Introduction
Thrombolytic therapy following the onset of stroke
has been demonstrated to prevent apoptosis of neural cells at risk
of injury; however, the re-establishment of blood circulation may
contribute to delayed secondary brain damage (1). Our previous study demonstrated that
reperfusion induced mitochondrial ultrastructural alterations and
dysfunction (2); mitochondria are
important in the regulation of intrinsic apoptosis (3). Previous in vivo and in
vitro models have demonstrated that postconditioning
significantly inhibits neuronal apoptosis and protects the brain
from reperfusion injury (4,5).
Therefore, it appears that inhibition of neuronal apoptosis is
important in postconditioning-initiated neuroprotection. However,
the mechanisms by which remote ischemic postconditioning (RIPostC)
protects neurons from reperfusion injury and the optimal
application time of RIPostC remain to be elucidated, in order for
this method to be translated into clinical practice.
The B-cell lymphoma 2 (Bcl-2) family of genes
regulates apoptosis and consists of two groups: Proapoptotic and
anti-apoptotic proteins. The ratio between proapoptotic and
anti-apoptotic members of the Bcl-2 family determines the fate of
the cell (6). Bcl-2 homology 3
(BH3) interacting-domain death agonist (BID) is a member of the
BH3-only subfamily, which is associated with proapoptotic proteins.
It has previously been demonstrated that BID may bridge the
crosstalk between extrinsic and intrinsic apoptotic pathways via
its cleavage by caspase-8 (7).
When cleaved, truncated BID (tBID) promotes apoptosis by activating
Bcl-2-associated X protein (Bax) and eliciting mitochondrial outer
membrane permeabilization (8). The
infarct sizes and apoptotic indexes in bid−/− brains are
greatly reduced under reperfusion conditions (9). BID is important in
reperfusion-induced neuronal apoptosis, and postconditioning may
significantly inhibit the apoptosis induced by ischemia/reperfusion
injury. Furthermore, RIPostC may share common mechanistic signaling
pathways with conventional postconditioning methods, although these
are performed in ischemic organs and not in distal limbs (10). Therefore, the present study
hypothesized that the BID-mediated mitochondrial apoptotic pathway
is an important target for RIPostC to prevent reperfusion-induced
neuronal apoptosis.
Materials and methods
Animal and experimental groups
All animal protocols used in the present study were
approved by the Harbin Medical University Committee on the
Guidelines for Animal Experiments (Harbin, China). All rats were
