Introduction
Heart failure follows hypertension as the end stage
of cardiac hypertrophy, and is associated with high mortality rates
worldwide. Therefore, reducing hypertension may ameliorate cardiac
hypertrophy and consequently prevent heart failure (1). The renin-angiotensin system serves an
important role in maintaining normal blood pressure, as well as the
development of hypertension and secondary hypertensive organ
disorders (2). Angiotensin II (Ang
II) receptor blockers (ARB), known as sartans, have been widely
used to treat hypertension in the clinic. Telmisartan is a
well-known nonpeptide ARB that selectively inhibits Ang II receptor
type 1 (AT1) without affecting additional receptor systems
(3,4). A significant number of reports have
demonstrated that telmisartan may be involved in reducing systolic
and diastolic blood pressure (BP) (5–7). In
addition, telmisartan has been reported to enhance renal blood
flow, thereby increasing renal function (8,9). In
diabetic and nondiabetic patients, telmisartan may improve insulin
sensitivity, reduce weight gain and prevent hepatic steatosis by
regulating caloric expenditure and lipid metabolism (10–12).
In addition, telmisartan may penetrate the blood-brain barrier in a
dose- and time-dependent manner and prevent cognitive decline
(13,14).
As well as reducing BP, a previous study
demonstrated that telmisartan may suppress cardiac hypertrophy in
TGR (mREN2)27 transgenic rats; a rat model of fulminant
hypertension (15). Telmisartan
monotherapy significantly reduced left atrial volume, alleviating
left ventricular hypertrophy (16). In the rat myocardial infarction
model, recovery of left ventricular function was improved with
telmisartan administration, indicating that telmisartan may prevent
unfavorable cardiac remodeling via the reduction of cardiac
hypertrophy and fibrosis (17).
However, it is thought that the effect of telmisartan on different
organs may be mediated by inhibition of the AT1 receptor (18).
Cardiac hypertrophy is characterized by cell
enlargement which involves physiological and pathological
hypertrophy (19). Pathological
cardiac hypertrophy is often coupled with interstitial and
perivascular fibrosis, as well as apoptosis and the release of
atrial natriuretic peptides (ANP) and brain/B-type natriuretic
peptides (BNP). Upon initiation of cardiac hypertrophy, concentric
hypertrophy is the primary phenotype that resists high afterload,
and is known as the adaptive phase. As cardiac damage progresses,
cell length increases, which leads to increased hypertrophy
(20). In cardiac hypertrophy,
nuclear factor of activated T-cells (NFAT) is considered to be an
important mediator of a number of signal-transduction pathways
involved in the coordination of pathological stimulation (21). In addition, NFAT may be stimulated
by the AT1 receptor (22).
However, it is unknown whether the effect of telmisartan on the
cardiomyocyte AT1 receptor blockade may extend to inactivation of
the NFAT pathway.
The aim of the present study was to clarify whether
telmisartan prevents cardiomyocyte hypertrophy by inhibiting NFAT
nuclear translocation, ANP/BNP release and cardiomyocyte
apoptosis.
Materials and methods
Animals
A total of 23 male C57/BL6 mice (age 8–10 weeks;
weight 20–23 g) were used for the purposes of this study. All mice
were purchased from Heze Better Biotechnology Co., Ltd. (Shangdong,
China) and had free access to normal chow diet and water,
temperature and humidity were kept at 22–24°C, 40–60% with a 12 h
light/12 h dark cycle. Mice were divided into 4 groups (5–7 mice
per cage) with the same average body weight. Out of these, two
groups were used for sham (control group) operations with the
administration of either saline (n=5) or telmisartan (n=5). Aortic
binding (AB) operations were performed on mice in the remaining two
groups, together with the administration of either saline (n=6) or
telmisartan (n=7). All mice were sacrificed at 4 weeks post-AB
operation.
AB model
The afterload-induced pressure model was generated
by AB under abdominal anesthesia as described previously (23). Briefly, the chest was first opened,
and AB of the aortic arch between the brachiocephalic artery and
left common carotid artery was performed using a 27-gauge needle as
the standard binding level to lead to the aortic arch narrow.
Following successful binding, the needle was removed and the chest
cavity was closed. The same operation without AB was performed on
mice in the sham group.
Neonatal rat cardiomyocyte primary
culture
Wistar rats (age, 1–3 days) were purchased from the
Experimental Animal Center of the Academy of Military Medical
Sciences (Beijing, China). Hearts were harvested under aseptic
conditions on a clean bench. Following 3 washes with sterilized
phosphate-buffered saline to remove excess blood, the hearts were
minced, washed with Hank's Balanced Salt Solution at 4°C and
digested in cardiomyocyte balance suspension liquid containing 0.1%
trypsin and 0.025% collagenase (Sigma-Aldrich; Merck KGaA;
Darmstadt, Germany) for 15 min. The undigested heart tissue was
re-digested 4 or 5 times. Following complete digestion,
cardiomyocytes were separated and isolated by centrifugation using
the Percoll gradient system (upper, 45.3% percoll; bottom, 58.5%
percoll; Sigma-Aldrich; Merck KGaA) as described previously
(24). The separated rat
cardiomyocytes were cultured in Dulbecco's modified Eagle's medium
containing 10% fetal bovine serum and penicillin (0.2%)
(Sigma-Aldrich; Merck KGaA) at a density of 1×105
cells/cm2 in a 5% CO2 incubator at 37°C. At 2
days following isolation, and when cardiomyocyte beating was
confirmed, the cells were used for the purposes of downstream
experiments. All animal experiments were approved by the Ethics
Review Committee of Harbin Medical University (Harbin, China).
