Introduction
Diffuse axonal injury (DAI) is the most common
pathological feature of traumatic brain injury (TBI) due to high
mortality and morbidity rates following injury (1). DAI typically results from head
rotational acceleration/deceleration following impact, and is
characterized by swellings or varicosities of axons and large
terminal bulbs (2,3). The clinical manifestation of DAI in
the long term includes: Disorders of consciousness, difficult
clinical diagnosis and bad prognosis, often resulting for vehicle
collisions, blast exposure and falls (4,5).
Although diffusion tensor imaging (DTI) has long been regarded as a
valuable technique for evaluating the white matter injury following
DAI in clinic (6–8), there remain no suitably effective
drugs to attenuate the formation and progression of DAI in
patients.
Neuronal apoptosis is a major contributor to the
secondary neuronal injury following TBI through multiple cellular
mechanisms, including Ca2+ overload, production of
reactive oxygen/nitrogen species (9–12)
and glutamate-mediated excitotoxicity (13,14).
Axonal injury is a common consequence of TBI (15). The characteristic axonal
pathological changes include axonal swelling and distortion, the
formation of axonal bulbs, and axotomy (16,17).
Therefore, treatment with an agent that has the ability to inhibit
the resulting neuronal apoptosis and axonal injury may potentially
improve multiple aspects of the response of the brain to
trauma.
FK506 (also known as tacrolimus) is an
immunosuppressive drug, frequently used in the field of
transplantation medicine in order to reduce allograft rejection.
Using a middle cerebral artery occlusion model, the neuroprotection
of FK506 was demonstrated to act via inhibition of apoptotic and
necrotic cell death, suppression of microglia activation and
alterations in cytokine expression, including interleukin (IL)-1β,
IL-6 and tumor necrosis factor (TNF)-α (18). A previous study identified that
FK506 may be able to inhibit neuronal apoptosis by maintaining
Bcl-2-associated agonist of cell death protein turnover, inhibiting
cytochrome C release from mitochondria (19), and inhibiting the release of
arachidonic acid (20). In
addition, studies have demonstrated that FK506 may attenuate the
axonal injury and improve axonal survival or regeneration by
inhibiting the calcineurin (CaN) activity or the post-traumatic
compound action potential after TBI (21–23).
Death-associated protein kinase 1 (DAPK1) is a novel
and specific cell death signaling molecule, which is directly
linked to glutamate receptor channels (24). It is a newly-identified
Ca2+/calmodulin-dependent serine/threonine protein
kinase (25) that serves a role in
several modes of cell death, including apoptosis and autophagy
(26).
Growth-associated protein-43 (GAP-43) is a specific
phosphatase protein in the vertebrate nerve cell membrane, which is
closely related to neural development, axonal plasticity and
synaptic remodeling (27). Several
studies have demonstrated that GAP-43 is upregulated in nerve
regeneration (28) and plasticity
following TBI in rat models (29).
Neurofilament-H (NF-H) is the most abundant protein component of
neurons and is released in large amounts from damaged or dying
neurons (30). NF-H is relatively
simple to detect as a predictive biomarker of the outcome following
TBI (31). Furthermore, NF-H only
exists in the axons, not in the neuronal cell bodies in
physiological condition. Following TBI, the neuronal cells
synthesize a large amount of NF-H, to adapt to the need of nerve
regeneration. Therefore, it could not only reflect the neuronal
function, but also reflect the situation of neurite regeneration
(30,31).
As a result of previous studies, the present study
hypothesized that FK506 may be therapeutically efficacious in
neuronal apoptosis and axonal injury following ischemia or nerve
trauma. To the best of our knowledge, the present study is the
first to have examined the possible functional role of FK506 in
neuronal apoptosis or regeneration following experimental DAI.
Neuronal apoptosis and axonal pathological alterations were
examined in a rat model, following induction of DAI via lateral
head rotation trauma. Furthermore, the protective effects of FK506
on brain injury following DAI were investigated.
Materials and methods
Model of DAI in vivo and
treatments
A total of 90 healthy, adult male Sprague-Dawley
rats (weight, 250–300 g, 8–10 weeks of age), with the same genetic
background, were obtained from the Animal Experimental Center of
Medical College of Xi'an Jiaotong University (License number SCXK
(Shaanxi) 2007-001). All animals received humane care in compliance
with the Guide for the Care and Use of Laboratory Animals from the
National Institutes of Health (publication no. 80–23). Experimental
and surgical procedures, as well as post-operative care were each
approved by the Biomedical Ethics Committee of Medical College of
Xi'an Jiaotong University (Xi'an, China). Animals were housed and
fed in a temperature- and humidity-controlled environment with a
standardized light-dark cycle (12 h/12 h). Food and water were
available ad libitum.
Rats were randomly divided into three groups: A
control group (Sham group, n=30), a DAI/vehicle-treated group
(DAI+Vehicle group, n=30) and a DAI/FK506-treated group (DAI+FK506
group, n=30). Each of the groups was randomly divided into three
subgroups that represented 1, 3 and 7 days post-injury (PI). The
present study involved the establishment of a DAI model in rats by
lateral head rotation device (32), which was modified from the
Xiaosheng et al (33,34).
All rats in DAI+Vehicle and DAI+FK506 groups after weighting were
anesthetized by intraperitoneal injection of chloral hydrate (30
mg/kg) and placed in the prone position. Following anesthesia, the
head of the rats were fixed in the rat instant head rotating injury
device, the rat head was horizontally secured to the lateral head
rotation device by two lateral ear bars, a head clip and an
anterior teeth hole, with its body 30° oblique to the top of the
laboratory table. For the injury group, following pushing the
trigger, the device rapidly rotated the rat head through a 90°
angle laterally (i.e., in the coronal plane). The rats were placed
in separated cages, maintaining the room temperature between 18 and
26°C and the indoor relative humidity at 40–70%. Primary coma was
observed in all injured rats. Rats that succumbed to their injuries
were excluded and later replaced by new rats. Control rats (Sham
group) only underwent anesthesia and were fixed to the device, but
were not subjected to injury.
Animals also received either FK506 or a vehicle
(0.9% sterile saline) delivered intravenously 30 min pre-DAI. A
single 3 mg/kg of FK506 in 0.9% sterile saline to a total volume of
1.0 ml was infused over a 10 min period to ensure that the rate of
injection did not significantly elevate MABP (21,22).
The vehicle was administrated using the same protocol. FK506
(Tacrolimus) was purchased from Abcam (Cambridge, UK; cat. no.
ab120223).
Embedding and sectioning
Euthanasia was conducted at 1, 3 and 7 days
post-injury following being freed from the injury device. Rats in
the Sham-operated group were euthanized at the same times. Half of
the rats (n=45) were sacrificed and perfused with 250 ml of normal
saline only. The brain stem and the hippocampus were collected for
western blotting. The remaining rats (n=45) were sacrificed and
perfused with 250 ml of normal saline followed by 400 ml of 4%
paraformaldehyde in 0.01 M PBS. The whole brain removed and
post-fixed in 4% paraformaldehyde solution, dehydrated via a graded
ethanol series, vitrified with dimethyl benzene, embedded with
paraffin and sectioned into 10 µm thick sections using a microtome.
A total of five sections, including the hippocampus tissue and
brain stem tissue from each animal, were randomly chosen and
mounted on poly-L-lysine coated slides (cat. no. P4981; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) for Glees-Marsland
Silver staining, terminal deoxynucleotidyl transferase dUTP nick
end labeling (TUNEL) staining, immunofluorescence and
immunohistochemical staining.
Immuohistochemistry
The brain sections were deparaffinized in xylene and
hydrated in a decreasing gradient of alcohol to distilled water.