handled according to the Guidelines for the Care and Use of
Laboratory Animals of the National Institutes of Health. Adult male
Sprague-Dawley rats (129 total; weight, 260–280 g; age, 6–8 weeks)
were provided by the laboratories of The Second Affiliated Hospital
of Harbin Medical University (Harbin, China), and housed under
controlled conditions (temperature, 27±2°C; humidity, 60–70%) under
a 12-h light/dark cycle. The rats were allowed ad libitum
access to food and water. Animals were randomly divided into five
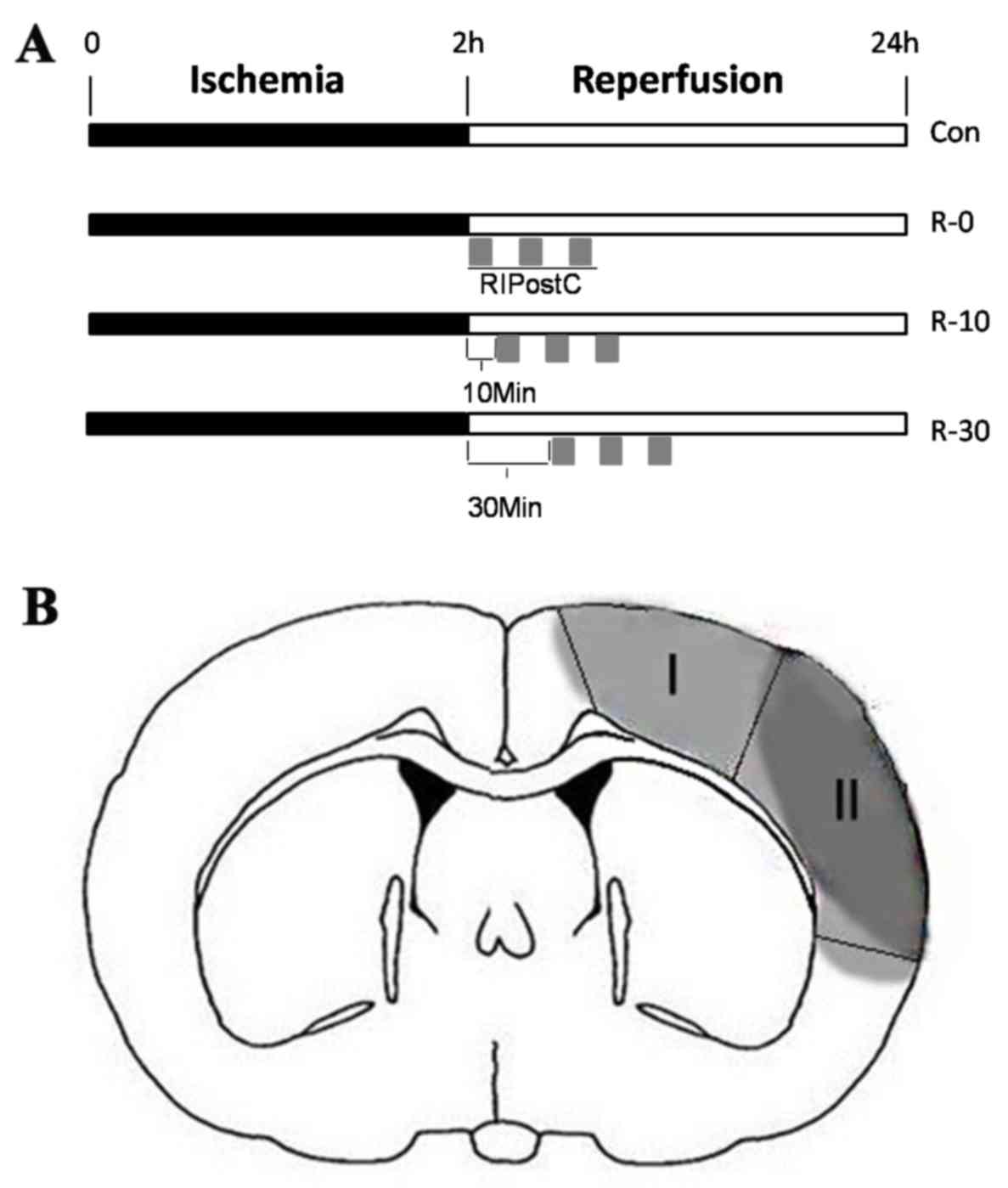
groups (sham, 5 rats; control, 49 rats; R-0, 49 rats; R-10, 13
rats; R-30, 13 rats), according to the RIpostC starting times: The
sham group, the control group (ischemia, 120 min), and three
RIPostC groups. In the three RIPostC groups, RIPostC was conducted
as follows: Three 10-min cycles of bilateral femoral artery
occlusion with an interval of 10 min reperfusion after 0, 10 or 30
min of brain reperfusion (R-0, R-10 and R-30 groups, respectively),
following administration of local anesthesia. The protocol for
RIPostC in each group is schematically presented in Fig. 1A. In order to exclude the effect of
RIPostC on the expression of apoptosis-related proteins in
non-ischemic reperfusion period, a RIPostC + sham group was added
as an additional control in the western blot analysis.
Transient middle cerebral artery
occlusion (MCAO) model
Focal cerebral ischemia was induced by MCAO as
previously described, with minor modifications (11). Briefly, 260–280 g male
Sprague-Dawley rats were anesthetized by intraperitoneal injection
of pentobarbital sodium (50 mg/kg in normal saline), and a caudal
ventral artery catheter was inserted for continuous arterial blood
pressure monitoring. Subsequently, the left external carotid artery
(ECA), internal carotid artery (ICA) and common carotid artery were
exposed. A 4–0 monofilament nylon thread (40–3734 PK 10; Doccol
Corporation, Sharon, MA, USA) with a silicon-rubber-coated tip was
then inserted into the ICA from a hole in the ECA; the suture was
advanced 16–18 mm through the ICA, until a mild resistance was felt
for 120 min. Blood flow was restored by gently withdrawing the
suture. During these procedures, the rectal temperature was
monitored continuously with a rectal probe; temperature was
maintained at 37±0.5°C by the use of a lamp and pad. The mean
arterial pressure and arterial blood gas values were recorded at 3
min prior to insertion of the suture (baseline), 60 min following
MCAO (ischemia), and 10 min following reperfusion in each group.