Telmisartan in vivo administration and
in vitro treatment
Telmisartan (Boehringer Ingelheim Shanghai
Pharmaceuticals, Co., Ltd., Shanghai, China) was dissolved in
saline prior to in vivo administration at a dose of 50
mg/kg/day. Saline or telmisartan were administered to the mice
once-a-day via oral gavage, commencing 5 days prior to AB
operations and continuing until they were sacrificed at 4 weeks
post-AB operation. For the treatment of neonatal primary rat
cardiomyocytes in vitro, telmisartan was first dissolved in
dimethyl sulfoxide. Three different concentrations of telmisartan
(5, 20 and 50 µM) were used for these experiments. In addition, Ang
II (10 µM; Sigma-Aldrich; Merck KGaA) was used to induce
cardiomyocyte hypertrophy in vitro. Rat cardiomyocytes were
treated with the vehicle (dimethyl sulfoxide), telmisartan and Ang
II for 12 h.
Morphological and histological
analyses
Mouse heart samples were fixed in 4% formaldehyde
overnight at room temperature and embedded in paraffin. Blocks were
cut into 5-µm thick sections and de-waxed in xylene for 10 min and
stained with hematoxylin and eosin (H&E) at room temperature.
Briefly, the sections were placed in distilled water, stained with
alum hematoxylin at room temperature for 10 min, rinsed under
running tap water, differentiated with 0.3% acid alcohol at room
temperature for ~30 sec, rinsed in running tap water again and
rinsed in Scott's tap water substitute (Sigma-Aldrich; Merck KGaA)
prior to rinsing in tap water again. The sections were subsequently
stained with eosin for 2 min. Following that, sections were
observed (Olympus FluoView™ FV1000) to evaluate cardiac morphology
and measure cardiomyocyte size. The thickness of the anterior,
posterior, lateral and septum heart walls was measured, and the
average of these four parameters was calculated as the average wall
thickness. Similarly, the internal dimensions (presented as an
average of the horizontal and vertical measurements) of left
ventricles were measured and an average value was calculated as
average internal dimensions. To investigate cardiomyocyte size, ~50
cardiomyocytes stained by H&E were measured and the average
value was calculated as the average area of myocytes per sample.
All of these measurements were performed using ImageJ software
(v1.48; National Institutes of Health, Bethesda, MD, USA). Heart
weight was measured by an experimental scale.
TUNEL assay
TUNEL assay was performed on heart sections (5-µm)
with a TdT-FragEL DNA Fragmentation Detection kit to quantify
apoptosis. Counterstaining with fluorescence mounting medium
containing DAPI (blue; Thermo Fisher Scientific, Inc., Waltham, MA,
USA) was performed to visualize normal nuclei. Sections were
observed by use of a fluorescence microscope Olympus FluoView™
FV1000. Measurements of the apoptotic nuclei percentage were
obtained from the border zone area of sections from each heart and
8 fields were randomly selected from the border zone of each heart
and the average TUNEL-positive percentage (%) was calculated.
Immunocytochemical analysis of NFATc3
nuclear translocation
Cardiomyocytes were collected and cultured on glass
bottom coverslips in 4-well dishes that had been previously coated
with collagen II, as described above for rat cardiomyocyte primary
culture. Following treatment with Ang II or telmisartan, cells were
fixed with 10% acetone at 4°C for 20 min. After the glass slides
were washed by TBS with 0.5% Tween-20, 3 times, double
immunofluorescent staining was performed using a specific antibody
against NFATc3 (cat. no. sc-8405, 1:500; Santa Cruz Biotechnoloy,
Inc., Dallas, TX, USA) at room temperature for 2 h, followed by the
addition of a donkey anti-rabbit IgG (red; Alexa Fluor®
488; cat. no. ab150077; 1:500; Abcam, Cambridge, UK) for 1 h at
room temperature. Finally, following 3 washes with TBS, slides were
mounted in fluorescence mounting medium containing DAPI (blue;
Thermo Fisher Scientific, Inc.). Cells were observed by use of a
fluorescence microscope, Olympus FluoView™ FV1000. °C.
Reverse transcription-quantitative
polymerase chain reaction (PCR)
Total RNA was extracted from the mouse hearts using
TRIzol (Invitrogen; Thermo Fisher Scientific, Inc.), and total RNA
(2,000 ng) was used, cDNA (50 ng/µl) was synthesized using oligo
(dT) primers and the Transcriptor First Strand cDNA Synthesis kit
(cat. no. 04896866001; Roche Applied Science, Penzberg, Germany).
PCR amplifications were performed using LightCycler® 480
SYBR-Green I Master (cat. no. 04887352001; Roche Applied Science).
Target gene expression levels were normalized to the expression of
the gene encoding the 18S ribosomal RNA subunit by the ΔΔCq method,
as described (25). The primers
used were as follows: Myosin heavy chain α isoform (Myh6), forward,
5′-AGATCATCAAGGCCAAGGCA-3′, and reverse,
5′-CGCTGGGTGGTGAAATCATT-3′; GATA binding protein 4 (Gata4),
forward, 5′-AGCTCCGTGTCCCAGACG-3′, and reverse,
5′-TCTGTGGAGACTGGCTGACG-3′; collagen I, forward,
5′-ATGTTCAGCTTTGTGGACCTC-3′, and reverse,
5′-TCCCTCGACTCCTACATCTTC-3′; collagen III, forward,
5′-TGGTTTCTTCTCACCCTTCTTC-3′, and reverse,
5′-TGCATCCCAATTCATCTACGT-3′.
Western blot analysis
Mouse ventricular cytoplasmic protein was extracted
using lysis buffer (50 mM Tris-HCl, pH 7.5, containing 150 mM NaCl,
25 mM EDTA, 0.25% sodium deoxycholate, and 1 mM dithiothreitol).
The nuclear protein precipitate was extracted using
radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris-HCl, pH
7.5, containing 150 mM NaCl, 1 mM EDTA and 1% NP-40). Total protein
was extracted from cultured cells using RIPA lysis buffer
containing 2% phenylmethylsulfonyl fluoride, 10% complete protease
inhibitor cocktail, 10% PhosSTOP (Sigma-Aldrich; Merck KGaA), 5%
NaF and 1% Na3VO4. The protein concentration
was determined using a Pierce BCA Protein assay kit (cat. no.