Endogenous peroxidase activity was blocked with 3%
H2O2 for 5 min, followed by a brief rinse in
distilled water and a 15 min wash in PBS. Sections were placed in
0.01 mol/l citrate buffer (pH 7.2) and heated in a microwave oven
at 95°C for 30 min. Sections were cooled at room temperature for 20
min and rinsed again in PBS. Non-specific protein binding was
blocked by 30 min of incubation in normal goat serum (cat. no.
16210064; Gibco; Thermo Fisher Scientific, Inc.) at room
temperature, followed by incubation with primary antibodies: Rabbit
anti-DAPK1 monoclonal antibody (dilution, 1:500; cat. no. 3798-1;
Epitomics, Burlingame, CA, USA), mouse anti-NF-H monoclonal
antibody (dilution, 1:200; cat. no. 2836; Cell Signaling
Technology, Inc., Danvers, MA, USA) and mouse anti-GAP-43
monoclonal antibody (dilution, 1:500; cat. no. sc-33705; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA) for 24 h at 4°C, followed by
a 15 min wash in PBS. Sections were then incubated with goat
anti-rabbit (dilution, 1:200; cat. no. 31460; Thermo Fisher
Scientific, Inc.) or goat anti-mouse IgG-biotin (dilution, 1:200;
cat. no. 31431; Thermo Fisher Scientific, Inc.) for 30 min at 37°C,
and sections were washed with PBS for 15 min following each step.
Diaminobenzidine was used as the chromogen, and hematoxylin was
used as the counterstain. Sections incubated with PBS in the
absence of primary antibodies were used as negative controls.
Microscopic observation of the stained sections was performed by an
experienced pathologist blind to the experimental conditions. The
immunoreactivity of all of the molecular markers was analyzed using
Image-Pro Plus 6.0 software (Media Cybernetics, Inc., Rockville,
MD, USA) in five microscopic fields (magnification, ×200).
Glees-Marsland Silver staining
The brain paraffin-embedded sections at 1 day
post-injury were examined by Glees-Marsland Silver staining methods
as previously described (35,36).
Nerve fibers were stained black. In each slice, five microscopic
fields (magnification, ×400) were randomly selected and their
images captured.
TUNEL staining
The brain paraffin-embedded sections were also
examined by TUNEL assay using the DeadEnd colorimetric TUNEL system
detection kit (cat. no. G7130; Promega Corporation, Madison, WI,
USA) as described previously (37). Brain tissue sections were initially
deparaffinized with xylene, rehydrated through descending
concentrations of ethanol and rinsed for 15 min in 0.1 M PBS and
then treated with 20 µg/ml of Proteinase K for 20 min at room
temperature. Samples were treated with 3%
H2O2 in methanol for 20 min to inactivate
endogenous peroxidase. Following washing with PBS, specimens were
incubated in the rTdT reaction mixture (100 µl; combining 98 µl of
Equilibration Buffer, 1 µl of Biotinylated Nucleotide Mix and 1 µl
of rTdT Enzyme) at 4°C overnight. Following incubation, all the
sections were rinsed in PBS and incubated with horseradish
peroxidase (dilution, 1:500) for 30 min at room temperature. Then,
the sections were washed extensively with PBS for 5 min and treated
with DAB solution (30 mg DAB and 200 µl
H2O2/100 ml PBS) for 10 min at room
temperature in the dark. Following washing under running water, all
the sections were counterstained with hematoxylin for 30 sec.
Finally, the sections were dehydrated in increasing graded ethanol,
cleared in xylene and mounted with a cover slip. With this method,
apoptotic nuclei were identified by the presence dark brown
staining.
Immunofluorescence staining
The brain tissue sections were de-paraffinized in
xylene and hydrated in a decreasing gradient of alcohol to
distilled water as above. Then sections were blocked with 10%
normal serum blocking solution species the same as the secondary
antibody, containing 3% (w/v) BSA, 0.1% Triton X-100, and 0.05%
Tween-20 2 h at room temperature in order to avoid unspecific
staining. The brain sections were incubated with primary antibodies
for anti-NF-H mouse monoclonal antibody (dilution, 1:400; cat. no.
2836; Cell Signaling Technology, Inc.) overnight at 4°C. Then
sections were incubated with fluorescein isothiocyanate conjugated
(FITC)-secondary antibody (goat anti-mouse; 1:500, cat. no. A24513;
Invitrogen; Thermo Fisher Scientific, Inc.) for 2 h at room
temperature. Sections were covered with DAPI (0.1 mg/ml in PBS;
cat. no. ab104139; Abcam, Cambridge, MA, USA) for 40 min at room
temperature and subsequently examined with a Leica fluorescence
microscope (DM5000B; Leica Microsystems GmbH, Wetzlar,
Germany).
Western blot analysis
Total protein was isolated from rat brain stem and
hippocampus tissues using ice-cold radioimmunoprecipitation assay
buffer (cat. no. 89900; Thermo Fisher Scientific, Inc). Total
protein concentrations were measured using the BCA Protein Assay
kit (cat. no. 23229; Thermo Fisher Scientific, Inc.). Samples (60
µg/lane) were separated on an SDS gel (8% for DAPK1 or 10% for
GAP43 and β-actin) and electrotransferred onto polyvinylidene
difluoride membranes. Following incubation for 2 h in a blocking
solution [5% non-fat milk in 20 mM Tris-HCl, 150 mM NaCl, 0.1%
Tween-20 (TBST)], membranes were blotted with primary antibodies
against DAPK1 (dilution, 1:1,000; cat. no. 3798-1; Epitomics),
GAP-43 (dilution, 1:1,000; cat. no. sc-33705; Santa Cruz
Biotechnology, Inc.) or β-actin (dilution, 1:1,000; cat. no.
ab8226; Abcam) overnight at 4°C. Following extensive washing with
TBST buffer, membranes were incubated with HRP-conjugated
anti-rabbit/mouse secondary antibodies (1:1,000; cat. no.
31460/31430; Thermo Fisher Scientific, Inc.) for 1 h at room
temperature. Specific bands were detected using a ECL Western
Blotting Detection System (cat. no. WBKLS0100; EMD Millipore,
Billerica, MA, USA). Western blotting was performed in at least six
independent experiments. Membranes were scanned and the density of
the bands analyzed with Quantity one software (version 4.62;
Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Statistical analysis
SPSS software (version, 18.0; SPSS Inc., Chicago,
IL, USA) was used for statistical analyses. All data were presented
as mean ± standard error of the mean (n=5). Comparisons among
multiple groups were performed using one-way analysis of variance.
Comparisons between the two groups were performed using Fisher's
Least Significant Difference test. P<0.05 was considered to
indicate a statistically significant difference.
Results
General observation
No animals succumbed to the treatments in the Sham
groups, and no obvious neurological deficits were observed.
However, a series of neurological symptoms had been discovered in
the rats following DAI, including the disturbance of consciousness,
reduction of physical activity, a weakened response to stimulation,
drowsiness, instability in walking, weakened balance ability, loss
of weight and even paralysis of limbs. Both Glees-Marsland silver
staining and immunocytochemical experiments for NF-H demonstrated
well changes of the axonal pathology. The rat brain tissue of the
normal control group under the light microscope demonstrated that
the nerve axons were smooth, with uniform thickness and arranged
closely in the cerebral white matter, however the DAI model group
in cerebral white matter present visible tortuous and swollen
axons, even the axonal fracture and axonal ball formation (Fig. 1A and B). These results suggested
that the DAI model was successfully established, allowing its use
in the subsequent experiment. In addition, the pathological changes
in axons were attenuated significantly in the FK506 group when
compared with the DAI+Vehicle group (both P<0.05; Fig. 1C and D).