Rats in the sham group underwent the same procedure, with the
exception of occlusion. Rats in the control group underwent
ischemia/reperfusion without RIPostC. Prior to the induction of
RIPostC, rats were allowed to regain consciousness, and the
neurological deficit score was determined by a blinded observer
based on Longa grade point standards (12). These standards were as follows: 0,
no neurological deficits; 1, failure to fully extend the right
forepaw; 2, circling to the right; 3, falling to the right; and 4,
unable to walk spontaneously and exhibiting depressed levels of
consciousness. In this experiment, the rats that scored 2 or 3 were
adopted for further study.
Measurement of neurobehavioral scores
and infarct volume
At 24 h after reperfusion, a revision of the
18-point scoring table proposed by Garcia et al (13) was used for neurobehavioral
evaluation by a blinded observer. The 18-point scoring table was
based on the following six tests: i) Spontaneous activity; ii)
symmetry in the movement of four limbs; iii) forepaw outstretching;
iv) climbing; v) body proprioception; and vi) response to vibrissal
stimulation. The scores were summed up at the end of the evaluation
(minimum score, 3; maximum score, 18). Following anesthesia with
pentobarbital sodium (30 mg/kg), the mice were rapidly decapitated.
The brains were frozen at −20°C for 15 min and were then cut into
five 2 mm-thick coronal sections, which were stained with 2%
5-triphenyl-2H tetrazolium chloride (Sigma-Aldrich; Merck KGaA
Millipore, Darmstadt Germany) at 37°C for 15 min (14). The infarct volume was analyzed
using Image-Pro Plus software version 5.1 (Media Cybernetics Inc.,
Rockville, MD, USA). The volume of the infarct area was calculated
as the percentage of the infarct volume relative to whole brain
volume (15).
Terminal deoxynucleotidyl transferase
dUTP nick end labeling (TUNEL) staining
Following 24 h of reperfusion, brain cell apoptosis
in the peri-infarct cortex (n=5/group) was assessed in situ
via TUNEL staining, as previously described (16). The infarct core and peri-infarct
zone were defined according to well-established protocols in rodent
models of unilateral proximal MCAO (17) (Fig
1B). TUNEL staining was quantitatively evaluated using the
method described by Wang et al (18). Positively stained cells were
counted in 10 random peri-infarct cortexes per rat, and the total
number of positively stained cells in these pixels was then counted
and expressed as cells/mm2.
Western blot analysis
At 3, 6, 12 and 24 h following reperfusion, the
brain samples were collected, and protein isolation from cortical
tissues, based on total cell extracts or subcellular fractionation
(cytosolic), was performed as previously described (19). A total of 20 mg tissue was
homogenized and lyzed in radioimmunoprecipitation assay buffer on
ice and the protein concentration of samples was determined using
the Bradford protein assay kit (both from Beyotime Institute of
Biotechnology, Shanghai, China). Western blotting was performed as
previously described (20), and
the following primary antibodies were incubated at 4°C overnight:
anti-BID (1:200; cat. no. sc-6358; Santa Cruz Biotechnology, Inc.,
Dallas, TX, USA), anti-cytochrome c (1:300; cat. no.
ab13575) and anti-β-actin (1:2,000; cat. no. ab8226) (both from
Abcam, Cambridge, UK). The polyvinylidene fluoride membranes were
incubated with horseradish peroxidase-conjugated rabbit anti-goat
immunoglobulin (Ig) G (1:1,000; cat. no. ZB-2306) and goat
anti-mouse IgG secondary antibodies (1:1,000; cat. no. ZB-2305)
(both from ZSGB-BIO, Beijing, China) for 1 h at room temperature.
The expression of β-actin in the same membrane served as an
internal reference. Blots were detected using a luminescent image
analyzer and semi-quantified using Image Quant TL version 7.0 (both
from GE Healthcare Biosciences, Pittsburgh, PA, USA).
