Introduction
Shear stress is the frictional force of blood over
the surface of the endothelium. Physiological shear stress has been
suggested to serve atheroprotective roles by promoting the
production of nitric oxide (NO) and inhibiting apoptosis (1,2).
Physiological shear stress appears to serve essential roles in the
production of NO by mediating the phosphorylation of endothelial
nitric oxide synthase (eNOS). Low shear stress (LSS), an
atheroprone factor, upregulates the expression of proinflammatory
factors, including adhesion molecules, chemoattractant chemokines
and cytokines (3), thereby
enhancing injury-induced inflammation. Previous studies have
demonstrated that LSS (<5 dyn/cm2) in the
vasculature, including the inner curvatures of coronary arteries
and near bifurcations, promotes atherogenesis by inhibiting eNOS
phosphorylation in such regions (4,5).
Several potential phosphorylation sites exist on eNOS, among which
Ser1177, Ser633 and Thr495 are most extensively investigated
(6). Phosphorylation of eNOS at
different regulatory sites serves different roles in the regulation
of enzyme activation in response to several stimuli. eNOS can be
activated by phosphorylation at Ser1177 or Ser633, and inhibited by
phosphorylation at Thr495 in the calmodulin binding domain
(7).
It has been generally accepted that exposure of
endothelial cells to physiological shear stress stimulates the
production of NO from eNOS. However, the molecular mechanisms by
which shear stress regulates NO remain controversial. Shear stress
activates not only protein kinase B (Akt), but also numerous other
target kinases, including protein kinase A (PKA), protein kinase C
(PKC), serum- and glucocorticoid- inducible kinase, and p70S6
kinase (8,9). Akt, PKA, PKC or AMP-activated kinase,
which were reported to phosphorylate eNOS at Ser1177, Ser633 and
Thr495, modulate the specific activation of eNOS and NO synthase in
endothelial cells subjected to physiological shear stress (1,10,11).
By contrast, another previous study demonstrated that shear stress
phosphorylates eNOS-Ser1179 in a
Phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K)- and
PKA-dependent manner, without involving Akt (12).
Although numerous previous reports have focused on
the effect of a physiological shear stress of 12 and 15
dyn/cm2 on different eNOS serine/threonine
phosphorylation sites and signaling pathways, little information is
available regarding their phosphorylation in response to LSS
(1,7,10,11).
Earlier in vitro studies from our laboratory indicated that
the possible involvement of LSS-induced human vascular endothelial
cell apoptosis is via Akt signaling (13). Due to the differential effects of
the phosphorylation at Ser1177, Thr495 and Ser633 on eNOS
activation, the present study firstly aimed to determine how these
three phosphorylation levels change in response to LSS by protein
kinase or other signaling pathways. Secondly, the present study
investigated whether LSS-induced changes in eNOS phosphorylation
altered endothelial NO release in the presence and absence of
various signaling inhibitors or activators. The present study aimed
to further clarify the mechanism of LSS-induced endothelial injury.
The results may assist when exploring novel markers to protect
endothelial function under LSS.
Materials and methods
Cell culture
Human umbilical vein endothelial cells (HUVECs) were
purchased from American Type Culture Collection (Manassas, VA,
USA). The cells were maintained in Dulbecco's modified Eagle medium
(Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
containing 1 g/l glucose and 10% fetal bovine serum (Gibco; Thermo
Fisher Scientific, Inc.) without antibiotics at 37°C in a 5%
CO2 incubator. HUVECs at passage 4–9 were used for the
following experiments.
LSS studies
The parallel flow chamber was produced by Shanghai
Medical Instrument School, as previously described (14). Briefly, by sandwiching a silicon
gasket between two stainless steel plates, cells grown to
confluence on coverslips were placed the lower plate and subjected
to fluid flow powered by a reciprocal pump at 60 times/min. The
value of shear stress was obtained by modulating the proportion of
fluid volume passing the flow chamber. The shear stress used in the
present experiments was 2 dyn/cm2.
Reagent and supplies
The ERK1/2 inhibitor, PD98059, and cAMP-dependent
PKA activator, 8-Bromo-cAMP, were purchased from Sigma-Aldrich (St.
Louis, MO, USA). Primary monoclonal rabbit antibodies against
phosphorylated p-eNOS -Thr495/Ser1177 (cat. no. 9574/cat. no.
9571), eNOS (cat. no. 9586), p-ERK1/2 (Thr202/Tyr204) (cat. no.
4377), ERK1/2 (cat. no. 4695), p-Akt-Thr308/Ser473 (cat. no.
4058/cat. no. 4056), Akt (cat. no. 4691) and GAPDH (cat. no. 2118),
and the Akt inhibitor, perifosine (cat. no. 14240), were obtained
from Cell Signaling Technology, Inc. (Beverly, MA, USA). The
primary monoclonal mouse antibody against p-eNOS-Ser633 (cat. no.
612664) was purchased from BD Biosciences (Franklin Lakes, NJ,
USA). All antibodies were used at a dilution of 1:1,000.
SDS-PAGE and immunoblotting
The cells were lysed on ice in a cocktail of
radioimmunoprecipitation assay buffer [50 mM Tris-HCl (pH 7.5), 75
mM NaCl, 15 mM EGTA, 1 mM dithiothreitol, 0.1% Tween-20, 60 mM
glycerophosphate, 1 mM NaF, 0.2 mM sodium orthovanadate and 2 mM
sodium pyrophosphate; Beyotime Institute of Biotechnology,
Shanghai, China], containing proteinase inhibitor (Sigma-Aldrich)
and phosphatase inhibitor (Roche Diagnostics, Basel, Switzerland).