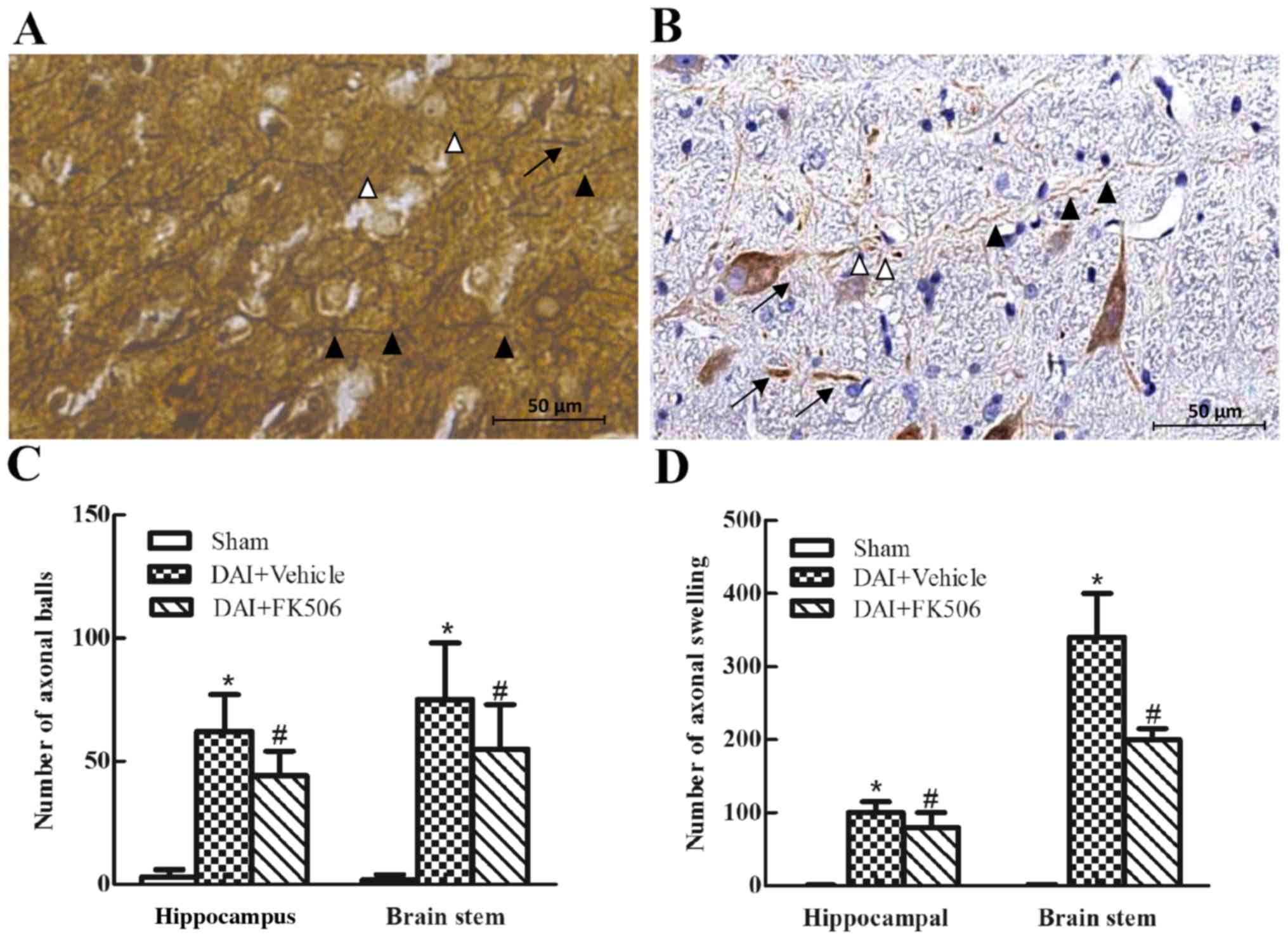 | Figure 1.Immunohistochemical and
Glees-Marsland staining of the hippocampal CA1 region and
brainstem. The axons became varicose, swollen and presented the
formation of axonal retraction balls in the injured group
(magnification, ×400). (A) The axonal pathological alterations
following DAI, as conducted by Glees-Marsland silver staining
(white arrowheads, axonal retraction balls; black arrowheads,
axonal varicose; black triangles, axonal swelling). (B) The axonal
pathological alterations following DAI as conducted by
immunohistochemical staining on NF-H (white arrowheads, axonal
retraction balls; black arrowheads, axonal varicose; black
triangles, axonal swelling). (C) Bar chart presenting the number of
axonal retractions balls in the field of view of the hippocampal
CA1 region and brainstem in Sham, DAI+Vehicle and DAI+FK506 groups.
(D) Bar chart presenting the amount of axonal swelling in the field
of view of the hippocampal CA1 region and brainstem in Sham,
DAI+Vehicle and DAI+FK506 groups. *P<0.05 vs. Sham group;
#P<0.05 vs. DAI+Vehicle group. DAI, diffuse axonal
injury; NF-H, neurofilament-H. |
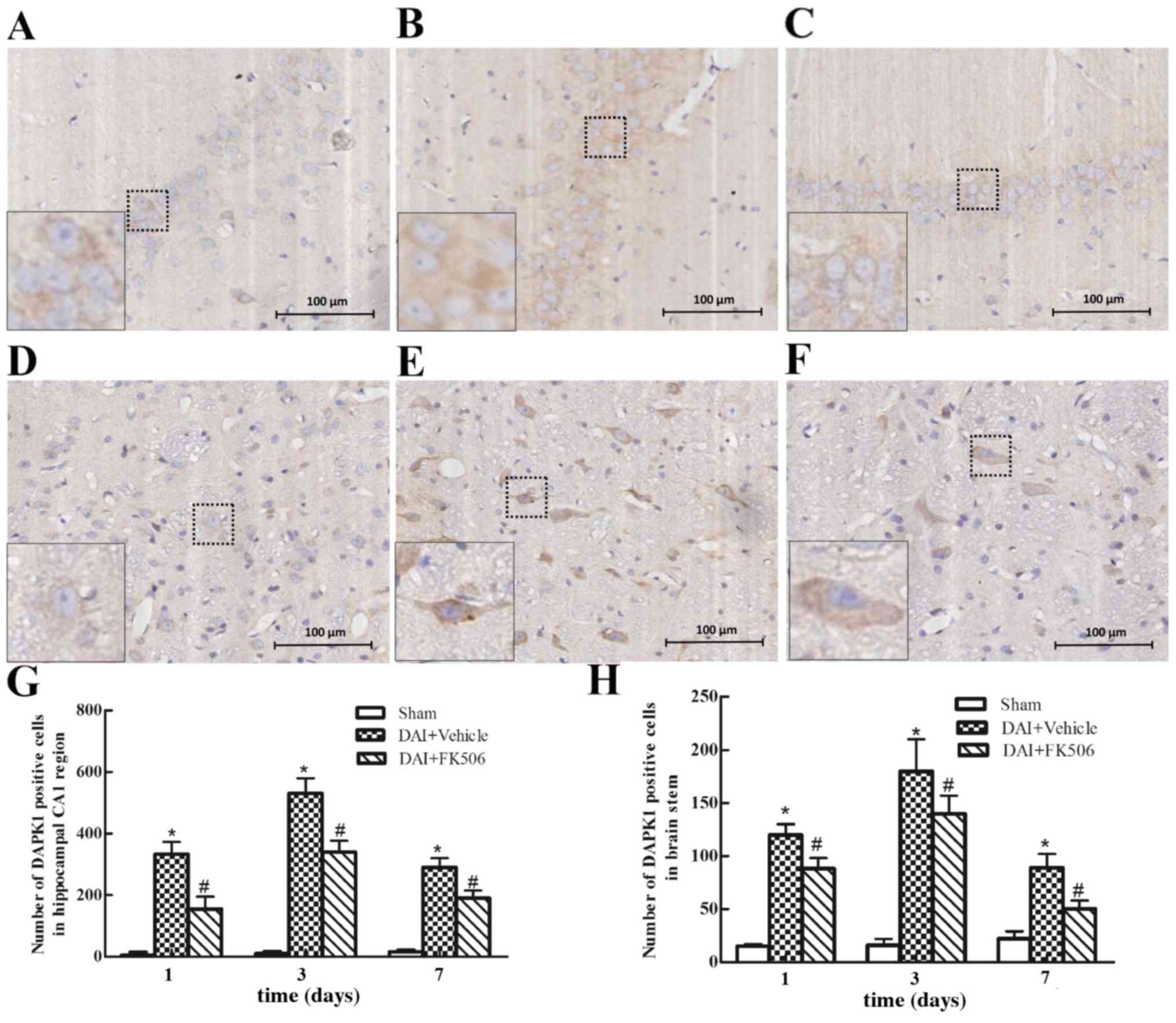
Immunocytochemical analysis of
DAPK1
The expression of DAPK1 through immunohistochemical
analysis was examined (Fig. 2).