Immunofluorescence analysis
Coronal sections were used for immunofluorescence
staining. The procedures for immunofluorescent staining were
conducted as previously described (11). Briefly, the sections were first
blocked with 5% bovine serum albumin (BSA; Beyotime Institute of
Biotechnology) for 30 min followed by overnight incubation at 4°C
with the primary antibody, anti-BID (1:100; cat. no. sc-6358; Santa
Cruz Biotechnology, Inc.), diluted in BSA. After washing three
times with PBS, sections were incubated with a secondary antibody
(1:300; cat. no. bs-0294M-RBITC; BIOSS, Beijing, China) conjugated
to a fluorochrome at room temperature for 1 h. Following the final
wash, the sections were placed on coverslips using a fluorescent
mounting medium (Beyotime Institute of Biotechnology). Double
immunofluorescent staining was conducted in a linear fashion as
previously described (21).
Following staining with the first set of primary and secondary
antibodies as aforementioned, the second set of primary antibodies
used were as follows: anti-cytochrome c oxidase (COX)IV
(1:200; cat. no. ab153709) and anti-neuronal nuclear antigen (NeuN;
1:500; cat. no. ab177487) (both from Abcam) and corresponding
secondary antibodies (1:400; cat. no. bs-0295d-FITC; BIOSS)
conjugated to a different fluorochrome, were applied in the same
manner as previously described. This procedure was conducted with
caution, to prevent cross-reaction of the secondary antibodies with
the two sets of primary antibodies, which were prepared in various
species. DAPI was used as a nuclear counterstain. Images were
obtained by fluorescence microscopy (Nikon Corporation, Tokyo,
Japan) and microscopic examination was conducted at excitation
wavelengths of 540 nm, and emission wavelengths of 605 nm (for red
fluorochrome) and 465/515 nm (for green fluorochrome).
Statistical analysis
Statistical analyses were conducted using SPSS
software, version 21.0 for Windows (IBM SPSS, Armonk, NY, USA).
Data are presented as the mean ± standard deviation of 3
independent experiments. One-way analysis of variance was conducted
to determine significance followed by Bonferroni's post hoc
correction for multiple comparisons. P<0.05 was considered to
indicate a statistically significant difference.
Results
RIPostC mediates neuroprotective
effects
Infarct volume and neuroethology score
The present study did not detect any significant
differences among the various time points for any of the
physiological parameters, including rectal temperature, mean
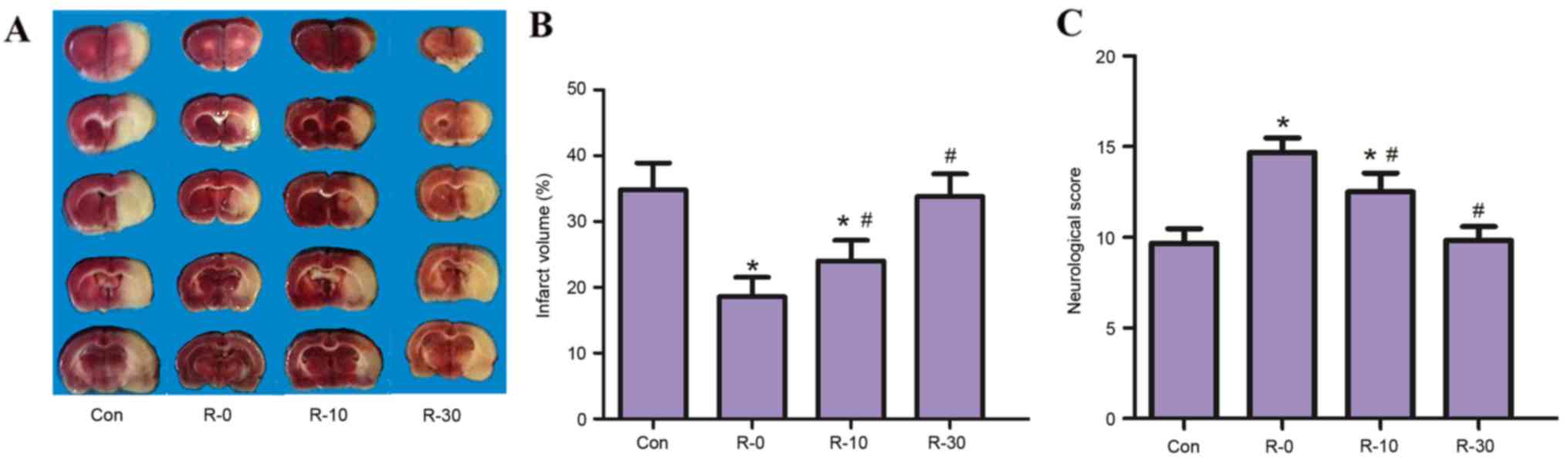
arterial pressure and arterial blood gas tension (Table I). It was demonstrated that RIPostC
