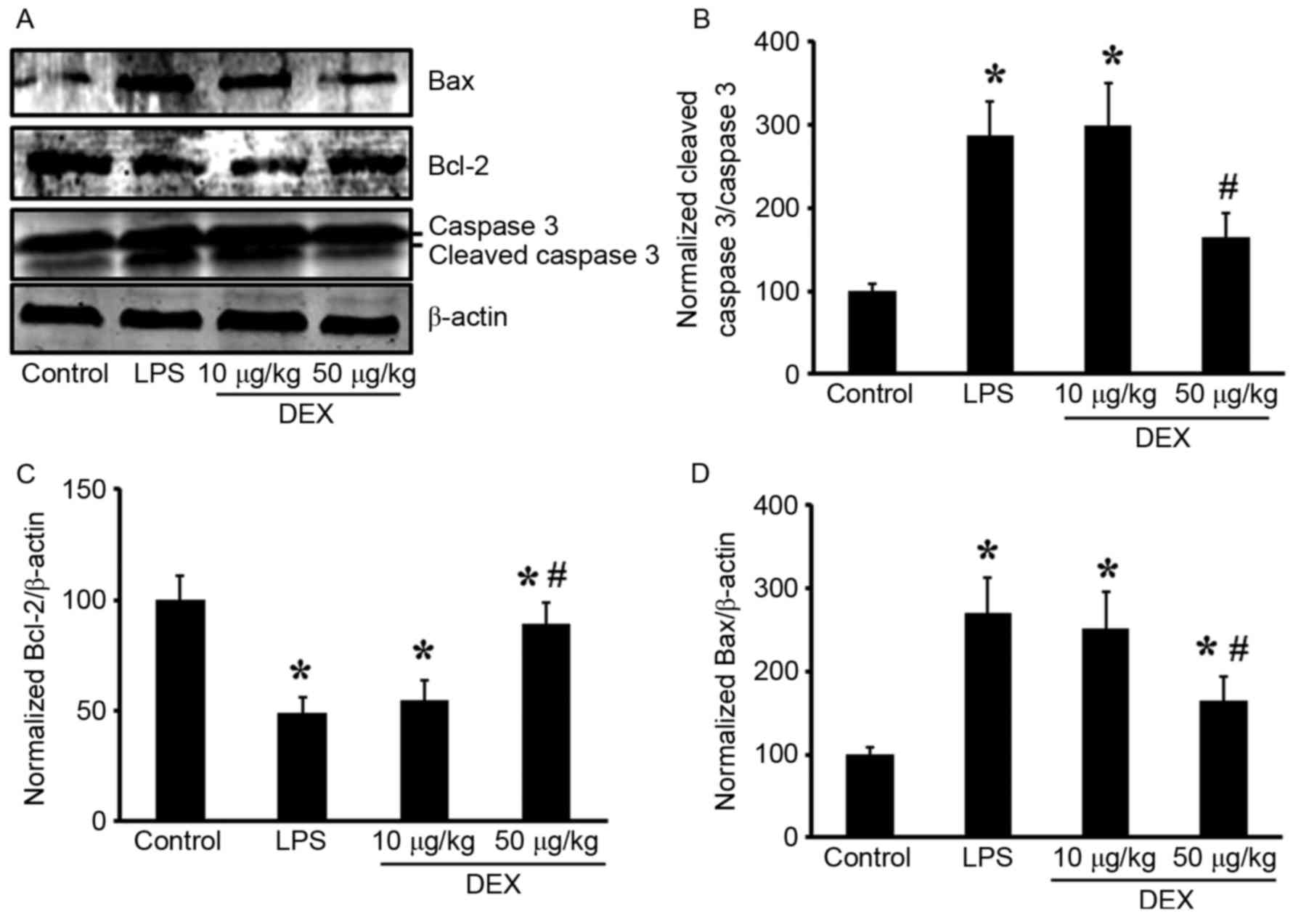
group for 10 µg/l DEX group). In the lungs, pretreatment with DEX
(50 µg/kg) inhibited LPS-induced cleaved caspase 3 upregulation
(Fig. 5A and B; P<0.05 compared
with the LPS group for 50 µg/kg DEX group; P>0.05 compared with
the LPS group for 10 µg/kg DEX group).