Homogenates were centrifuged at 12,000 × g for 20 min at 4°C. The
protein concentrations were quantified using a bicinchoninic acid
protein assay kit, according to the manufacturer's protocol (KeyGen
Biotech. Co., Ltd., Nanjing, China).
Aliquots of cell lysates were resolved on a 10%
SDS-PAGE gels and transferred onto a polyvinylidene difluoride
membrane (Millipore, Bedford, MA, USA). The membranes were blocked
with 5% FBS in TBST for 2 h prior to incubation with a 1:1,000
dilutions of primary antibody overnight at 4°C and then with a
1:2,000 dilutions of secondary antibody conjugated with alkaline
phosphatase (cat. no. 7074; Cell Signaling Technology, Beverly, MA,
USA) for 1 h at room temperature. The protein bands were detected
using the Immobilon Western HRP Substrate Peroxide Solution (cat.
no. WBKLS0500; EMD Millipore, Billerica MA, USA). The intensities
of immunoreactive bands were analyzed using Image J software
version 1.43 (National Institutes of Health, Bethesda, MD,
USA).
Detection of NO
4,5-diaminofluorescein diacetate (DAF-2DA), a
membrane-permeable probe, enters cells and is converted into a
product with green fluorescence in the presence of NO. Ice-cold
phosphate-buffered saline (PBS) was incubated with DAF-2DA (Life
Technologies; Thermo Fisher Scientific, Inc.) at 37°C for 30 min in
the dark. Following incubation, the cells were washed twice with
ice-cold PBS and their nuclei were labeled with DAPI. Images were
captured using a fluorescence microscope (Olympus IX71; Olympus,
Tokyo, Japan) and analyzed using Image J software version 1.43. A
total of five fields of view were used per sample. DAPI was used
for the total cell count in each field of view. Background
fluorescence, which was subtracted from the fluorescent intensity
values, was determine by imaging unstained cells. The fluorescent
intensity values in other groups were normalized to that of the
control group, where the value was set to 1.0.
Statistical analysis
Statistical analysis was performed using SPSS 16.0
(SPSS, Inc. Chicago, IL, USA) with one-way analysis of variance
followed by the least significant difference test. The data are
presented as the mean ± standard error of the mean of at least
three independent experiments P<0.05 was considered to indicate
a statistically significant difference.
Results
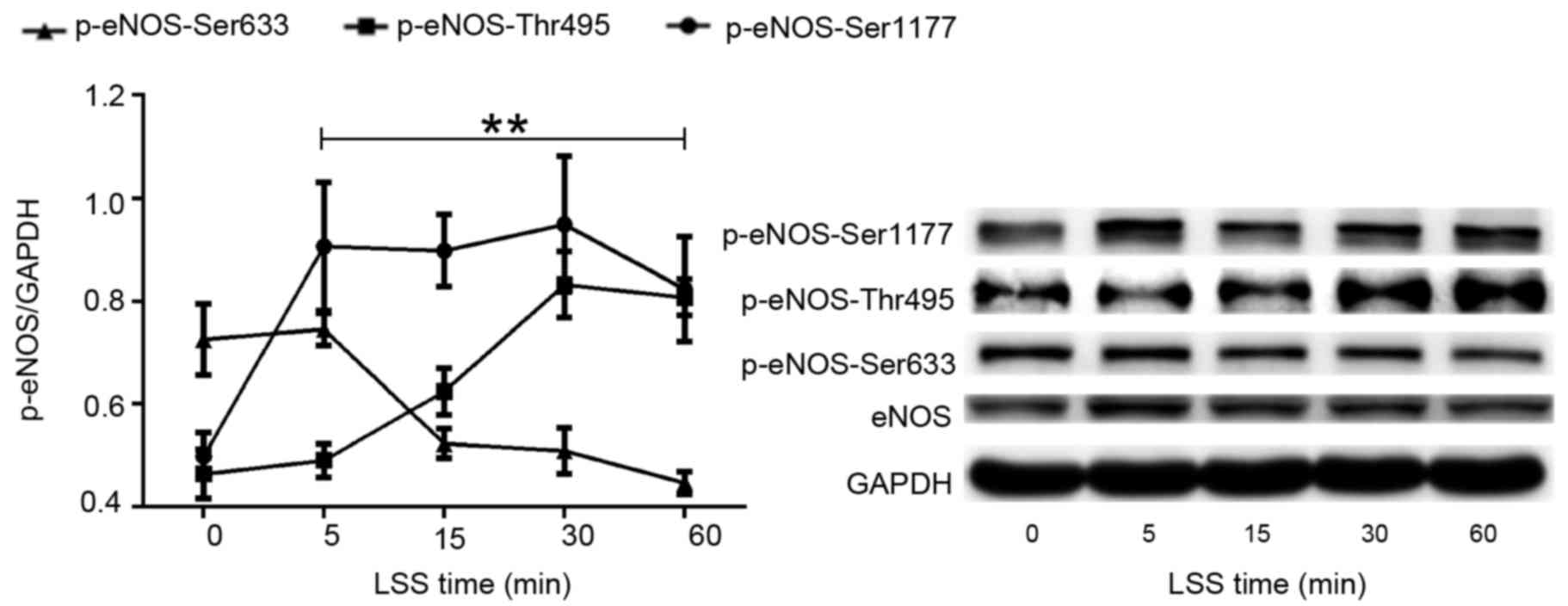
LSS alters the phosphorylation of eNOS
at Ser1177, Thr495 and Ser633
The present study first set out to investigate the
effect of LSS on the phosphorylation of eNOS at various amino acid
residues in HUVECs. A parallel plate flow chamber was used to mimic
shear stress at 2 dyn/cm2, and the cells were treated
for 5, 15, 30 and 60 min. Cells without LSS treatment served as the
control. Western blot analyses were performed using antibodies
specific to eNOS phosphorylation at Ser1177, Thr495 and Ser633. As
shown in Fig. 1, LSS stimulated
the phosphorylation of eNOS-Ser1177, which was similar to the
effect of physiological shear stress on Ser1177. Phosphorylation of
eNOS-Ser1177 was apparent as early as 5 min after LSS onset and
reached maximum by 30 min.