The results demonstrated that DAPK1 was primarily localized to the
cytoplasm of the hippocampal CA1 and brain stem regions. DAPK1
exhibited low expression in the Sham-treated group (Fig. 2A and D). In the DAI+Vehicle group,
the positive cell number of DAPK1 was significantly increased at 24
h post-injury when compared with the Sham group, reaching a peak at
day 3, before decreasing at day 7 (all P<0.05; Fig. 2B, E, G and H). The intervention by
FK506 significantly decreased the positive cell number of DAPK1 at
each time point, when compared with the DAI+Vehicle group (all
P<0.05; Fig. 2C, F, G and
H).
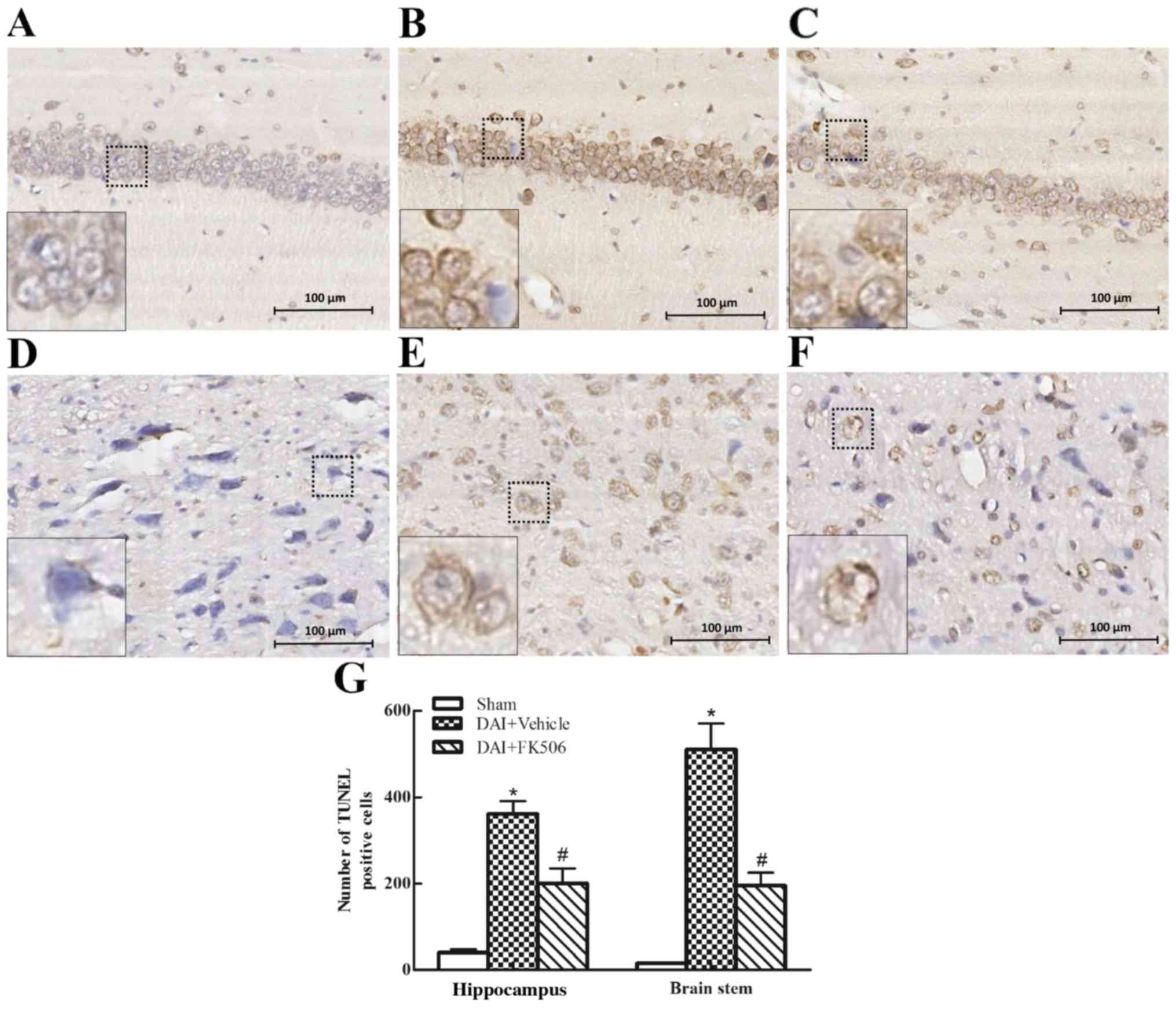
TUNEL Staining
TUNEL staining labels apoptotic cells directly, with
normal cells stained in blue, and apoptotic cells stained in brown
(Fig. 3). The results indicated
that there were ~0 positive neurons in the hippocampus and brain
stem of the Sham group (Fig. 3A and
D). The number of TUNEL-positive cells in the DAI+Vehicle group
was increased compared with the Sham groups (Fig. 3B, E and G), but decreased in the
FK506 group (Fig. 3C, F and
G).