23225; Pierce; Thermo Fisher Scientific, Inc.). Proteins (20 µg)
were electrophoresed by 10% SDS-PAGE (cat. no. NP0301BOX;
Invitrogen; Thermo Fisher Scientific, Inc.) and subsequently
electrotransferred to a polyvinylidene difluoride membrane (cat.
no. IPVH00010; Merck Millipore). Membranes were blocked in TBS with
0.5% Tween-20 containing 5% skim milk for 60 min at room
temperature. Membranes were subsequently incubated with the
following primary antibodies overnight at 4°C: NFATc3 (cat. no.
sc-8405; 1:1,000), NFATc4 (cat. no. sc-13036; 1:1,000), ANP (cat.
no. sc-20158; 1:2,000), BNP (cat. no. sc-67455; 1:2,000), caspase 3
(cat. no. sc-7148; 1:1,000), GAPDH (cat. no. sc-365062; 1:2,000)
and lamin (cat. no. sc-20680; 1:2,000). All primary antibodies were
purchased from Santa Cruz Biotechnology, Inc. The membrane was
incubated at room temperature for 1 h with Peroxidase-affiniPure
goat anti-mouse IgG (H+L; cat. no. 115-035-003; 1:2,000; Jackson
ImmunoResearch Laboratories, Inc., West Grove, PA, USA) or
Peroxidase-affiniPure goat anti-rabbit IgG (H+L; cat. no.
111-035-003; 1:2,000; Jackson ImmunoResearch Laboratories, Inc.). A
FluorChem E (Cell Biosciences Pty, Ltd., Heidelberg, VIC,
Australia) imaging system was used to visualize the signals. GAPDH
was used as a loading control.
Statistical analysis
Results are expressed as the mean ± standard
deviation. Comparisons between 2 groups were analyzed using a
Student's t-test. Comparisons among >2 groups were analyzed
using one-way analysis of variance and Turkey's multiple
comparisons test. Statistical analysis was performed using GraphPad
Prism (v6.0; GraphPad Software, Inc., La Jolla, CA, USA) P<0.05
was considered to indicate a statistically significant
difference.
Results
Telmisartan inhibited cardiac
hypertrophy
In order to determine whether telmisartan inhibits
cardiac hypertrophy, an in vivo study using a mouse AB model
was performed. At 4 weeks following the AB operation, mice
administered with saline exhibited a hypertrophied heart with
increased wall thickness, an enlarged left ventricular dimension
and increased heart weight (HW)/body weight (BW) ratio, when
compared with the saline-treated sham group (Fig. 1A, C and E). By contrast, mice
administered with telmisartan exhibited reduced cardiac
hypertrophy, with a significantly reduced wall thickness, smaller
left ventricular dimensions and a lower HW/BW ratio when compared
with the saline-treated AB group (Fig.
1A, C and E). In addition, cardiomyocyte size was significantly
lower in the hearts of mice in the telmisartan-treated group when
compared to that of the saline-treated AB group (Fig. 1B and D). The mRNA expression levels
of cardiac hypertrophy markers, Myh6 and Gata4, as well as the
markers of cardiac fibrosis, collagen I and collagen III were
increased when compared with the sham telmisartan-treated AB group,
but at a significantly lower level when compared with the
saline-treated AB group (Fig. 1F and