When HUVECs were exposed to LSS, a significant
increase in the phosphorylation at Thr495 was observed in a
time-dependent manner. LSS-stimulated phosphorylation of Thr495 was
later than that of Ser1177. Any significant increase of
phosphorylation of eNOS-Thr495 was observed at 15 min LSS exposure
and remained elevated for the 60 min (Fig. 1).
However, the effect of LSS on eNOS-Ser633 was
completely different compared with that of eNOS-Ser1177 and
-Thr495. Fig. 1 suggested that 15
min LSS exposure was required to observe a significant decrease of
the phosphorylation of eNOS-Ser633. This decrease in expression
remained for at least 60 min.
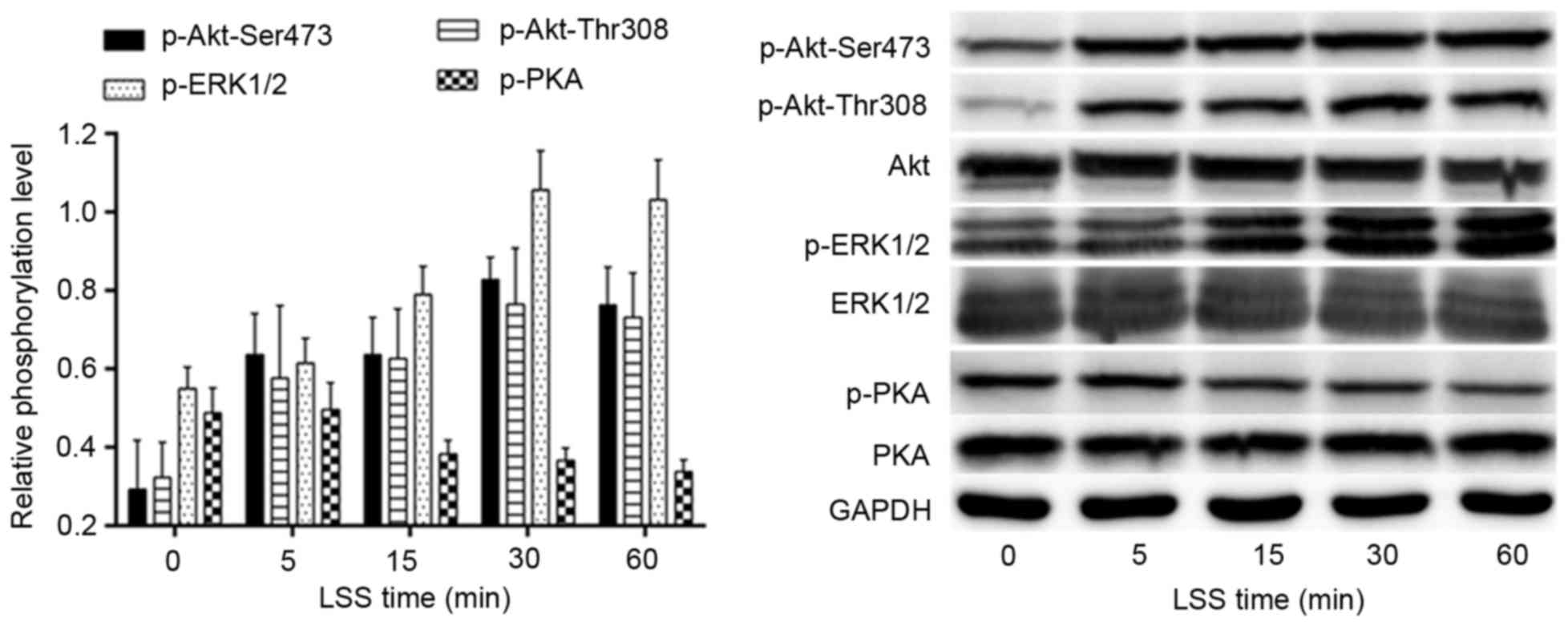
LSS regulates the phosphorylation of
Akt-Thr308/Ser473, ERK1/2 and PKA
The effects of LSS on the phosphorylation of Akt,
ERK1/2 and PKA were determined by western blotting using antibodies
specific for each phosphorylated site at various time points. As
shown in Fig. 2, the LSS-dependent
phosphorylation of Akt-Thr308, as well as Akt-Ser473, began to
increase at 5 min LSS and the phosphorylation of ERK1/2 occurred
later than 5 min LSS. The phosphorylation of Akt-Ser473/Thr308 and
ERK1/2 peaked at 30 min; however, PKA demonstrated an adverse
tendency with Akt and ERK1/2 in the same time course. The
phosphorylation of PKA was reduced after 5 min LSS. The lowest
value may actually be observed outside the 60 min time course.