applied at the moment of reperfusion significantly reduced infarct
volume in MCAO rats. All RIPostC groups, with the exception of the
R-30 group (33.80±4.22%), exhibited significantly reduced infarct
volumes at 24 h following reperfusion (control, 35.3±5.13%; R-0,
18.24±3.01%; R-10, 24.25±3.62%; P<0.05; Fig. 2A and B), this effect was
particularly evident in the R-0 group. In addition, a similar
result was observed with regards to neuroethology score; the
18-point scores of rats in the R-0 and R-10 groups were
significantly higher compared with in the control group.
Furthermore, the R-0 group exhibited significantly higher
neurological scores compared with the R-10 group (control,
9.67±0.81; R-0, 14.66±0.62; R-10, 12.83±0.68; R-30, 9.83±0.72;
P<0.05; Fig. 2C).
 | Table I.Physiological parameters. |
Table I.
Physiological parameters.
|
| Group |
|---|
|
|
|
|---|
| Parameter | Sham | Con | R-0 | R-10 | R-30 |
|---|
| Temperature (°C) |
|
|
|
|
|
|
Baseline | 37.00±0.10 | 36.90±0.30 | 36.80±0.20 | 37.00±0.10 | 36.90±0.10 |
|
Ischemia | 36.90±0.20 | 36.80±0.20 | 36.80±0.10 | 36.90±0.10 | 36.80±0.20 |
| Reperfusion | 37.00±0.10 | 36.80±0.10 | 37.00±0.10 | 36.80±0.20 | 36.80±0.30 |
| MAP (mmg) |
|
|
|
|
|
|
Baseline | 96.00±5.00 | 96.00±4.00 | 99.00±1.00 | 98.00±3.00 | 96.00±4.00 |
|
Ischemia | 97.00±3.00 | 98.00±2.00 | 97.00±3.00 | 97.00±5.00 | 95.00±5.00 |
| Reperfusion | 98.00±2.00 | 99.00±2.00 | 96.00±3.00 | 95.00±5.00 | 98.00±2.00 |
| PaO2
(mmH) |
|
|
|
|
|
|
Baseline | 165.00±3.00 | 161.00±5.00 | 163.00±3.00 | 159.00±8.00 | 162.00±3.00 |
|
Ischemia | 159.00±7.00 | 163.00±4.00 | 162.00±5.00 | 161.00±6.00 | 165.00±2.00 |
| Reperfusion | 160.00±4.00 | 162.00±7.00 | 164.00±3.00 | 167.00±1.00 | 159.00±9.00 |
| PaCO2
(mmHg) |
|
|
|
|
|
|
Baseline | 37.00±4.00 | 38.00±2.00 | 37.00±4.00 | 38.00±2.00 | 37.00±2.00 |
|
Ischemia | 36.00±3.00 | 36.00±4.00 | 36.00±6.00 | 36.00±5.00 | 37.00±3.00 |
| Reperfusion | 37.00±3.00 | 37.00±2.00 | 36.00±5.00 | 37.00±3.00 | 38.00±1.00 |
| pH |
|
|
|
|
|
|
Baseline | 7.40±0.02 | 7.38±0.01 | 7.39±0.01 | 7.38±0.02 | 7.36±0.03 |
|
Ischemia | 7.39±0.01 | 7.37±0.02 | 7.39±0.01 | 7.37±0.02 | 7.38±0.01 |
| Reperfusion | 7.38±0.02 | 7.39±0.01 | 7.38±0.02 | 7.38±0.01 | 7.36±0.02 |
TUNEL-positive cells
The present study aimed to determine whether RIPostC
is associated with a decrease in neuronal apoptosis in the
peri-infarct cortex. TUNEL staining was nearly negative in the sham
group; however, a large number of TUNEL-positive cells was observed
in the control and R-30 groups. Only a few TUNEL-positive cells
were observed in other RIPostC groups, in particular, very few
TUNEL-positive cells were detected in the R-0 group (Fig. 3).