G). These results indicated that telmisartan may inhibit
cardiac hypertrophy and fibrosis in a mouse model of cardiac
afterload.
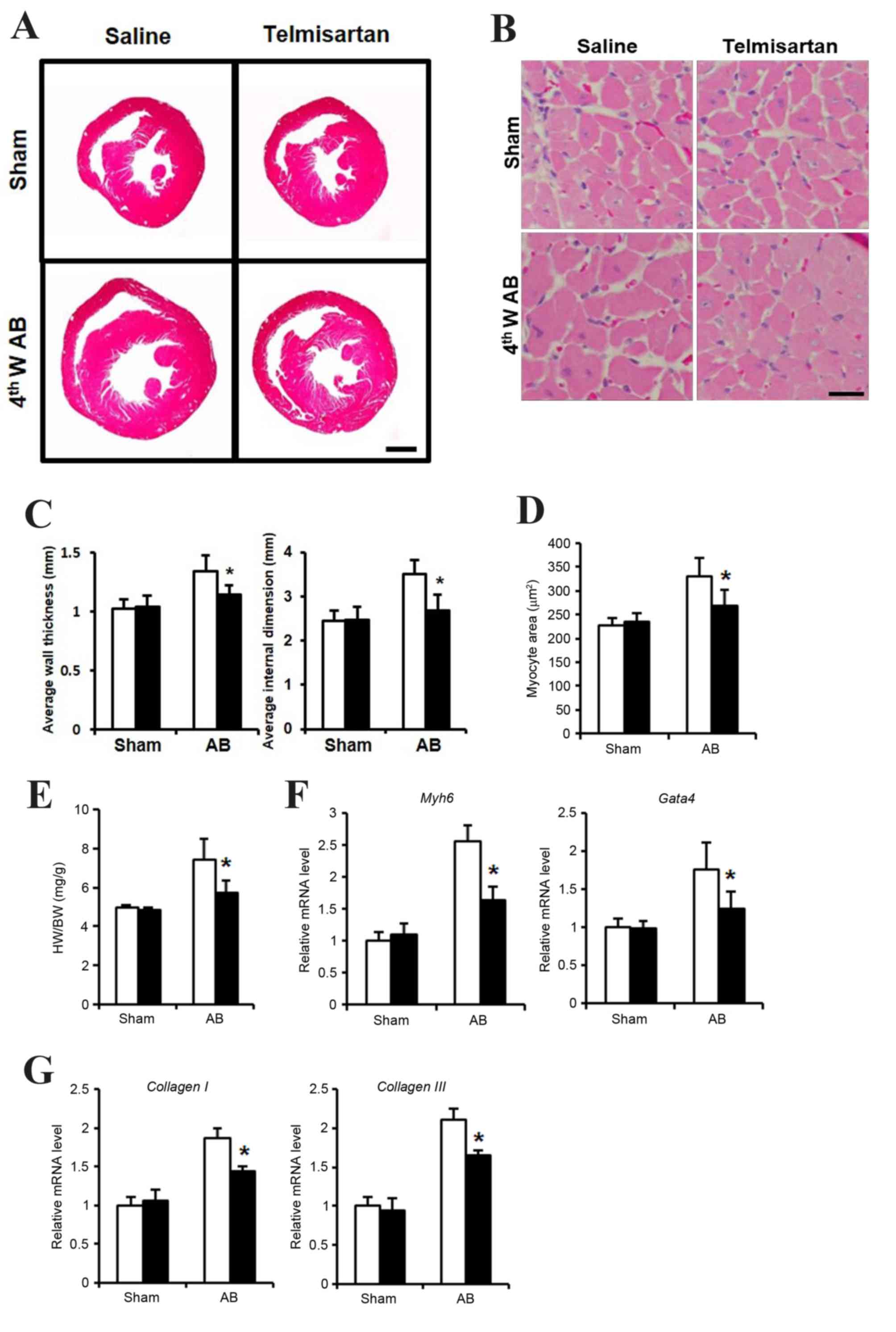 | Figure 1.Cardiac hypertrophy phenotype of
telmisartan treatment in a mouse AB model. (A) H&E staining of
whole hypertrophic hearts at 4 weeks following AB operation (n=5–7;
scale bar, 1 mm). (B) H&E staining sections were analyzed
further to observe cellular hypertrophy at 4 weeks following the AB
operation (n=5; scale bar, 25 µm). (C) Average heart wall thickness
(left panel) and average left ventricular internal dimension (right
panel) according to the H&E staining images. (D) Quantification
of hypertrophied cardiomyocyte size. (E) The HW/BW (heart
weight/body weight) ratio as a cardiac hypertrophy index. The mRNA
levels of (F) Myh6 and Gata4 as markers of cardiac hypertrophy, and
(G) collagen I and collagen III as markers of cardiac fibrosis, as
determined by reverse transcription-quantitative polymerase chain
reaction. Data are presented as the mean ± standard deviation.
*P<0.05 vs. the saline-treated AB group. AB, aortic binding;
H&E, hematoxylin and eosin staining; 4thW AB, 4 weeks post-AB
operation; HW, heart weight; BW, body weight; Myh6, myosin heavy
chain a isoform; GATA4, GATA binding protein 4. |
Telmisartan inhibited NFAT nuclear
translocation and ANP/BNP expression
NFAT is an important nuclear transcriptional factor
involved in cardiac hypertrophy (26). Thus, the authors investigated
whether telmisartan inhibited NFAT nuclear translocation. There are
five subtypes of NFAT in cardiomyocytes, however, it is thought
that NFATc3 and NFATc4 are the most highly expressed in
cardiomyocytes, and therefore NFATc3 and NFATc4 may be the two most
important subtypes (27).
Following the extraction of cardiomyocyte nuclear protein, the
expression levels of NFATc3 and NFATc4 were significantly increased
in the AB group when compared with the sham group (P<0.01;
Fig. 2A and B). However, this
expression was significantly reduced in the telmisartan-treated AB
group when compared with the untreated AB group (P<0.05)
(Fig. 2A and B).
Activated NFAT has been reported to stimulate the
expression of ANP and BNP (28,29).
Thus, the present study investigated whether ANP/BNP expression may
be inhibited by telmisartan treatment. ANP and BNP expression
significantly increased in the hearts of mice from the AB group
when compared with the sham group (P<0.01; Fig. 2C and D). However, telmisartan
administration significantly lowered ANP and BNP levels when
compared to the untreated AB group (P<0.01; Fig. 2C and D). The inhibitory effect of
telmisartan on ANP expression was marginally more pronounced than
its effect on BNP protein levels (Fig.
2C and D).
Telmisartan inhibited cardiac
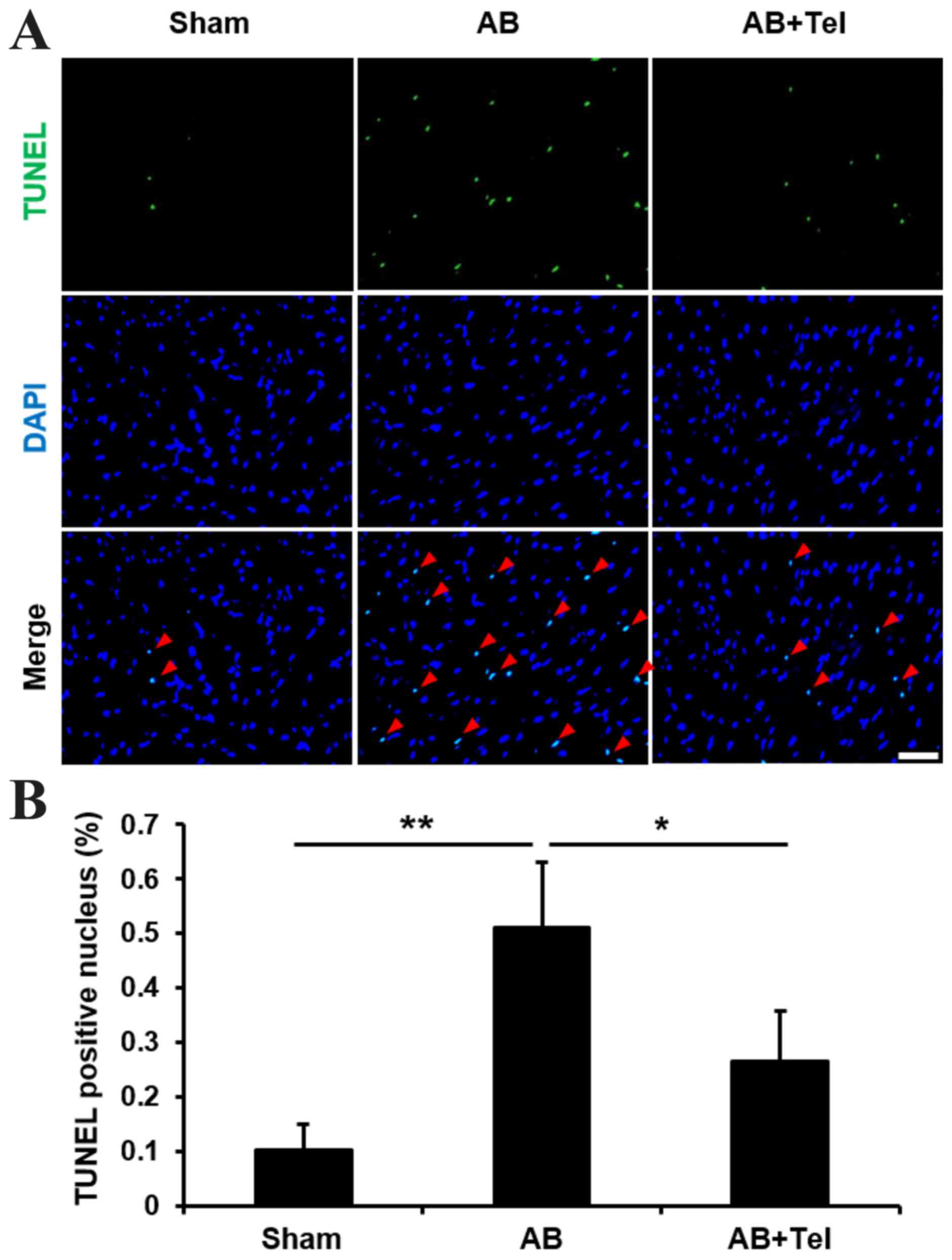
apoptosis
As ANP and BNP reportedly induce apoptosis (30,31),
and cardiac afterload may induce damaging levels of apoptosis in