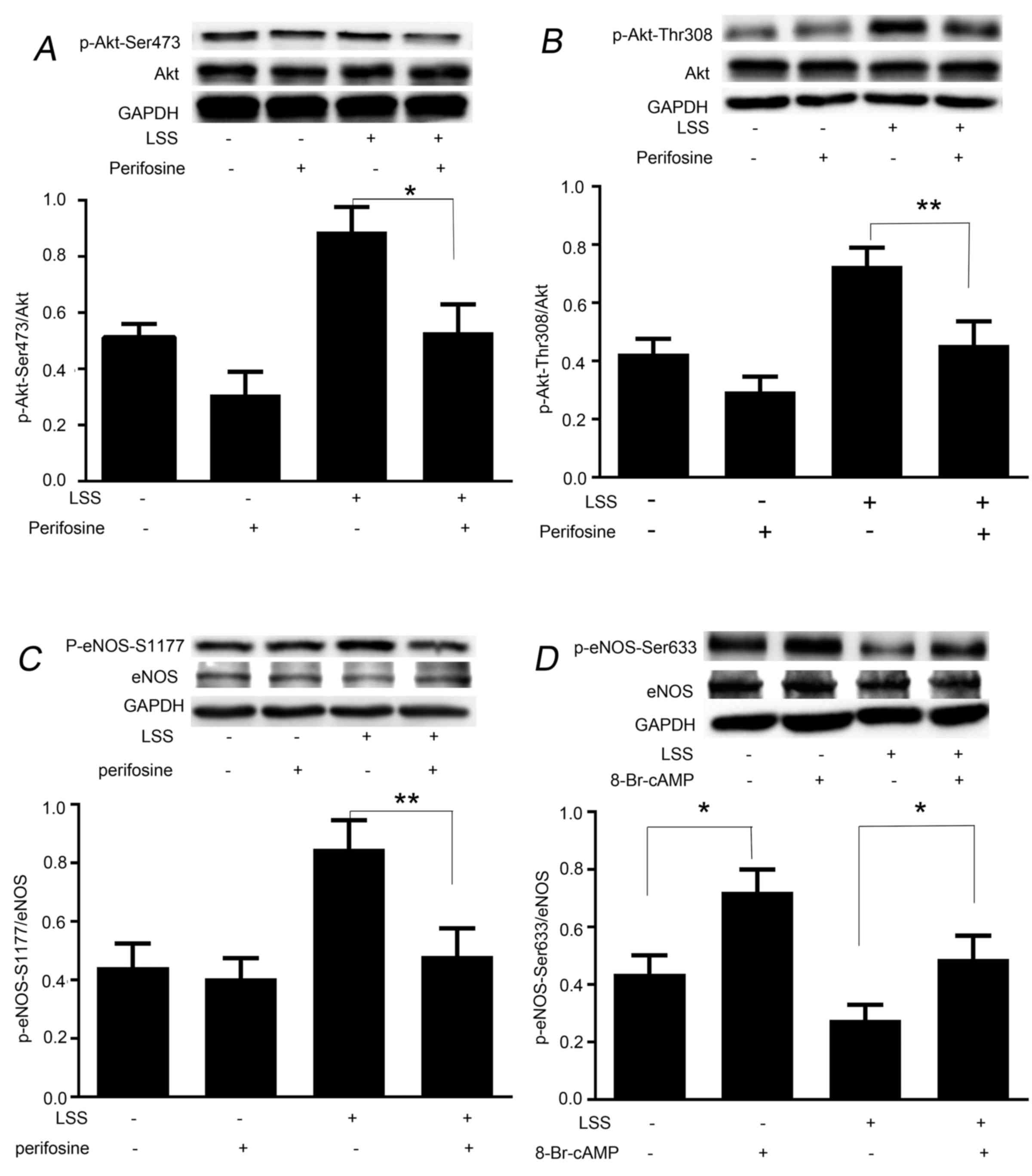
LSS induces the phosphorylation of eNOS-Ser1177 and
eNOS-Ser633 in a protein kinase-dependent manner. The present study
next aimed to determine whether phosphorylation of eNOS at Ser1177
and dephosphorylation at Ser633 by LSS are regulated in a protein
kinase-dependent manner. Perifosine, an inhibitor of Akt-Ser473 and
-Thr308, was used to treat the cells prior to the initiation of LSS
for 30 min. Western blot analysis revealed that treatment of HUVECs
with perifosine was effective in depressing LSS-dependent
phosphorylation of not only Akt-Ser473 (Fig. 3A), but also Akt-Thr308 (Fig. 3B). Perifosine also significantly
inhibited the LSS-dependent phosphorylation of eNOS-Ser1177
(Fig. 3C); however, did not
suppress LSS-induced attenuation of phosphorylation of eNOS-Ser633
(data not shown). As shown in Fig.
3D, treatment of the cells with 8-Br-cAMP for 30 min
significantly recovered the phosphorylation level of eNOS-Ser633.
In addition, 8-Br-cAMP significantly elevated static
phosphorylation of eNOS-Ser633 and -Ser1179; however, not
eNOS-Thr495 (data not shown).