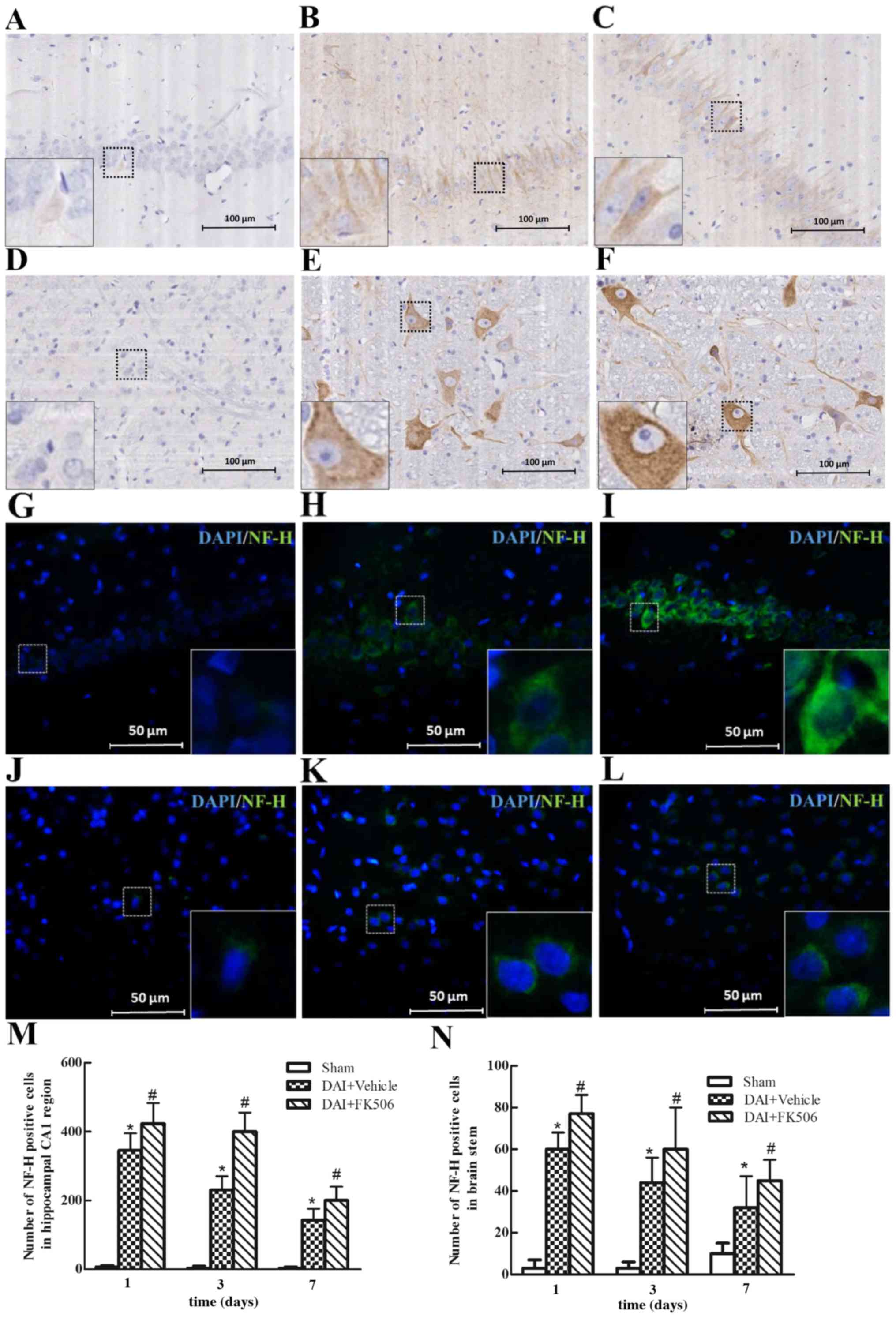
Immunocytochemical analysis for
NF-H
The expression of NF-H was evaluated through
immunohistochemical and immunofluorescence staining (Fig. 4). The results indicated that NF-H
was mainly localized in the axons in the hippocampal CA1 region, as
well as the brain stem region. NF-H was lowly expressed in the
Sham-treated group (Fig. 4A, D, G, J
and M and N). Following DAI, the expression of positive NF-H
was increased and to peak at 1 day post-injury, then gradually
reduced at days 3 and 7 (Fig. 4B, E,
H, K, M and N). However, following the intervention of FK506,
this may increase the expression of positive NF-H at each time
point, and could even be able to maintain a high-level expression
of NF-H at day 3 post-injury (Fig. 4C,
F, I, L, M and N).
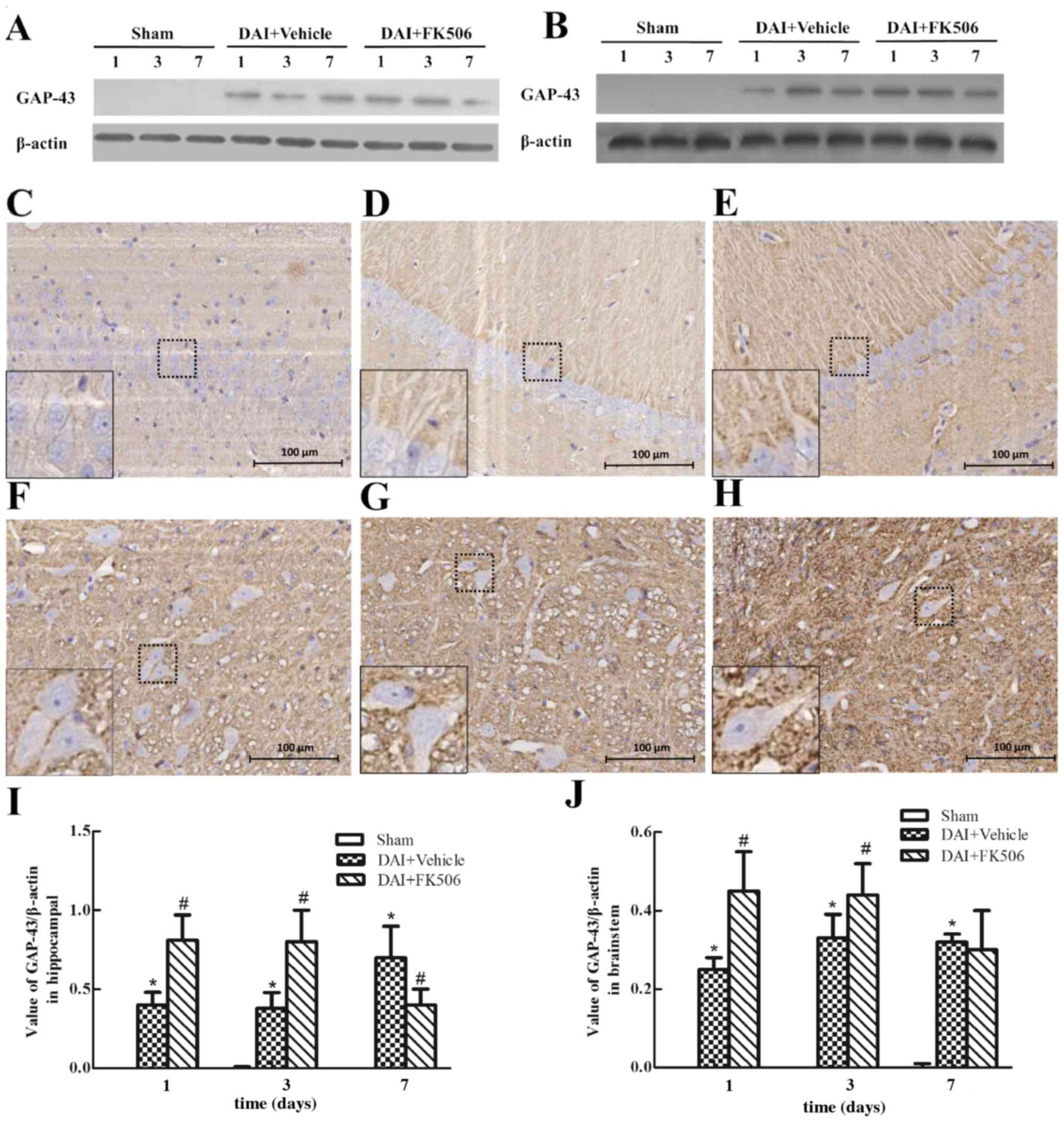
Expression of GAP-43 after DAI
Western blot analysis revealed a significant
upregulation of GAP-43 in the DAI group when compared with the
control and Sham groups (Fig. 5).