Additionally, the present study investigated the
changes in expression levels of the Bcl-2 family of proteins (Bax
and Bcl-2) in lung tissue. LPS inhalation resulted in
downregulation of the Bcl-2 protein, and DEX prevented this
decrease (P<0.05 compared with the LPS group for 50 µg/kg DEX
group; P>0.05 compared with the LPS group for 10 µg/kg DEX
group; Fig. 5A and C). Expression
of the pro-apoptotic protein Bax was upregulated by LPS induction
and inhibited by DEX pretreatment (P<0.05 compared with the LPS
group for 50 µg/kg DEX group; P>0.05 compared with the LPS group
for 10 µg/kg DEX group; Fig. 5A and
D). These results indicated that intratracheal administration
LPS induced lung cell apoptosis, which can be significantly
alleviated by treatment with DEX.
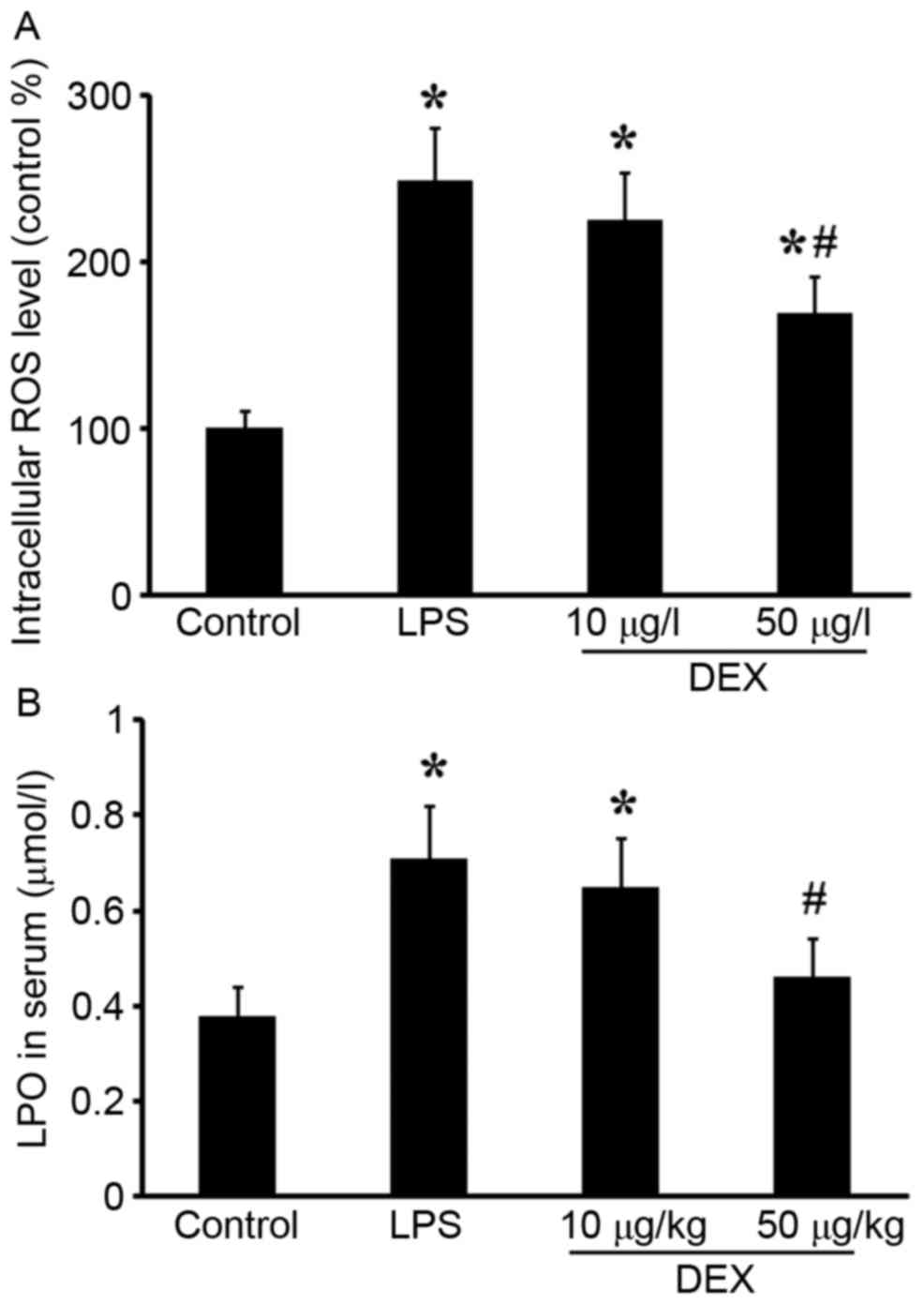
DEX alleviates LPS-induced
oxidation
Oxidative stress is one of the most important
mediators of mitochondrial dysfunction and apoptotic signaling.