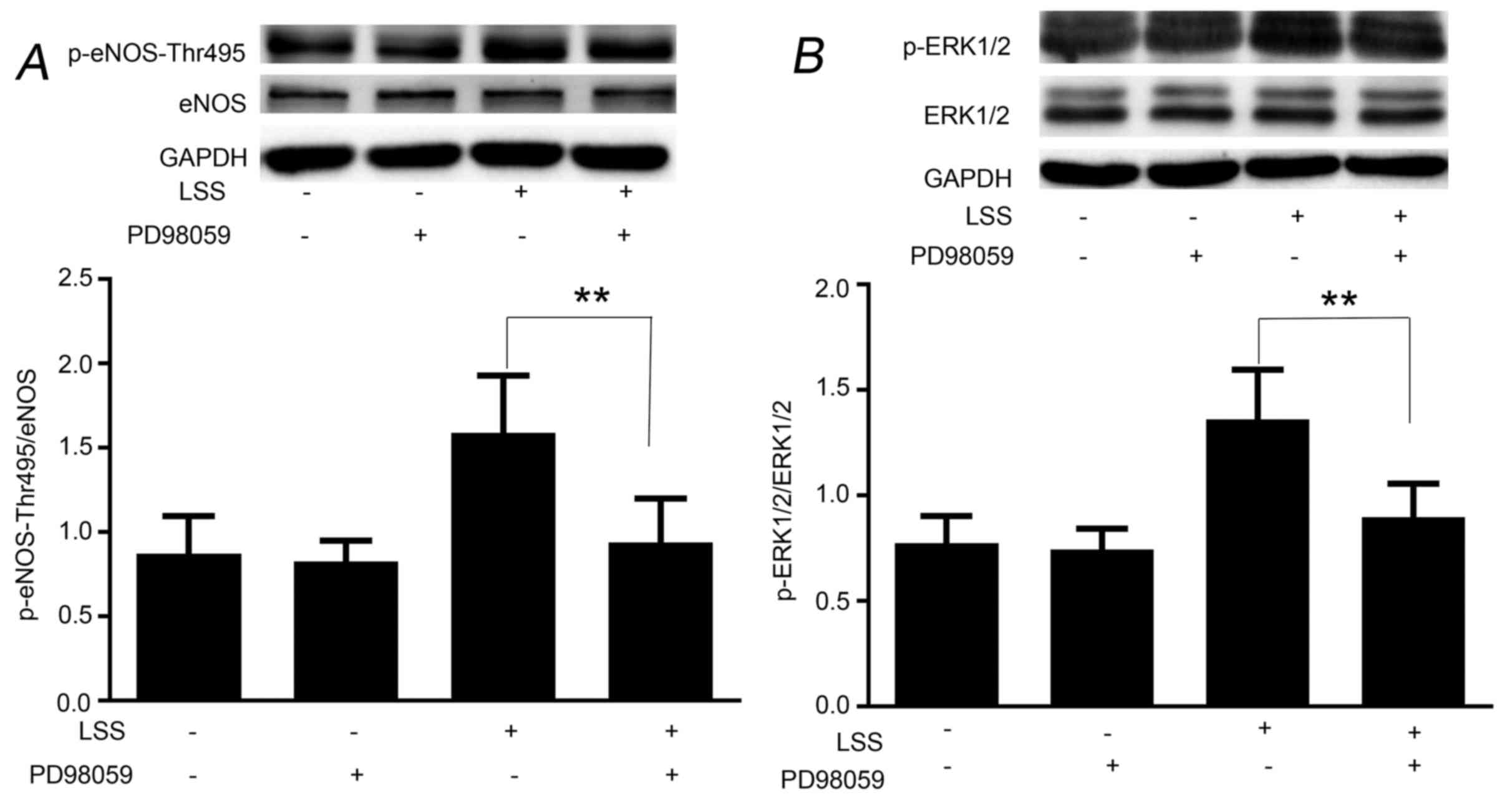
LSS phosphorylates eNOS-Thr495 via the
ERK1/2 pathway
To examine whether the LSS-evoked ERK1/2 is upstream
of eNOS-Thr495, HUVECs were pretreated with the ERK1/2 inhibitor,
PD98059, for 30 min. As shown in Fig.
4, PD98059 was able to reduce the phosphorylation of
eNOS-Thr495 (Fig. 4A) by
inhibiting the phosphorylation of ERK1/2 (Fig. 4B); however, did not reduce the
phosphorylation of eNOS-Ser1177 in response to LSS (data not
shown). The basic phosphorylation levels of eNOS-Ser1177 and
eNOS-Thr495 remained unchanged by PD98059 (data not shown).
ERK1/2/eNOS-Thr495 is a critical
signaling pathway for LSS-mediated NO synthase decrease
NO production under LSS was measured using the
NO-specific fluorescent dye DAF-2 DA. The results demonstrated that
LSS rapidly stimulated NO release in a transient manner with a
maximum at 5 min; however, after 5 min, NO production was observed
a time-dependent attenuation (Fig. 5A
and B). A total of 30 min pretreatment with PD98059 completely
inhibited LSS-induced suppression of NO production (Fig. 5C). In order to clarify the
signaling mechanisms of eNOS-Ser1177 and eNOS-Ser633 on NO
production mediated by LSS, the HUVECs were pretreated with
perifosine or 8-Br-cAMP for 30 min prior to a 30 min exposure to
LSS. The results demonstrated that 8-Br-cAMP significantly
abolished negative effect of LSS on NO production to a certain
degree, although NO production was not restored to the control
levels (Fig. 5D). Perifosine
modestly aggravated LSS-induced NO drop (Fig. 5E).