heart tissues (32), the
anti-apoptotic effects of telmisartan were investigated in the
present study. The terminal deoxynucleotidyl transferase dUTP nick
end labeling (TUNEL) stain revealed that the percentage of
TUNEL-positive nuclei induced by cardiac afterload were
significantly reduced by telmisartan administration when compared
with the untreated AB group (P<0.05; Fig. 3). These results indicate that
telmisartan may inhibit NFAT nuclear translocation, ANP and BNP
expression and cardiomyocyte apoptosis to protect against cardiac
afterload-induced cardiac hypertrophy.
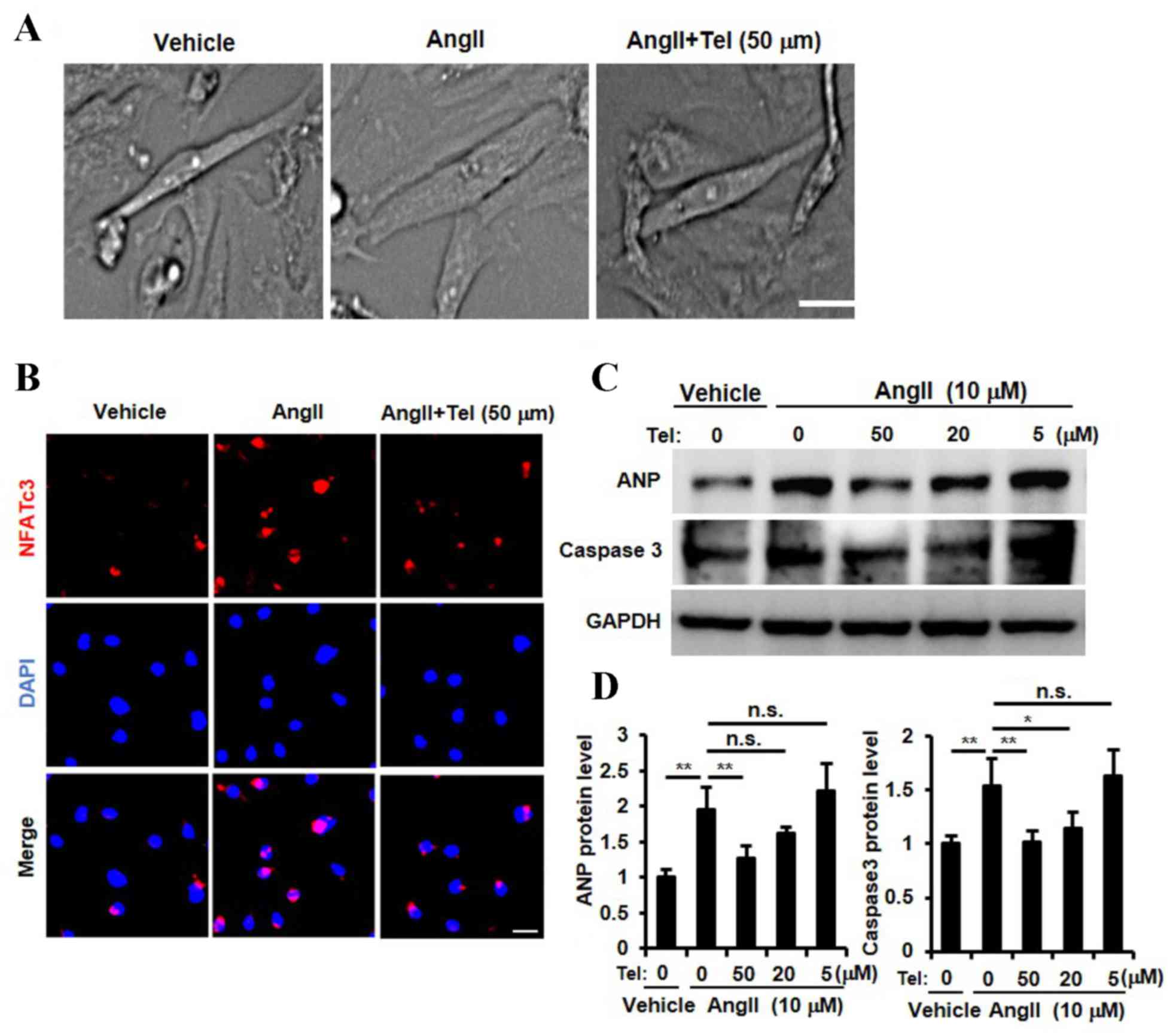
Telmisartan suppressed cardiomyocyte
hypertrophy, NFAT nuclear translocation, ANP expression and
cardiomyocyte apoptosis in vitro
The results presented so far provide evidence to
suggest that telmisartan may inhibit cardiac hypertrophy, NFATc3
and NFATc4 nuclear translocation, ANP expression and cardiac cell
apoptosis in a mouse model of cardiac afterload. In order to
investigate whether these biochemical effects are detectable in
cardiomyocytes directly, a primary neonatal rat cardiomyocyte
culture was established and stimulated with Ang II. As shown in
Fig. 4A, telmisartan appeared to
inhibit Ang II-induced cardiomyocyte hypertrophy, which was
consistent with the results observed in the in vivo mouse AB
model (Fig. 1A-E). In order to
determine whether telmisartan may inhibit the nuclear translocation
of NFAT, ANP expression and cardiomyocyte apoptosis, cardiomyocytes
were stimulated with three different concentrations of telmisartan
(5, 10 and 50 µm). A high concentration of telmisartan (50 µm)
inhibited Ang II-induced NFATc3 nuclear translocation (Fig. 4B). In addition, ANP expression was
significantly inhibited by telmisartan in a dose-dependent manner
(P<0.01, 50 µM vs. 0 µM telmisartan; Fig. 4C and D). Furthermore, the Ang
II-stimulated increase in caspase 3 protein expression levels,
which is a known marker for apoptosis (33), was inhibited by telmisartan in a
dose-dependent manner (P<0.01, 50 µM vs. 0 µM telmisartan;
Fig. 4C and D). These results
indicated that telmisartan may inhibit NFAT nuclear translocation,
ANP expression, Ang II-induced cardiomyocyte apoptosis and suppress
cardiomyocyte hypertrophy in vitro.
Discussion
Angiotensin-converting enzyme inhibitors (ACEIs) and
ARBs are important agents in the treatment of hypertension,
however, ~20% of patients tolerate ARBs but are unable tolerate
ACEIs (34). This indicates that
ARBs may be more effective for broader clinical use. In a previous
study involving the AB mouse cardiac hypertrophy model, the AT1
receptor was activated without the involvement of Ang II (35). Candesartan, olmesartan and Losartan
were reported to inhibit pressure overload-induced cardiac
hypertrophy even in the absence of Ang II (36), and telmisartan directly inhibited
cardiomyocyte hypertrophy in primary rat cardiomyocyte cultures
(37). These reports indicate that