In vitro, a fluorescent probe, DCFH-DA, was used as a
specific marker for quantitative mitochondrial ROS accumulation.
LPS-induced mitochondrial ROS production was evidenced by increased
intensity of DCF fluorescence in LPS-challenged rats compared with
the control animals. This increase was attenuated by pretreatment
with DEX (50 µg/l) (P<0.05 compared with the LPS group for 50
µg/l DEX group; P>0.05 compared with the LPS group for 10 µg/l
DEX group; Fig. 6A). To evaluate
the oxidative stress in vivo, LPO levels were determined in
rat serum. The LPO levels significantly increased in the
LPS-challenged rats, which was subsequently reduced by pretreatment
with DEX (50 µg/kg) (P<0.05 compared with the LPS group for 50
µg/kg DEX group; P>0.05 compared with the LPS group for 10 µg/kg
DEX group; Fig. 6B).
Discussion
Gram-negative organisms account for ~50% of
infections predisposing to ALI, often in the setting of pneumonia
or sepsis (27). LPS endotoxin is
the critical mediator of the organ dysfunction and mortality
associated with severe Gram-negative infections (29,30).
It is well-established that intratracheal administration of LPS can
induce a model of ALI (4,27). In the present study, to investigate
whether DEX attenuates sepsis-induced ALI, ALI rat model was
established through intratracheal injection of LPS (5 mg/kg),
according to a previous report (4). The present study observed significant
lung injuries and dysfunction following LPS administration,
evidenced by deterioration of histopathology, increased vascular
permeability, W/D weight ratio of the lung and decreased
PaO2/FIO2, which is consistent
with other studies (2,4). In previous studies, the protective
effect of DEX against ALI has been demonstrated in several models
(16,17,19).
Following pretreatment of DEX (50 µg/kg), LPS-induced ALI was
attenuated, which was reflected by improved histopathological
changes, vascular permeability, lung water content and
PaO2/FIO2.
DEX has been reported to exert protective effect
against ischemia/reperfusion injury in isolated rat hearts and may
be associated with inhibiting the opening of mPTP (24). In addition, DEX has been
demonstrated to induce human neutrophil apoptosis, which is
associated with the caspase cascade and loss of MMP (26). To explore the mechanism behind the
attenuation of ALI performed by DEX, the mitochondrial function and
mitochondrial-dependent apoptosis were investigated in the present
study. A reduction in MMP was demonstrated through JC-1 staining.
LPS-induced MMP dissipation was reflected by the fluorescence
changes in vivo and in vitro, which was subsequently
improved by DEX treatment. MMP is dissipated in response to the
arrest of the function of the respiratory complexes (I–V), which
contributes to an inhibition of ATP biosynthesis. In vitro,
DEX inhibited LPS-induced cellular ATP decrease.