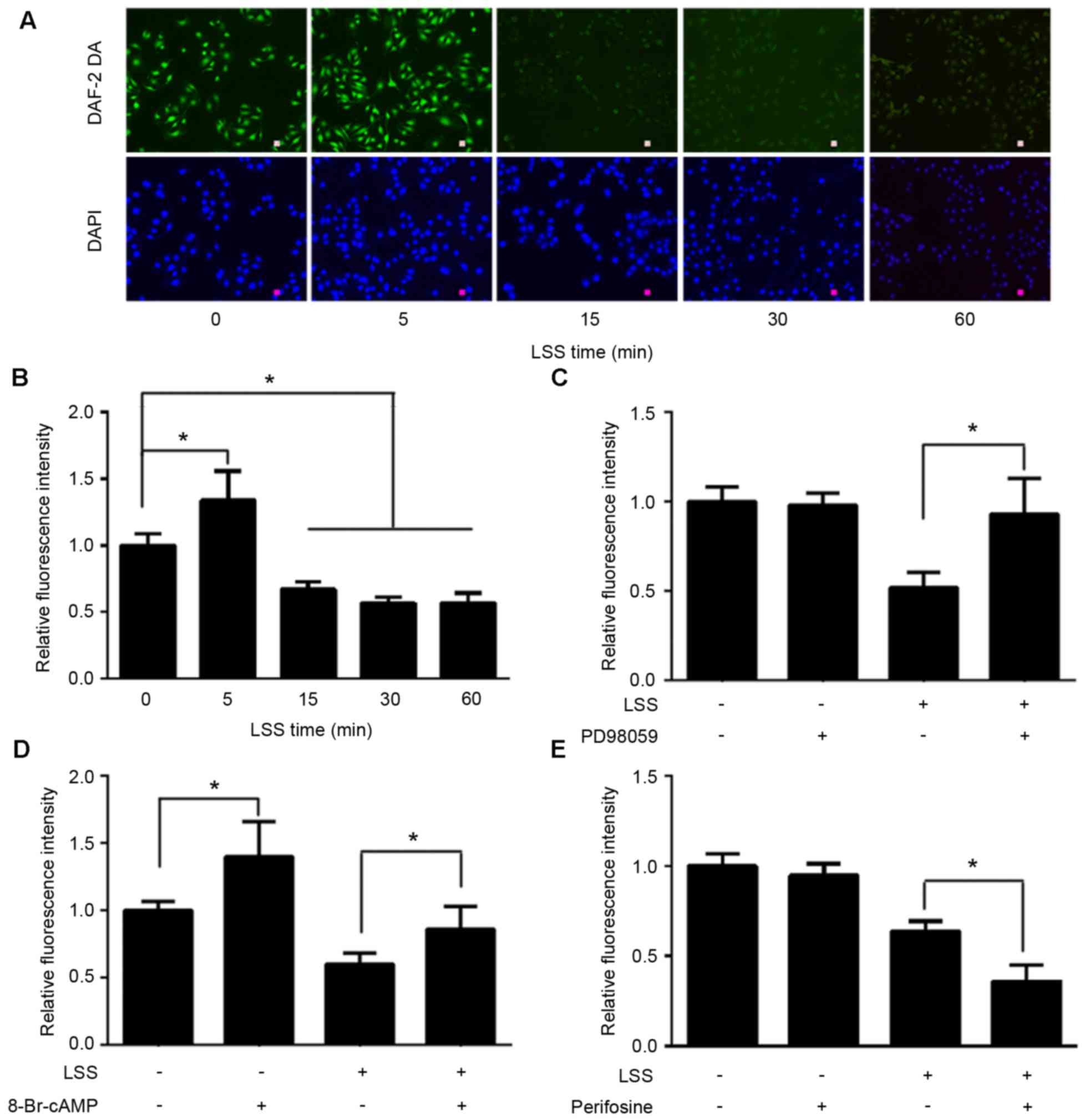 | Figure 5.Effect of LSS on NO production and
signaling mechanisms in HUVECs. (A and B) HUVECs were exposed to
LSS for 0, 5, 15, 30 and 60 min. NO were stained using the
NO-specific fluorescent dye DAF-2 DA (scale bar, 25 µm). Although
LSS caused a transient NO output growth at 5 min, NO production
demonstrated a notable time-depended decrease from 15 min in
response to LSS. The HUVECs were pretreated with vehicle (DMSO) or
30 µmol/l ERK1/2 inhibitor, PD98059, and the NO production was
assessed. (C) NO output, which was inhibited by LSS treatment, was
completely recovered to the control levels following treatment with
PD98059. (D) The PKA activator, 8-Br-cAMP (1 mmol/l), not only
partially inhibited LSS-induced NO production decline, but also
stimulated static HUVECs NO production, which was not observed
following PD98059 and perifosine treatment. (E) The reduction in NO
production was intensified following treatment with 40 µmol/l Akt
inhibitor, perifosine. The data are presented as the mean ±
standard error of the mean of 3–4 independent experiments
(*P<0.01). HUVECs, human umbilical vein endothelial cells; LSS,
low shear stress; NO, nitric oxide; PKA, protein kinase A; Akt,
protein kinase B. |
Discussion
Our previous findings demonstrated that LSS induces
human vascular endothelial cell apoptosis via Akt signaling
(13). The present study
investigated the phosphorylation of eNOS at Ser1177, Thr495 and
Ser633, and the possible mechanisms in HUVECs in response to LSS.
The results revealed that activation of Akt/eNOS-Ser1177 can
completely reverse LSS-induced NO synthase decrease via the
PKA/eNOS-Ser633 pathway. Downregulation of endothelial NO
predominantly depended on phosphorylation of eNOS-Thr495 via an
ERK1/2 mechanism, since PD98059 completely inhibited the NO
downregulation induced by LSS.
It is well known that phosphorylation of
eNOS-Ser1177 stimulates eNOS activation in response to various
physiological stimuli, including bradykinin, shear stress and
vascular endothelial growth factor, which activate eNOS in a
Ca2+-dependent or Ca2+-independent manner
(10,11). It has been previously demonstrated
that physiological shear stress, as an atheroprotective factor,
increases the phosphorylation of eNOS-Ser1177 and NO production
(12). The present data suggested
LSS, as a atheroprone factor, also stimulated the phosphorylation
of eNOS-Ser1177, while NO level decreased after a transient
increase at 5 min LSS. The present study accounted the alternation
of NO level for the differential change of eNOS multi-site
phosphorylation. Transient increase of NO is likely to be a
protective effect of endothelial cells at the beginning of harmful
stimuli.