telmisartan may serve an effective role in inhibiting cardiac
hypertrophy.
Although NFAT has been reported as a universal
factor for the induction of cardiac hypertrophy, it is unclear
whether NFAT activation may be involved in the suppression of
cardiac hypertrophy by telmisartan. In the present study,
telmisartan inhibited cardiomyocyte hypertrophy in a mouse model of
cardiac afterload, with a reduction in cardiomyocyte size and
reduced expression of cardiomyocyte hypertrophy and cardiac
fibrosis markers, which is consistent with previous reports
(38). However, it was previously
reported that telmisartan does not exert its inhibitory effects on
cardiomyocyte hypertrophy following 2 weeks of administration in
the absence of Ang II (36), which
implies that telmisartan may require additional time to exert its
inhibitory effects.
NFAT is thought to be important for inducing
cardiomyocyte hypertrophy (39).
The authors of the present study therefore hypothesized that
telmisartan may inhibit NFAT activation. The results demonstrated
that telmisartan inhibited the cardiac overload-induced activation
of NFATc3 and NFATc4. Previous studies have demonstrated that
extracellular signal-regulated kinases (Erk) 1/2 and NFAT form a
complex in cardiomyocytes. Erk1/2 directly regulate NFAT DNA
binding activation, and exert their effects on NFAT synergy without
increasing NFAT translocation and translation. Erk1/2 and NFAT are
together required to induce cardiac hypertrophy (40,41).
Although the inhibition of NFAT nuclear translocation by
telmisartan was only investigated in the present study, it remains
formally possible that Erk1/2 and NFAT form a complex to exert
their cardiomyocyte hypertrophy-inducing effects. The inhibition of
NFAT nuclear translocation may be dependent on Erk1/2 inhibition,
however, the present study did not investigate this possibility.
Nevertheless, the Erk1/2-NFAT complex may additionally be inhibited
by telmisartan.
Previous studies have revealed that peroxisome
proliferator-activated receptor-γ (PPARγ) activation may be
involved in the underlying mechanisms of telmisartan-induced
inhibition of cardiomyocyte hypertrophy (42), and PPARγ and its ligand may further
inhibit the nuclear translocation of NFAT (43). These studies indicate that
telmisartan-induced inhibition of NFAT nuclear translocation may be
dependent on telmisartan-induced PPARg activation.
NFAT has been observed to participate in
pathological cardiac hypertrophy (39), and activated NFAT promotes ANP or
BNP release and induces cell apoptosis (41,44).
The present study demonstrated that telmisartan inhibited ANP/BNP
expression and apoptosis in the heart and cardiomyocytes. Notably,
telmisartan did not inhibit BNP expression as effectively as ANP
expression. Therefore, it is possible that an additional pathway
involving NFAT and BNP exists. Alternatively, telmisartan may have
inhibited NFAT as well as an additional complex involving NFAT.