Mitochondrial-dependent apoptosis is activated
through the mitochondrial release of cytochrome c, AIF and
Smac (28). Cytochrome c,
once in the cytosol, acts to release the apoptosome assembly from
the apoptotic protease-activating factor 1, ATP and procaspase-9,
leading to cellular morphological and functional alterations via
the activation of caspase 3 and caspase 7 (13). To further elucidate the present
hypothesis of mitochondrial mediated apoptosis, cytosolic
cytochrome c levels were estimated with a cytochrome
c ELISA kit. It was demonstrated that DEX improved
LPS-induced mitochondrial release of cytochrome c into the
cytoplasm. Additionally, the present results demonstrated that DEX
prevents LPS-induced caspase 3 activation in vivo and in
vitro.
Mitochondrial regulation of apoptosis is also
regulated by members of the Bcl-2 family of proteins (31,32).
The Bcl family consists of both antiapoptotic (Bcl-2, Bcl-xL) and
proapoptotic (BAK, Bax) factors. The pro-apoptotic members of this
family, including Bax, trigger the release of mitochondrial
apoptogenic factors into the cytoplasm by acting on the mPTP,
thereby leading to caspases activation. While the antiapoptotic
members serve a contrasting role to prevent apoptosis. In the
present study, the expression levels of Bax and Bcl-2 were detected
by western blotting in the rat samples. The results suggested that
LPS-induced upregulation of Bax and downregulation of Bcl-2, which
were improved by DEX treatment.
Oxidative stress is one of the most important
mediators of mitochondrial dysfunction and apoptotic signaling
(33,34). mPTP are major targets of ROS, from
which Smac and cytochrome c are released into the cytoplasm
after mPTP opening (7,35). The protective effect of DEX has
been demonstrated in other diseases. In the present study, DEX
inhibits LPS-induced oxidative stress, as evidenced by decreased
cellular ROS and serum LPO levels following LPS stimulation. It is
quite possible that mitochondrial oxidative stress serves an
important role in the release of cytochrome c into the
cytoplasm. DEX, by its antioxidant activity, may have prevented
this effect.
In conclusion, the present study has provided
evidence to suggest that DEX exerted significant attenuation of
LPS-induced ALI in a rat model. It was also revealed that the
potential mechanism of this action is through amelioration of
oxidative stress, mitochondrial dysfunction and
mitochondrial-dependent apoptosis.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (no. 81500066).
References
|
1
|
Aschner Y, Zemans RL, Yamashita CM and
Downey GP: Matrix metalloproteinases and protein tyrosine kinases:
Potential novel targets in acute lung injury and ARDS. CHEST.
146:1081–1091. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Xu Y, Ito T, Fushimi S, Takahashi S,
Itakura J, Kimura R, Sato M, Mino M, Yoshimura A and Matsukawa A:
Spred-2 deficiency exacerbates lipopolysaccharide-induced acute
lung inflammation in mice. PLOS One. 9:e1089142014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ito Y, Correll K, Schiel JA, Finigan JH,
Prekeris R and Mason RJ: Lung fibroblasts accelerate wound closure
in human alveolar epithelial cells through hepatocyte growth
factor/c-Met signaling. Am J Physiol Lung Cell Mol Physiol.
307:L94–L105. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gonzales JN, Gorshkov B, Varn MN, Zemskova
MA, Zemskov EA, Sridhar S, Lucas R and Verin AD: Protective effect
of adenosine receptors against lipopolysaccharide-induced acute
lung injury. Am J Physiol Lung Cell Mol Physiol. 306:L497–L507.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Martínez-González I, Roca O, Masclans JR,
Moreno R, Salcedo MT, Baekelandt V, Cruz MJ, Rello J and Aran JM:
Human mesenchymal stem cells overexpressing the IL-33 antagonist
soluble IL-1 receptor-like-1 attenuate endotoxin-induced acute lung
injury. Am J Respir Cell Mol Biol. 49:552–562. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang W, Xie Q, Zhou X, Yao J, Zhu X, Huang
P, Zhang L, Wei J, Xie H, Zhou L and Zheng S: Mitofusin-2 triggers
mitochondria Ca2+ influx from the endoplasmic reticulum to induce
apoptosis in hepatocellular carcinoma cells. Cancer Lett.