Physiological shear stress was reported to
phosphorylate eNOS-Ser1179 by a PI3K mechanism in bovine aortic
endothelial cells (15). However,
other experiments have demonstrated that eNOS-Ser1179 was not
directly phosphorylated by PI3K. It was concluded that PI3K caused
activation of phosphoinositide-dependent kinase-1 (PDK1), which in
turn stimulated downstream PKA, which directly or indirectly
phosphorylated eNOS-Ser1179 in bovine aortic endothelial cells
(12). Whether the PI3K/PDK1
pathway regulates PKA and other protein kinases, in response to
LSS, remains to be determined. In the present study, LSS stimulated
the phosphorylation of PI3K within 5 min. Inhibiting PI3K with
wortmannin or LY294002 suggested that PI3K was not involved in
regulating eNOS phosphorylation at Ser1177, Ser633 or Thr495 (data
not shown).
Another significant finding of the present study was
that LSS promoted the dephosphorylation of eNOS-Ser633 and PKA in a
time-dependent manner. The PKA activator, 8-br-cAMP, restored the
level of phosphorylation of eNOS-Ser633 inhibited by LSS. This
result illustrated that eNOS-Ser633 as a protective amino acid
residue is dephosphorylated via the dephosphorylation of PKA by
LSS. Numerous experiments under physiological stimuli, including
shear stress, have suggested that PKA increased the level of
phosphorylation at the site of eNOS-Ser633 or other sites, and NO
production (16–19). These results suggested that the
effects of shear stress on the phosphorylation of eNOS-Ser633 and
PKA depend on shear stress value. In other words, phosphorylation
of eNOS-Ser633 begins to reduce with the decline of the value of
shear stress.
The present data provided certain insights how LSS
has an effect on the phosphorylation of eNOS-Thr495. The ERK1/2
inhibitor, PD98059, completely prevented the phosphorylation of
eNOS-Thr495 caused by LSS. It demonstrated LSS-stimulated
phosphorylation of eNOS-Thr495 is via ERK1/2 pathway. However
phosphorylation of eNOS-Ser1177 promoted NO output and
dephosphorylation of eNOS-Ser633 downregulated NO output under LSS.
In addition, NO output recovered to almost control degree with
PD98059 inhibiting the phosphorylation of eNOS-Thr495. These
findings suggested that LSS-induced function of eNOS-Ser1177 and
-Ser633 is cancelled out by each other. NO synthase decrease
following LSS predominantly depends on the phosphorylation of
eNOS-Thr495 modulated by ERK1/2. The effect of physiological shear
stress on the phosphorylation of eNOS-Thr495 indicated certain
contradictory results in certain previous studies (16,20).
Barauna et al (20)
revealed that inhibition of shear stress-induced ERK activation
leads to increasing eNOS activation by eNOS-Thr495
dephosphorylation in human saphenous vein endothelial cells.
However, other findings suggest no changes on the phosphorylation
status of eNOS-Thr497 by shear stress in bovine aortic endothelial
cells (16). It is possible that
the different species of cells are one of underlying reasons for
such differences, and also different stimuli appear to elicit
distinct changes of phosphorylation of eNOS-Thr495 and -Ser1177.
Although, phosphorylation of eNOS-Thr495 and -Ser1177 were
increased by LSS in the present study, NO release was abolished in
a time-dependent manner after 5 min LSS. A simple interpretation of
this result is that phosphorylation of eNOS-Thr495 is a predominant
factor that promotes endothelial injury induced by LSS.
Certain previous experiments have demonstrated that
phosphorylation of eNOS at Thr495 is PKC-dependent (21–24).
In order to elucidate a potential upstream signaling molecule of
ERK1/2, PKC, another member of the protein kinase familiar, was
investigated with phospho-specific anti-PKC antibodies (data not
shown); however no stimulus-related changes in the association of
classical PKC and eNOS-Thr495 were observed. One reason is that PKC
has nothing to do with the phosphorylation of eNOS-Thr495 promoted
by LSS. The other reason is that other subtypes of PKC that are
excluded in the present study are involved in phosphorylating
eNOS-Thr495.
In conclusion, the present study found that LSS
changes the phosphorylation of eNOS at Ser1177, Thr495 and Ser633.
Phosphorylation of eNOS-Ser1177 and dephosphorylation of eNOS-Ser63
under LSS are regulated in a protein kinase-dependent manner. The
activation of Akt is responsible for the phosphorylation of
eNOS-Ser1177 and elevation of NO in 5 min. LSS-stimulated NO
release via Akt/eNOS-Ser1177 is neutralized by dephosphorylation of
eNOS-Ser633, derived from deactivated PKA. Changes in the
phosphorylation at eNOS-Thr495 resulted in a decrease in NO and
endothelial injury. This may explain why LSS is an atheroprone
factor. The present results provided important insights and
suggested that ERK1/2 inhibition can restore endothelial NO
production under LSS.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (no. 81270191). The authors
would like to thank Professor Yu-Lin Gu (Nature-Think Company,
Shanghai, China) for technical support of the mechanics of the
parallel flow chamber.
References
|
1
|
Tarbell JM, Shi ZD, Dunn J and Jo H: Fluid
mechanics, arterial disease, and gene expression. Annu Rev Fluid
Mech. 46:591–614. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Resnick N, Yahav H, Shay-Salit A, Shushy
M, Schubert S, Zilberman LC and Wofovitz E: Fluid shear stress and
the vascular endothelium: For better and for worse. Prog Biophys
Mol Biol. 81:177–199. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chatzizisis YS, Coskun AU, Jonas M,
Edelman ER, Feldman CL and Stone PH: Role of endothelial shear
stress in the natural history of coronary atherosclerosis and
vascular remodeling: Molecular, cellular, and vascular behavior. J
Am Coll Cardiol. 49:2379–2393. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gimbrone MA Jr, Topper JN, Nagel T,
Anderson KR and Garcia-Cardeña G: Endothelial dysfunction,
hemodynamic forces, and atherogenesis. Ann N Y Acad Sci.