In addition, a gradual increase of GAP-43 expression was observed
until it reached a peak 7 days post-injury in the hippocampus and
following DAI. When FK506 was administered, western blot analysis
revealed that FK506 induced a upregulation of GAP-43 expression
when compared with the levels detected in the DAI and vehicle group
(Fig. 5A, B, I and J). The
expression of GAP-43 in the normal rat brain was low, as
demonstrated by immunohistochemistry staining. Following DAI, there
was observation of GAP-43 protein assembled in the membrane of
neuronal cells and neuropil, and the expression of GAP-43 was
highest at day 7 in the hippocampus and at day 3 in the brainstem.
In addition, administration of the FK506 caused the peak of the
expression to advance to day 1 post-injury and maintained a
high-level of expression at day 3 (Fig. 5C-H).
Discussion
In DAI, the axonal degeneration resulted from the
stretching and shearing force, caused by the rapid and sudden
acceleration or deceleration of the brain tissue during a traumatic
event. The secondary injury manifests as a series of dynamic
metabolic imbalances, including loss of ionic homeostasis, injury
of reactive oxygen, production of excitotoxicity and overload of
Ca2+, which ultimately resulted in neuronal cell death
and axonal injury (10,32,38,39).
DAI in the brain may cause neurologic and cognitive impairments,
reflected in consciousness and personality alteration. There have
been few treatments that have been proven to be effective for DAI
patients (38). In the present
study, the molecular mechanism underlying the neuroprotective
actions of FK506 was investigated, examining the neuronal cell
death and axonal injury in the affected rat brain. The DAI model
for the current study was based on the DAI modeling device
developed by Xiaosheng et al (33,34),
which was established by instant rotational acceleration (32). In addition, axonal pathological
changes in the hippocampus and brainstem were observed using
Glees-Marsland silver staining and immunohistochemistry staining of
NF-H. Specific pathological alterations were observed in axons
associated with DAI, including nerve axonal varicose, swelling,
fracture and retraction. The reticular formation of brainstem has
the function of maintaining the biological consciousness, so the
degree of axonal injury in the DAI-affected brain may seriously
affect the degree of consciousness, which is consistent with the
severity of the disorder in the clinic. However, in the present
study, FK506 was identified to significantly reduce the axonal
pathological changes following DAI, which may be associated with
the activity of CaN and MPT (21).
It has been recognized that apoptotic cell death
serves a key role in neuronal cell death in TBI (40,41),
and that limiting the apoptotic cascades following DAI may result
in decreased mortality rates, improve functional outcome scores,
shorter hospital stays and decrease the charges for healthcare
(10,38,39).
In the present study, the aim was to evaluate the effect of FK506
on the apoptotic cell death pathway following DAI, the expression
of DAPK1 was analyzed, and measured the number of TUNEL-positive
cells in the hippocampal CA1 region and brainstem. The authors
identified that the expression of DAPK1 was significantly increased
following DAI, and the apoptosis of neurons in hippocampus and
brainstem was significantly increased at the same time. Previous
studies indicated that DAPK1 serves a significant role in several
modes of cell death, including apoptosis and autophagy (26). The apoptosis induced by DAPK1 is
involved in Fas- (42),
interferon-γ-, TNF- (43),
ceramide- (26), and p53-
(44,45) mediated apoptosis, as well as in the
disruption of matrix survival signals and suppression of
integrin-mediated cell adhesion. DAPK1 appeared to function early
in the apoptotic pathway prior to the commitment of the cells to
apoptosis. In the developing and adult central nervous system,
DAPK1 mRNA is widely expressed in proliferative regions within the
cerebral cortex and hippocampus (43). In addition, inhibition of DAPK1
with a selective inhibitor attenuates hypoxia-ischemia-induced
acute brain injury (45). DAPK1
controls a range of key signaling and cell death pathways as a
molecular switch, and also has been suggested to have an important
role in excitotoxicity (45–47).
Emerging evidence suggests that inhibition of DAPK1, which prevents
excessive NMDA receptor (R) activation without interfering with
physiological functions, provides neuroprotection in animal models
of adult stroke (24,48). NMDAR activation influences neuronal
proliferation and survival and is involved in synapse formation,
function and plasticity. In cell signal transduction, the CaN
activated by Ca2+ and CaM was necessary for the
dephosphorylation of DAPK1 in neural cell apoptosis (49–51),
so the activation of DAPK1 was required the CaN. Fortunately, CaN
is the target protein of the immunosuppressant FK506 (52). In the present study, the authors
hypothesized the neuronal cell apoptosis observed following the
induction of experimental DAI, and its possible association with
the upregulation of DAPK1. FK506 was able to inhibit the activity
of DAPK1. Ameliorating the effect of FK506 primarily reduced the
amount of neuronal cell death and the expression of DAPK1.
Therefore, FK506 was able to inhibit the activation and the
expression of DAPK1 and decrease neuronal cell death, which serves
a crucial role in neuroprotection following DAI. The process began
as early as 1 day post-injury, which indicated that FK506 could
inhibit the neuronal apoptosis in the early stage of DAI
injury.
NF-H is a structural protein that constitutes a
skeletal structure of the nervous cell body and axis, and it may be
used as an indicator of the function of the neuron and an indirect
indicator of axon regeneration (53). NF-H only exists in the axon under
physiological conditions, not in the neuron body. However, the
neuron induces a large amount of synthesis of NF-H following the
TBI, in order to adapt to the demand of nerve regeneration
(31). This reflects the neural
function and may reflect the condition of axon regeneration. In our
study, the expression of NF-H in the FK506-intervention group was
significantly increased than that in DAI group, and the peak
expression appeared in advance and lasted longer. FK506 may have
increased the synthesis of NF-H, accelerated the regeneration of
nerve cells, and promoted the repair of nerve tissue following DAI.
The mechanism may be associated with the decrease of T lymphocyte
infiltration and destruction, and the inhibition of the apoptosis
of neurons. Furthermore, in the present study, immunohistochemistry
was carried out on NF-H to identify the pathological alterations to
axons following DAI, especially in the early stage of 1 day
post-injury. There was a large amount of axonal swelling and axonal
retraction ball formation in the damaged regions of the hippocampus
and brainstem following DAI. In the FK506-intervention groups, the
pathological alterations to axons were markedly improved. Although
how the precise mechanism of this axonal protection occurs is not
well understood, it has been assumed that FK506 may act by reducing
mitochondrial permeability transition, inhibiting CaN activity
(22) and increasing the amount of
the active form of the GAP-43 in neurons (54).
GAP-43 is a form of nervous tissue-specific protein
that is highly expressed in neurons during development and nerve
regeneration, which has proven to be implicated in neurite
outgrowth, long-term potentiation, signal transduction and
neurotansmitter release (28,55).
In addition, GAP-43 is a rapid transport membrane phosphoric acid
protein found in the growth cones of developing and sprouting CNS
axons, which is associated with neuronal sprouting, development,
differentiation and regeneration (29,56).
In the present study, GAP-43 participated in the process of CNS
plasticity in response to injury, which is a complex process
involving synaptic stability and axonal remodeling, and the level
of GAP-43 is additionally predicted to be upregulated in any
process that involves axonal membrane remodeling following injury
(29,57,58).
Therefore, the present study used GAP-43 as a marker of axonal
regeneration, and observed that the expression of GAP-43 was
remarkably increased at day 1 and gradually increased to a peak at
day 3 or 7, which indicated the altering process of neuronal
regeneration following DAI. Hence, GAP-43 may be synthesized in
large amounts following DAI, and then shuttled along the axon by
fast axonal transport to the site of injury to promote axonal
regeneration (56). Although
increased expression of GAP-43 has been identified in a DAI rat
model in the study, the mechanisms underlying the overexpression of
GAP-43 remain unclear. It was observed that in the FK506
intervention group, the expression of GAP-43 was higher than in the
DAI group at the same point-in-time, and the peak expression of
GAP-43 was observed relatively earlier, at day 1 post-injury. It
follows that FK506 promoted a large number of GAP-43 protein
synthesis at the injured neurons, and transported from the neuronal
bodies to axons, which promoted the regeneration of axons, and
accelerate the recovery of nerve function. The upregulation of
GAP-43 by FK506 may be associated with the inhibition of CaN
activation and promotion of GAP-43 moving from the binding domain
of CaN to the injured axons (59,60).