358:47–58. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sinha K, Das J, Pal PB and Sil PC:
Oxidative stress: The mitochondria-dependent and
mitochondria-independent pathways of apoptosis. Arch Toxicol.
87:1157–1180. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liu G, Zhang J, Chen H, Wang C, Qiu Y, Liu
Y, Wan J and Guo H: Effects and mechanisms of alveolar type II
epithelial cell apoptosis in severe pancreatitis-induced acute lung
injury. Exp Ther Med. 7:565–572. 2014.PubMed/NCBI
|
|
9
|
Bhandari V, Choo-Wing R, Lee CG, Zhu Z,
Nedrelow JH, Chupp GL, Zhang X, Matthay MA, Ware LB, Homer RJ, et
al: Hyperoxia causes angiopoietin 2-mediated acute lung injury and
necrotic cell death. Nat Med. 12:1286–1293. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kujoth GC, Hiona A, Pugh TD, Someya S,
Panzer K, Wohlgemuth SE, Hofer T, Seo AY, Sullivan R, Jobling WA,
et al: Mitochondrial DNA mutations, oxidative stress, and apoptosis
in mammalian aging. Science. 309:481–484. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Green DR and Kroemer G: The
pathophysiology of mitochondrial cell death. Science. 305:626–629.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Khalil WK, Assaf N, ElShebiney SA and
Salem NA: Neuroprotective effects of bee venom acupuncture therapy
against rotenone-induced oxidative stress and apoptosis. Neurochem
int. 80:79–86. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lakhani SA, Masud A, Kuida K, Porter GA
Jr, Booth CJ, Mehal WZ, Inayat I and Flavell RA: Caspases 3 and 7:
Key mediators of mitochondrial events of apoptosis. Science.
311:847–851. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen CH, Chen SJ, Su CC, Yen CC, Tseng TJ,
Jinn TR, Tang FC, Chen KL, Su YC, Lee KI, et al: Chloroacetic acid
induced neuronal cells death through oxidative stress-mediated
p38-MAPK activation pathway regulated mitochondria-dependent
apoptotic signals. Toxicology. 303:72–82. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Singh BK, Tripathi M, Chaudhari BP, Pandey
PK and Kakkar P: Natural terpenes prevent mitochondrial
dysfunction, oxidative stress and release of apoptotic proteins
during nimesulide-hepatotoxicity in rats. Plos One. 7:e342002012.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chen C, Zhang Z, Chen K, Zhang F, Peng M
and Wang Y: Dexmedetomidine regulates inflammatory molecules
contributing to ventilator-induced lung injury in dogs. J Surg Res.
187:211–218. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gu J, Chen J, Xia P, Tao G, Zhao H and Ma
D: Dexmedetomidine attenuates remote lung injury induced by renal
ischemia-reperfusion in mice. Acta Anaesthesiol Scand.
55:1272–1278. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cavalcanti V, Santos CL, Samary CS, Araújo
MN, Heil LB, Morales MM, Silva PL, Pelosi P, Fernandes FC, Villela
N and Rocco PR: Effects of short-term propofol and dexmedetomidine
on pulmonary morphofunction and biological markers in experimental
mild acute lung injury. Respir Physiol Neurobiol. 203:45–50. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sen V, Guzel A, Sen HS, Ece A, Uluca U,
Söker S, Doğan E, Kaplan İ and Deveci E: Preventive effects of
dexmedetomidine on the liver in a rat model of acid-induced acute
lung injury. Biomed Res Int. 2014:6218272014.PubMed/NCBI
|
|
20
|
Hwang L, Choi IY, Kim SE, Ko IG, Shin MS,
Kim CJ, Kim SH, Jin JJ, Chung JY and Yi JW: Dexmedetomidine
ameliorates intracerebral hemorrhage-induced memory impairment by
inhibiting apoptosis and enhancing brain-derived neurotrophic
factor expression in the rat hippocampus. Int J Mol Med.