902:230–239, 239–240. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cunningham KS and Gotlieb AI: The role of
shear stress in the pathogenesis of atherosclerosis. Lab Invest.
85:9–23. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ramadoss J, Pastore MB and Magness RR:
Endothelial caveolar subcellular domain regulation of endothelial
nitric oxide synthase. Clin Exp Pharmacol Physiol. 40:753–764.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kolluru GK, Siamwala JH and Chatterjee S:
eNOS phosphorylation in health and disease. Biochimie.
92:1186–1198. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Toker A and Newton AC: Cellular signaling:
Pivoting around PDK-1. Cell. 103:185–188. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Maroski J, Vorderwülbecke BJ, Fiedorowicz
K, Da S, ilva-Azevedo L, Siegel G, Marki A, Pries AR and Zakrzewicz
A: Shear stress increases endothelial hyaluronan synthase2 and
hyaluronan synthesis especially in regard to an atheroprotective
flow profile. Exp Physiol. 96:977–986. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Michell BJ, Harris MB, Chen ZP, Ju H,
Venema VJ, Blackstone MA, Huang W, Venema RC and Kemp BE:
Identification of regulatory sites of phosphorylation of the bovine
endothelial nitric-oxide synthase at serine 617 and serine 635. J
Biol Chem. 277:42344–42351. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dimmeler S, Fleming I, Fisslthaler B,
Hermann C, Busse R and Zeiher AM: Activation of nitric oxide
synthase in endothelial cells by Akt-dependent phosphorylation.
Nature. 399:601–605. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Boo YC, Sorescu G, Boyd N, Shiojima I,
Walsh K, Du J and Jo H: Shear stress stimulates phosphorylation of
endothelial nitric-oxide synthase at Ser1179 by Akt-independent
mechanisms: Role of protein kinase A. J Biol Chem. 277:3388–3396.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang J, Wang Z, Zuo G, Li B, Zhang J,
Tian N and Chen S: Low shear stress induces human vascular
endothelial cell apoptosis by activating Akt signal and increasing
reactive oxygen species. Nan Fang Yi Ke Da Xue Xue Bao. 33:313–317.
2013.PubMed/NCBI
|
|
14
|
Wang Z, Zhang J, Li B, Gao X, Liu Y, Mao W
and Chen SL: Resveratrol ameliorates low shear stressinduced
oxidative stress by suppressing ERK/eNOSThr495 in endothelial
cells. Mol Med Rep. 10:1964–1972. 2014.PubMed/NCBI
|
|
15
|
Devika NT and Ali BM Jaffar: Analysing
calcium dependent and independent regulation of eNOS in endothelium
triggered by extracellular signalling events. Mol Biosyst.
9:2653–2664. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Boo YC, Hwang J, Sykes M, Michell BJ, Kemp
BE, Lum H and Jo H: Shear stress stimulates phosphorylation of eNOS
at Ser(635) by a protein kinase A-dependent mechanism. Am J Physiol
Heart Circ Physiol. 283:H1819–H1828. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Walther S, Pluteanu F, Renz S, Nikonova Y,
Maxwell JT, Yang LZ, Schmidt K, Edwards JN, Wakula P, Groschner K,
et al: Urocortin 2 stimulates nitric oxide production in
ventricular myocytes via Akt- and PKA-mediated phosphorylation of
eNOS at serine 1177. Am J Physiol Heart Circ Physiol.
307:H689–H700. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hu Z, Xiong Y, Han X, Geng C, Jiang B, Huo
Y and Luo J: Acute mechanical stretch promotes eNOS activation in
venous endothelial cells mainly via PKA and Akt pathways. PLoS One.
8:e713592013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yang C, Talukder MA, Varadharaj S,
Velayutham M and Zweier JL: Early ischaemic preconditioning
requires Akt- and PKA-mediated activation of eNOS via serine1176
phosphorylation. Cardiovasc Res. 97:33–43. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Barauna VG, Mantuan PR, Magalhaes FC,
Campos LC and Krieger JE: AT1 receptor blocker potentiates
shear-stress induced nitric oxide production via modulation of eNOS
phosphorylation of residues Thr(495) and Ser(1177.). Biochem
Biophys Res Commun. 441:713–719. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen F, Kumar S, Yu Y, Aggarwal S, Gross
C, Wang Y, Chakraborty T, Verin AD, Catravas JD, Lucas R, et al:
PKC-dependent phosphorylation of eNOS at T495 regulates eNOS
coupling and endothelial barrier function in response to G+
-toxins. PLoS One. 9:e998232014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jiang X, Yang F, Tan H, Liao D, Bryan RM
Jr, Randhawa JK, Rumbaut RE, Durante W, Schafer AI, Yang X and Wang
H: Hyperhomocystinemia impairs endothelial function and eNOS
activity via PKC activation. Arterioscler Thromb Vasc Biol.
25:2515–2521. 2015. View Article : Google Scholar
|
|
23
|
Sakata K, Kondo T, Mizuno N, Shoji M,
Yasui H, Yamamori T, Inanami O, Yokoo H, Yoshimura N and Hattori Y:
Roles of ROS and PKC-βII in ionizing radiation-induced eNOS
activation in human vascular endothelial cells. Vascul Pharmacol.
70:55–65. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chu S and Bohlen HG: High concentration of
glucose inhibits glomerular endothelial eNOS through a PKC
mechanism. Am J Physiol Renal Physiol. 287:F384–F392. 2004.
View Article : Google Scholar : PubMed/NCBI
|