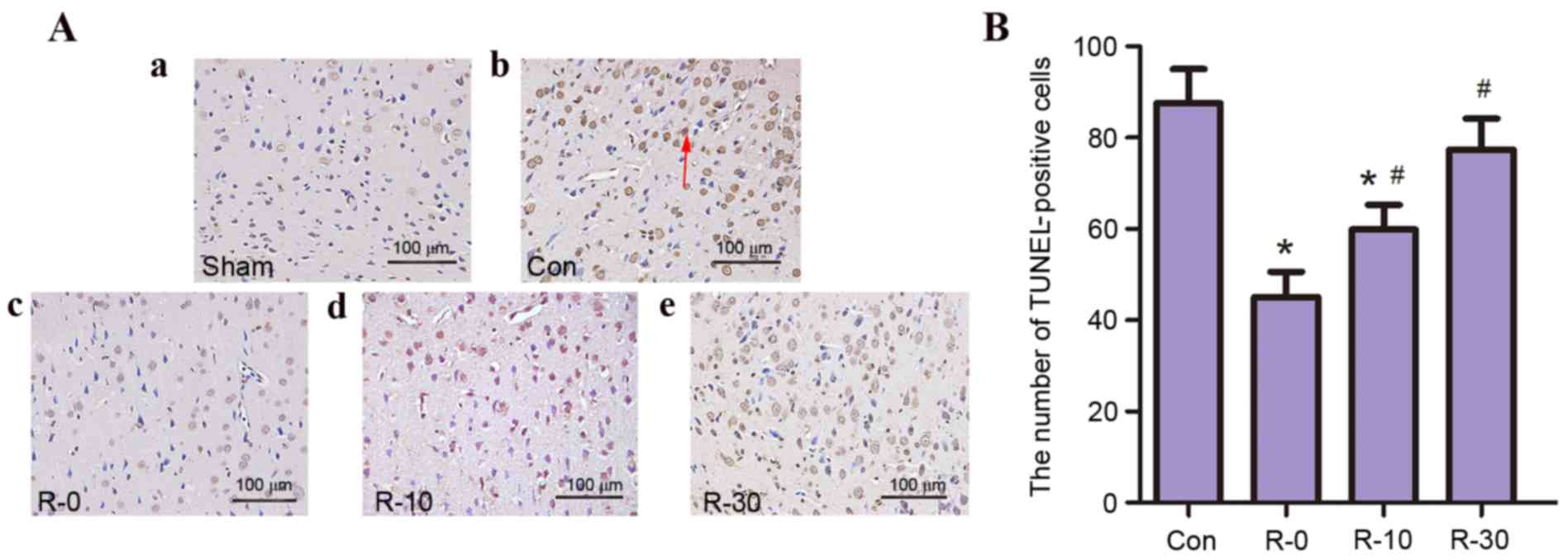 | Figure 3.RIPostC reduced the number of
TUNEL-positive cells following reperfusion (n=5/group). (A)
Representative photomicrographs of TUNEL staining in the
peri-infarct zone of rat brain sections from (a) sham, (b) control,
(c) R-0, (d) R-10 and (e) R-30 groups at 24 h following
reperfusion. The arrow indicates TUNEL-positive cells.
Magnification, ×400; scale bar, 100 µm. (B) Quantification of
TUNEL-positive cells. Data are presented as the mean ± standard
deviation. *P<0.05 vs. the control group; #P<0.05
vs. R-0 group. Con, control; RIPostC, remote ischemic
postconditioning; R-0, -10, -30, RIPostC 0, 10 and 30 min groups,
respectively; TUNEL, terminal deoxynucleotidyl transferase dUTP
nick end labeling. |
These results indicated that following application
of RIPostC at the early stage of reperfusion infarct volume
decreased, neuroethology score improved and the rate of apoptosis
was reduced; these effects were particularly evident in the R-0
group.