31:1047–1056. 2013.PubMed/NCBI
|
|
21
|
Zhang XY, Liu ZM, Wen SH, Li YS, Li Y, Yao
X, Huang WQ and Liu KX: Dexmedetomidine administration before, but
not after, ischemia attenuates intestinal injury induced by
intestinal ischemia-reperfusion in rats. Anesthesiology.
116:1035–1046. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Xiang H, Hu B, Li Z and Li J:
Dexmedetomidine controls systemic cytokine levels through the
cholinergic anti-inflammatory pathway. Inflammation. 37:1763–1770.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang X, Wang J, Qian W, Zhao J, Sun L,
Qian Y and Xiao H: Dexmedetomidine inhibits tumor necrosis
factor-alpha and interleukin 6 in lipopolysaccharide-stimulated
astrocytes by suppression of c-Jun N-terminal kinases.
Inflammation. 37:942–949. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jiang C, Xia M, Wang M and Chen S:
Dexmedetomidine preconditioning protects isolated rat hearts
against ischemia/reperfusion injuries and its mechanism. Zhejiang
Da Xue Xue Bao Yi Xue Ban. 42:326–330. 2013.(In Chinese).
PubMed/NCBI
|
|
25
|
Chiu KM, Lin TY, Lu CW and Wang SJ:
Inhibitory effect of glutamate release from rat cerebrocortical
nerve terminals by α2 adrenoceptor agonist dexmedetomidine. Eur J
Pharmacol. 670:137–147. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kishikawa H, Kobayashi K, Takemori K,
Okabe T, Ito K and Sakamoto A: The effects of dexmedetomidine on
human neutrophil apoptosis. Biomed Res. 29:189–194. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Xie K, Yu Y, Huang Y, Zheng L, Li J, Chen
H, Han H, Hou L, Gong G and Wang G: Molecular hydrogen ameliorates
lipopolysaccharide-induced acute lung injury in mice through
reducing inflammation and apoptosis. Shock. 37:548–555.
2012.PubMed/NCBI
|
|
28
|
Childs EW, Tharakan B, Hunter FA, Tinsley
JH and Cao X: Apoptotic signaling induces hyperpermeability
following hemorrhagic shock. Am J Physiol Heart Circ Physiol.
292:H3179–H3189. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kumpf O, Giamarellos-Bourboulis EJ, Koch
A, Hamann L, Mouktaroudi M, Oh DY, Latz E, Lorenz E, Schwartz DA,
Ferwerda B, et al: Influence of genetic variations in TLR4 and
TIRAP/Mal on the course of sepsis and pneumonia and cytokine
release: An observational study in three cohorts. Crit Care.
14:R1032010. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Saluk-Juszczak J and Wachowicz B: The
proinflammatory activity of lipopolysaccharide. Postepy Biochem.
51:280–287. 2005.(In Polish). PubMed/NCBI
|
|
31
|
Renault TT, Floros KV, Elkholi R, Corrigan
KA, Kushnareva Y, Wieder SY, Lindtner C, Serasinghe MN, Asciolla
JJ, Buettner C, et al: Mitochondrial shape governs BAX-induced
membrane permeabilization and apoptosis. Mol Cell. 57:69–82. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Llambi F and Green DR: Apoptosis and
oncogenesis: Give and take in the BCL-2 family. Curr Opin Genet
Dev. 21:12–20. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Liu Y, Zeng X, Hui Y, Zhu C, Wu J, Taylor
DH, Ji J, Fan W, Huang Z and Hu J: Activation of α7 nicotinic
acetylcholine receptors protects astrocytes against oxidative
stress-induced apoptosis: Implications for Parkinson's disease.
Neuropharmacology. 91:87–96. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Matés JM, Segura JA, Alonso FJ and Márquez
J: Oxidative stress in apoptosis and cancer: An update. Arch
Toxicol. 86:1649–1665. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang X, Song R, Chen Y, Zhao M and Zhao K:
Polydatin-a new mitochondria protector for acute severe hemorrhagic
shock treatment. Expert Opin Inv Drug. 22:169–179. 2013. View Article : Google Scholar
|