In conclusion, the results of the present study
indicated that neuronal cell apoptosis and axonal degeneration were
observed following experimental DAI. FK506 may serve an important
role in anti-apoptosis and attenuating axonal degeneration in
experimental DAI by inhibiting the activity of DAPK1. Novel data is
provided to suggest that FK506 may promote the axon formation and
nerve regeneration after experimental DAI by observing the
expressions of GAP-43 and NF-H. Collectively, we summarized that
FK506 could reduce the apoptosis of neurons and axon pathological
changes following DAI, and relieve the nerve tissue injury. In
addition, FK506 may accelerate the regeneration of neurons, and
promote the repair of injured brain tissue. However, a study of the
precise molecular mechanism of this process remains elusive and
further experiments are required to address this aspect.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant no. 30471774) and the
New Century Excellent Talent Support Project of Ministry of
Education (grant no. NCET-05-0831).
References
|
1
|
Smith DH, Hicks R and Povlishock JT:
Therapy development for diffuse axonal injury. J Neurotrauma.
30:307–323. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Johnson VE, Stewart W and Smith DH: Axonal
pathology in traumatic brain injury. Exp Neurol. 246:35–43. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Smith DH, Meaney DF and Shull WH: Diffuse
axonal injury in head trauma. J Head Trauma Rehabil. 18:307–316.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Struffert T, Axmann C and Reith W:
Craniocerebral trauma. 2: Intra-axial injuries, secondary injuries.
Radiologe. 43:1001–1014; quiz 1015–1016. 2003.(In German).
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Toupalík P, Klír P, Bouska I and Chadová
L: Immunohistochemical methods in the differential diagnosis of
primary traumatic and subsequent secondary cerebral changes. Soud
Lek. 45:18–21. 2000.(In Czech). PubMed/NCBI
|
|
6
|
Tavanti F, Coppola V, Romano A, Beccia M,
Giuliani G, Pierallini A and Bozzao A: Diffuse axonal injury with
selective involvement of the corticospinal tract. A diffusion
tensor imaging case study. Neuroradiol J. 27:397–399. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jing G, Yao X, Li Y, Xie Y, Li WX, Liu K,
Jing Y, Li B, Lv Y and Ma B: Mild hypothermia for treatment of
diffuse axonal injury: A quantitative analysis of diffusion tensor
imaging. Neural Regen Res. 9:190–197. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kwon HG and Jang SH: The usefulness of
diffusion tensor imaging in detection of diffuse axonal injury in a
patient with head trauma. Neural Regen Res. 7:475–478.
2012.PubMed/NCBI
|
|
9
|
Babaee A, Eftekhar-Vaghefi SH,
Asadi-Shekaari M, Shahrokhi N, Soltani SD, Malekpour-Afshar R and
Basiri M: Melatonin treatment reduces astrogliosis and apoptosis in
rats with traumatic brain injury. Iran J Basic Med Sci. 18:867–872.
2015.PubMed/NCBI
|
|
10
|
Wang JF, Li Y, Song JN and Pang HG: Role
of hydrogen sulfide in secondary neuronal injury. Neurochem Int.
64:37–47. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hilton GD, Stoica BA, Byrnes KR and Faden
AI: Roscovitine reduces neuronal loss, glial activation, and
neurologic deficits after brain trauma. J Cereb Blood Flow Metab.
28:1845–1859. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ma Y, Liu W, Wang Y, Chao X, Qu Y, Wang K
and Fei Z: VEGF protects rat cortical neurons from mechanical
trauma injury induced apoptosis via the MEK/ERK pathway. Brain Res
Bull. 86:441–446. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hardingham GE and Bading H: Synaptic
versus extrasynaptic NMDA receptor signalling: Implications for
neurodegenerative disorders. Nat Rev Neurosci. 11:682–696. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fontana Karklin AC, Fox DP, Zoubroulis A,
Mortensen Valente O and Raghupathi R: Neuroprotective effects of
the glutamate transporter activator
(R)-(−)-5-methyl-1-nicotinoyl-2-pyrazoline (MS-153) following
traumatic brain injury in the adult rat. J Neurotrauma.
33:1073–1083. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Armstrong RC, Mierzwa AJ, Marion CM and
Sullivan GM: White matter involvement after TBI: Clues to axon and
myelin repair capacity. Exp Neurol. 275:328–333. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tang-Schomer MD, Patel AR, Baas PW and
Smith DH: Mechanical breaking of microtubules in axons during
dynamic stretch injury underlies delayed elasticity, microtubule
disassembly and axon degeneration. FASEB J. 24:1401–1410. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Brizuela M, Blizzard CA, Chuckowree JA,
Dawkins E, Gasperini RJ, Young KM and Dickson TC: The
microtubule-stabilizing drug Epothilone D increases axonal
sprouting following transection injury in vitro. Mol Cell Neurosci.
66:129–140. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zawadzka M and Kaminska B: A novel
mechanism of FK506-mediated neuroprotection: Downregulation of
cytokine expression in glial cells. Glia. 49:36–51. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shichinohe H, Kuroda S, Abumiya T, Ikeda
J, Kobayashi T, Yoshimoto T and Iwasaki Y: FK506 reduces infarct
volume due to permanent focal cerebral ischemia by maintaining BAD
turnover and inhibiting cytochrome c release. Brain Res.
1001:51–59. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gabryel B, Bielecka A, Stolecka A,
Bernacki J and Langfort J: Cytosolic phospholipase A2
inhibition is involved in the protective effect of nortriptyline in
primary astrocyte cultures exposed to combined oxygen-glucose
deprivation. Pharmacol Rep. 62:814–826. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Singleton RH, Stone JR, Okonkwo DO,
Pellicane AJ and Povlishock JT: The immunophilin ligand FK506
attenuates axonal injury in an impact-acceleration model of
traumatic brain injury. J Neurotrauma. 18:607–614. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Marmarou CR and Povlishock JT:
Administration of the immunophilin ligand FK506 differentially
attenuates neurofilament compaction and impaired axonal transport
in injured axons following diffuse traumatic brain injury. Exp
Neurol. 197:353–362. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Reeves TM, Phillips LL, Lee NN and
Povlishock JT: Preferential neuroprotective effect of tacrolimus
(FK506) on unmyelinated axons following traumatic brain injury.
Brain Res. 1154:225–236. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tu W, Xu X, Peng L, Zhong X, Zhang W,
Soundarapandian MM, Balel C, Wang M, Jia N, Zhang W, et al: DAPK1
interaction with NMDA receptor NR2B subunits mediates brain damage
in stroke. Cell. 140:222–234. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bialik S and Kimchi A: The
death-associated protein kinases: Structure, function, and beyond.
Annu Rev Biochem. 75:189–210. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Pelled D, Raveh T, Riebeling C, Fridkin M,
Berissi H, Futerman AH and Kimchi A: Death-associated protein (DAP)
kinase plays a central role in ceramide-induced apoptosis in
cultured hippocampal neurons. J Biol Chem. 277:1957–1961. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kato S, Matsukawa T, Koriyama Y, Sugitani
K and Ogai K: A molecular mechanism of optic nerve regeneration in
fish: The retinoid signaling pathway. Prog Retin Eye Res. 37:13–30.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Liu F, Liao F, Li W, Han Y and Liao D:
Progesterone alters Nogo-A, GFAP and GAP-43 expression in a rat
model of traumatic brain injury. Mol Med Rep. 9:1225–1231.