Following this investigation there are two points
that require further investigation in future studies. Firstly,
whether telmisartan inhibits the Erk1/2 and NFAT complex, and
secondly, whether the telmisartan-mediated inhibition of NFAT
nuclear translocation is dependent on PPARg activation.
In conclusion, the present study demonstrated that
telmisartan suppressed cardiomyocyte hypertrophy in vivo and
in vitro, potentially by suppressing cardiomyocyte ANP/BNP
expression and apoptosis, which may be dependent on the inhibition
of NFAT nuclear translocation. These results may provide a novel
insight into the mechanism of telmisartan-induced cardiomyocyte
hypertrophy inhibition.
References
|
1
|
Frohlich ED, Apstein C, Chobanian AV,
Devereux RB, Dustan HP, Dzau V, Fauad-Tarazi F, Horan MJ, Marcus M,
Massie B, et al: The heart in hypertension. N Engl J Med.
327:998–1008. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Curtiss C, Cohn JN, Vrobel T and Franciosa
JA: Role of the renin-angiotensin system in the systemic
vasoconstriction of chronic congestive heart failure. Circulation.
58:763–770. 1978. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
McClellan KJ and Markham A: Telmisartan.
Drugs. 56:1039–1046. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wienen W, Hauel N, Van Meel JC, Narr B,
Ries U and Entzeroth M: Pharmacological characterization of the
novel nonpeptide angiotensin II receptor antagonist, BIBR 277. Br J
Pharmacol. 110:245–252. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Amerena J, Pappas S, Ouellet JP, Williams
L and O'Shaughnessy D: ABPM comparison of the anti-hypertensive
profiles of telmisartan and enalapril in patients with
mild-to-moderate essential hypertension. J Int Med Res. 30:543–552.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Saha L: Comparison of the efficacy and
tolerability of telmisartan and enalapril in patients of mild to
moderate essential hypertension. Indian J Pharmacol. 43:3602011.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Neutel JM, Littlejohn TW, Chrysant SG and
Singh A: Telmisartan Study Group: Telmisartan/Hydrochlorothiazide
in comparison with losartan/hydrochlorothiazide in managing
patients with mild-to-moderate hypertension. Hypertens Res.
28:555–563. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wienen W and Entzeroth M: Effects on
binding characteristics and renal function of the novel,
non-peptide angiotensin II antagonist BIBR277 in the rat. J
Hypertens. 12:119–128. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Makino H, Haneda M, Babazono T, Moriya T,
Ito S, Iwamoto Y, Kawamori R, Takeuchi M and Katayama S: INNOVATION
Study Group: Prevention of transition from incipient to overt
nephropathy with telmisartan in patients with type 2 diabetes.
Diabetes Care. 30:1577–1578. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Derosa G, Ragonesi PD, Mugellini A,
Ciccarelli L and Fogari R: Effects of telmisartan compared with
eprosartan on blood pressure control, glucose metabolism and lipid
profile in hypertensive, type 2 diabetic patients: A randomized,
double-blind, placebo-controlled 12-month study. Hypertens Res.
27:457–464. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Honjo S, Nichi Y, Wada Y, Hamamoto Y and
Koshiyama H: Possible beneficial effect of telmisartan on glycemic
control in diabetic subjects. Diabetes Care. 28:4982005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nagel JM, Tietz AB, Göke B and Parhofer
KG: The effect of telmisartan on glucose and lipid metabolism in
nondiabetic, insulin-resistant subjects. Metabolism. 55:1149–1154.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gohlke P, Weiss S, Jansen A, Wienen W,
Stangier J, Rascher W, Culman J and Unger T: AT1 receptor
antagonist telmisartan administered peripherally inhibits central
responses to angiotensin II in conscious rats. J Pharmacol Exp
Ther. 298:62–70. 2001.PubMed/NCBI
|
|
14
|
Mogi M, Li JM, Tsukuda K, Iwanami J, Min
LJ, Sakata A, Fujita T, Iwai M and Horiuchi M: Telmisartan
prevented cognitive decline partly due to PPAR-gamma activation.
Biochem Biophys Res Commun. 375:446–449. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Böhm M, Lippoldt A, Wienen W, Ganten D and
Bader M: Reduction of cardiac hypertrophy in TGR(mREN2)27 by
angiotensin II receptor blockade. Mol Cell Biochem.
163–164:217–221. 1996. View Article : Google Scholar
|
|
16
|
Mattioli AV, Zennaro M, Bonatti S, Bonetti
L and Mattioli G: Regression of left ventricular hypertrophy and
improvement of diastolic function in hypertensive patients treated
with telmisartan. Int J Cardiol. 97:383–388. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Maejima Y, Okada H, Haraguchi G, Onai Y,
Kosuge H, Suzuki J and Isobe M: Telmisartan, a unique ARB, improves
left ventricular remodeling of infarcted heart by activating PPAR
gamma. Lab Invest. 91:932–944. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Siragy H: Angiotensin II receptor
blockers: Review of the binding characteristics. Am J Cardiol.
84:3S–8S. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
D'Ascenzi F, Pelliccia A, Corrado D,
Cameli M, Curci V, Alvino F, Natali BM, Focardi M, Bonifazi M and
Mondillo S: Right ventricular remodelling induced by exercise
training in competitive athletes. Eur Heart J Cardiovasc Imaging.