2014.PubMed/NCBI
|
|
29
|
Benowitz LI and Routtenberg A: GAP-43: An
intrinsic determinant of neuronal development and plasticity.
Trends Neurosci. 20:84–91. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang YM, Zhang YQ, Cheng SB, Chen SX,
Chen AL and Tang CZ: Effect of acupuncture on proliferation and
differentiation of neural stem cells in brain tissues of rats with
traumatic brain injury. Chin J Integr Med. 19:132–136. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zurek J and Fedora M: The usefulness of
S100B, NSE, GFAP, NF-H, secretagogin and Hsp70 as a predictive
biomarker of outcome in children with traumatic brain injury. Acta
Neurochir (Wien). 154:93–103. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li Y, Song J, Liu X, Zhang M, An J, Sun P,
Li D, Jin T and Wang J: High expression of STIM1 in the early
stages of diffuse axonal injury. Brain Res. 1495:95–102. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Xiao-Sheng H, Sheng-Yu Y, Xiang Z, Zhou F
and Jian-ning Z: Diffuse axonal injury due to lateral head rotation
in a rat model. J Neurosurg. 93:626–633. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Xiao-Sheng H, Gui-Tao Y, Xiang Z and Zhou
F: A morphological study of diffuse axonal injury in a rat model by
lateral head rotation trauma. Acta Neurol Belg. 110:49–56.
2010.PubMed/NCBI
|
|
35
|
Marsland TA, Glees P and Erikson LB:
Modification of the Glees silver impregnation for paraffin
sections. J Neuropathol Exp Neurol. 13:587–591. 1954. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ng HK, Mahaliyana RD and Poon WS: The
pathological spectrum of diffuse axonal injury in blunt head
trauma: Assessment with axon and myelin strains. Clin Neurol
Neurosurg. 96:24–31. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Maroon H, Walshe J, Mahmood R, Kiefer P,
Dickson C and Mason I: Fgf3 and Fgf8 are required together for
formation of the otic placode and vesicle. Development.
129:2099–2108. 2002.PubMed/NCBI
|
|
38
|
Jia X, Cong B, Wang S, Dong L, Ma C and Li
Y: Secondary damage caused by CD11b+ microglia following diffuse
axonal injury in rats. J Trauma Acute Care Surg. 73:1168–1174.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Logsdon AF, Lucke-Wold BP, Turner RC,
Huber JD, Rosen CL and Simpkins JW: Role of microvascular
disruption in brain damage from traumatic brain injury. Compr
Physiol. 5:1147–1160. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Dressler J and Vemuganti R: Apoptosis and
gene expression after TBI. Leg Med (Tokyo). 11:(Suppl 1). S54–S55.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Mao Z, Song Z, Li G, Lv W, Zhao X, Li B,
Feng X and Chen Y: 8-hydroxy-2-(di-n-propylamino)tetralin
intervenes with neural cell apoptosis following diffuse axonal
injury. Neural Regen Res. 8:133–142. 2013.PubMed/NCBI
|
|
42
|
Aberg M, Johnell M, Wickstrom M and
Siegbahn A: Tissue Factor/FVIIa prevents the extrinsic pathway of
apoptosis by regulation of the tumor suppressor Death-Associated
Protein Kinase 1 (DAPK1). Thromb Res. 127:141–148. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Yoo HJ, Byun HJ, Kim BR, Lee KH, Park SY
and Rho SB: DAPk1 inhibits NF-κB activation through TNF-α and
INF-γ-induced apoptosis. Cell Signal. 24:1471–1477. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Martoriati A, Doumont G, Alcalay M,
Bellefroid E, Pelicci PG and Marine JC: dapk1, encoding an
activator of a p19ARF-p53-mediated apoptotic checkpoint, is a
transcription target of p53. Oncogene. 24:1461–1466. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Pei L, Shang Y, Jin H, Wang S, Wei N, Yan
H, Wu Y, Yao C, Wang X, Zhu LQ and Lu Y: DAPK1-p53 interaction
converges necrotic and apoptotic pathways of ischemic neuronal
death. J Neurosci. 34:6546–6556. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Tian J, Cheng J, Zhang J, Ye L, Zhang F,
Dong Q, Wang H and Fu F: Protection of pyruvate against glutamate
excitotoxicity is mediated by regulating DAPK1 protein complex.
PLoS One. 9:e957772014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Lai TW, Zhang S and Wang YT:
Excitotoxicity and stroke: Identifying novel targets for
neuroprotection. Prog Neurobiol. 115:157–188. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Shamloo M, Soriano L, Wieloch T, Nikolich
K, Urfer R and Oksenberg D: Death-associated protein kinase is
activated by dephosphorylation in response to cerebral ischemia. J
Biol Chem. 280:42290–42299. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Fujita Y and Yamashita T: Role of DAPK in
neuronal cell death. Apoptosis. 19:339–345. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Nair S, Hagberg H, Krishnamurthy R,
Thornton C and Mallard C: Death associated protein kinases:
Molecular structure and brain injury. Int J Mol Sci.
14:13858–13872. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Schumacher AM, Velentza AV and Watterson
DM: Death-associated protein kinase as a potential therapeutic
target. Expert Opin Ther Targets. 6:497–506. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Wang HL, Du YW, Xiang BQ, Lin WL and Wei
Q: The regulatory domains of CNA have different effects on the
inhibition of CN activity by FK506 and CsA. IUBMB Life. 59:388–393.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Shaw G, Yang C, Ellis R, Anderson K,
Mickle Parker J, Scheff S, Pike B, Anderson DK and Howland DR:
Hyperphosphorylated neurofilament NF-H is a serum biomarker of
axonal injury. Biochem Biophys Res Commun. 336:1268–1277. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Bavetta S, Hamlyn PJ, Burnstock G,
Lieberman AR and Anderson PN: The effects of FK506 on dorsal column
axons following spinal cord injury in adult rats: Neuroprotection
and local regeneration. Exp Neurol. 158:382–393. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Huang HL, Li JM and Zhao YN: Effect of
shenxiong huayu capsule on cerebral ischemia/reperfusion injury and
the expression of GAP43 in hippocampal CA1 of rats. Zhongguo Zhong
Xi Yi Jie He Za Zhi. 34:185–190. 2014.(In Chinese). PubMed/NCBI
|
|
56
|
Williams RR, Venkatesh I, Pearse DD,
Udvadia AJ and Bunge MB: MASH1/Ascl1a leads to GAP43 expression and
axon regeneration in the adult CNS. PLoS One. 10:e01189182015.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Hulsebosch CE, DeWitt DS, Jenkins LW and
Prough DS: Traumatic brain injury in rats results in increased
expression of Gap-43 that correlates with behavioral recovery.
Neurosci Lett. 255:83–86. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Li H, Dokas LA, Godfrey DA and Rubin AM:
Remodeling of synaptic connections in the deafferented vestibular
nuclear complex. J Vestib Res. 12:167–183. 2002.PubMed/NCBI
|
|
59
|
Baumgärtel K and Mansuy IM: Neural
functions of calcineurin in synaptic plasticity and memory. Learn
Mem. 19:375–384. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Boczek T, Ferenc B, Lisek M and Zylinska
L: Regulation of GAP43/calmodulin complex formation via
calcineurin-dependent mechanism in differentiated PC12 cells with
altered PMCA isoforms composition. Mol Cell Biochem. 407:251–262.
2015. View Article : Google Scholar : PubMed/NCBI
|