17:301–307. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bernardo BC, Weeks KL, Pretorius L and
McMullen JR: Molecular distinction between physiological and
pathological cardiac hypertrophy: Experimental findings and
therapeutic strategies. Pharmacol Ther. 128:191–227. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Heineke J and Molkentin JD: Regulation of
cardiac hypertrophy by intracellular signalling pathways. Nat Rev
Mol Cell Biol. 7:589–600. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Irani RA, Zhang Y, Blackwell SC, Zhou CC,
Ramin SM, Kellems RE and Xia Y: The detrimental role of angiotensin
receptor agonistic autoantibodies in intrauterine growth
restriction seen in preeclampsia. J Exp Med. 206:2809–2822. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rockman HA, Ross RS, Harris AN, Knowlton
KU, Steinhelper ME, Field LJ, Ross J Jr and Chien KR: Segregation
of atrial-specific and inducible expression of an atrial
natriuretic factor transgene in an in vivo murine model of cardiac
hypertrophy. Proc Natl Acad Sci USA. 88:8277–8281. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chlopcíková S, Psotová J and Miketová P:
Neonatal rat cardiomyocytes-a model for the study of morphological,
biochemical and electrophysiological characteristics of the heart.
Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 145:49–55.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Molkentin JD: Calcineurin-NFAT signaling
regulates the cardiac hypertrophic response in coordination with
the MAPKs. Cardiovasc Res. 63:467–475. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Pu WT, Ma Q and Izumo S: NFAT
transcription factors are critical survival factors that inhibit
cardiomyocyte apoptosis during phenylephrine stimulation in vitro.
Circ Res. 92:725–731. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tokudome T, Horio T, Kishimoto I, Soeki T,
Mori K, Kawano Y, Kohno M, Garbers DL, Nakao K and Kangawa K:
Calcineurin-nuclear factor of activated T cells pathway-dependent
cardiac remodeling in mice deficient in guanylyl cyclase A, a
receptor for atrial and brain natriuretic peptides. Circulation.
111:3095–3104. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Liang F, Lu S and Gardner DG:
Endothelin-dependent and -independent components of
strain-activated brain natriuretic peptide gene transcription
require extracellular signal regulated kinase and p38
mitogen-activated protein kinase. Hypertension. 35:188–192. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wu CF, Bishopric NH and Pratt RE: Atrial
natriuretic peptide induces apoptosis in neonatal rat cardiac
myocytes. J Biol Chem. 272:14860–14866. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Suenobu N, Shichiri M, Iwashina M, Marumo
F and Hirata Y: Natriuretic peptides and nitric oxide induce
endothelial apoptosis via a cGMP-dependent mechanism. Arterioscler
Thromb Vasc Biol. 19:140–146. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Diep QN, El Mabrouk M, Yue P and Schiffrin
EL: Effect of AT(1) receptor blockade on cardiac apoptosis in
angiotensin II-induced hypertension. Am J Physiol Heart Circ
Physiol. 282:H1635–H1641. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Porter AG and Jänicke RU: Emerging roles
of caspase-3 in apoptosis. Cell Death Differ. 6:99–104. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Telmisartan Randomised AssessmeNt Study in
ACE iNtolerant subjects with cardiovascular Disease (TRANSCEND)
Investigators. Yusuf S, Teo K, Anderson C, Pogue J, Dyal L, Copland
I, Schumacher H, Dagenais G and Sleight P: Effects of the
angiotensin-receptor blocker telmisartan on cardiovascular events
in high-risk patients intolerant to angiotensin-converting enzyme
inhibitors: A randomised controlled trial. Lancet. 372:1174–1183.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zou Y, Akazawa H, Qin Y, Sano M, Takano H,
Minamino T, Makita N, Iwanaga K, Zhu W, Kudoh S, et al: Mechanical
stress activates angiotensin II type 1 receptor without the
involvement of angiotensin II. Nat Cell Biol. 6:499–506. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Li L, Zhou N, Gong H, Wu J, Lin L, Komuro
I, Ge J and Zou Y: Comparison of angiotensin II type 1-receptor
blockers to regress pressure overload-induced cardiac hypertrophy
in mice. Hypertens Res. 33:1289–1297. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chang WH, Yan JJ, Li X, Guo HY and Liu Y:
Effects of telmisartan on angiotensin II-induced cardiomyocyte
hypertrophy and p-ERK1/2 phosphorylation in
rat cultured cardiomyocytes. Asian Biomed. 5:459–465. 2011.
|
|
38
|
Muller P, Kazakov A, Semenov A, Jagoda P,
Friedrich EB, Böhm M and Laufs U: Ramipril and telmisartan exhibit
differential effects in cardiac pressure overload-induced
hypertrophy without an additional benefit of the combination of
both drugs. J Cardiovasc Pharmacol Ther. 18:87–93. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wilkins BJ, Dai YS, Bueno OF, Parsons SA,
Xu J, Plank DM, Jones F, Kimball TR and Molkentin JD:
Calcineurin/NFAT coupling participates in pathological, but not
physiological, cardiac hypertrophy. Circ Res. 94:110–118. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sanna B, Bueno OF, Dai YS, Wilkins BJ and
Molkentin JD: Direct and indirect interactions between
calcineurin-NFAT and MEK1-extracellular signal-regulated kinase 1/2
signaling pathways regulate cardiac gene expression and cellular
growth. Mol Cell Biol. 25:865–878. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Robbs BK, Lucena PI and Viola JP: The
transcription factor NFAT1 induces apoptosis through cooperation
with Ras/Raf/MEK/ERK pathway and upregulation of TNF-α expression.
Biochim Biophys Acta. 1833:2016–2028. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Yamagishi S and Takeuchi M: Telmisartan is
a promising cardiometabolic sartan due to its unique
PPAR-gamma-inducing property. Med Hypotheses. 64:476–478. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Bao Y, Li R, Jiang J, Cai B, Gao J, Le K,
Zhang F, Chen S and Liu P: Activation of peroxisome
proliferator-activated receptor gamma inhibits endothelin-1-induced
cardiac hypertrophy via the calcineurin/NFAT signaling pathway. Mol
Cell Biochem. 317:189–196. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Molkentin JD, Lu JR, Antos CL, Markham B,
Richardson J, Robbins J, Grant SR and Olson EN: A
calcineurin-dependent transcriptional pathway for cardiac
hypertrophy. Cell. 93:215–228. 1998. View Article : Google Scholar : PubMed/NCBI
|