RIPostC-mediated effects on tBID and
cytochrome c levels
Expression of tBID and cytochrome c
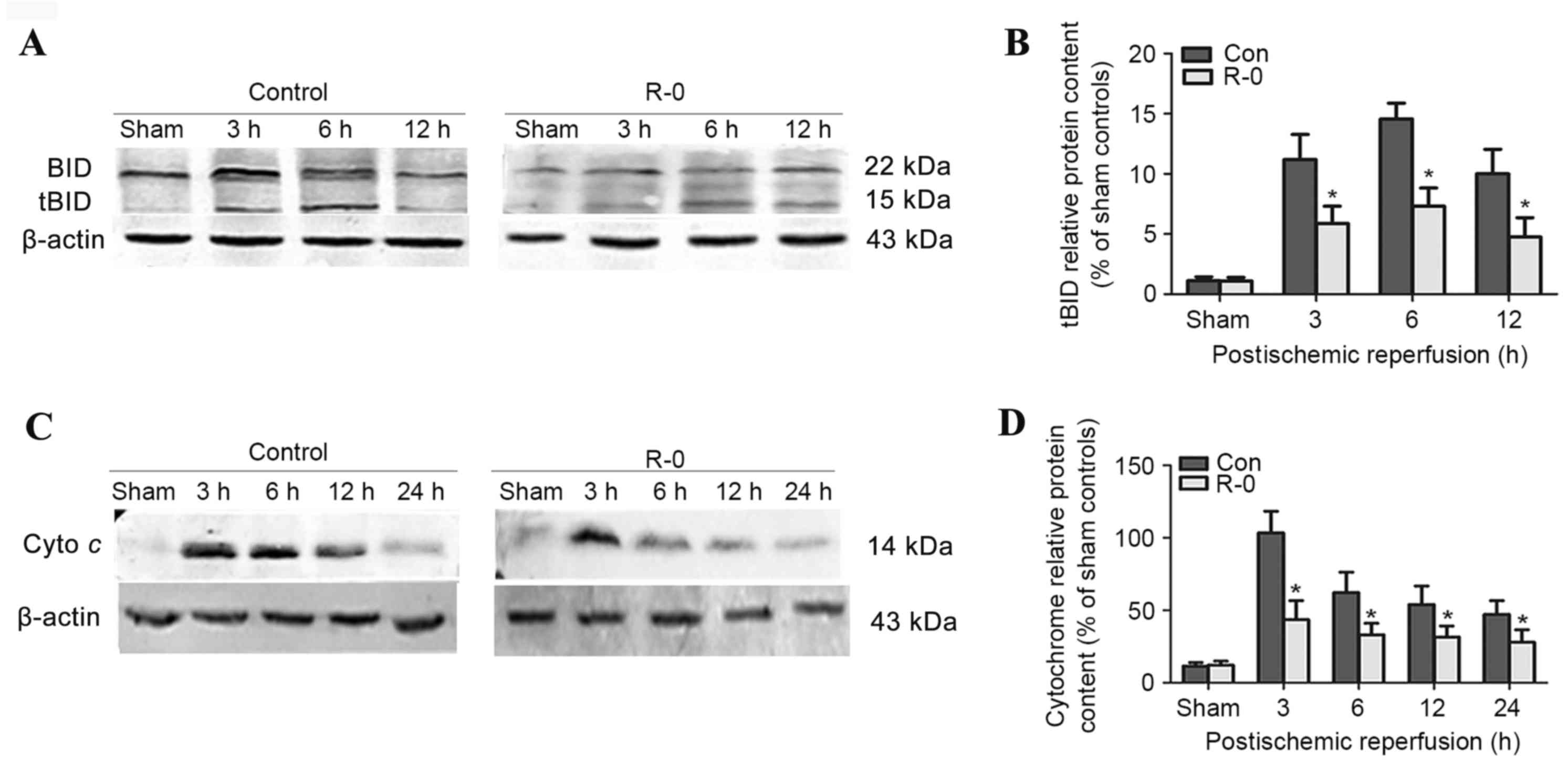
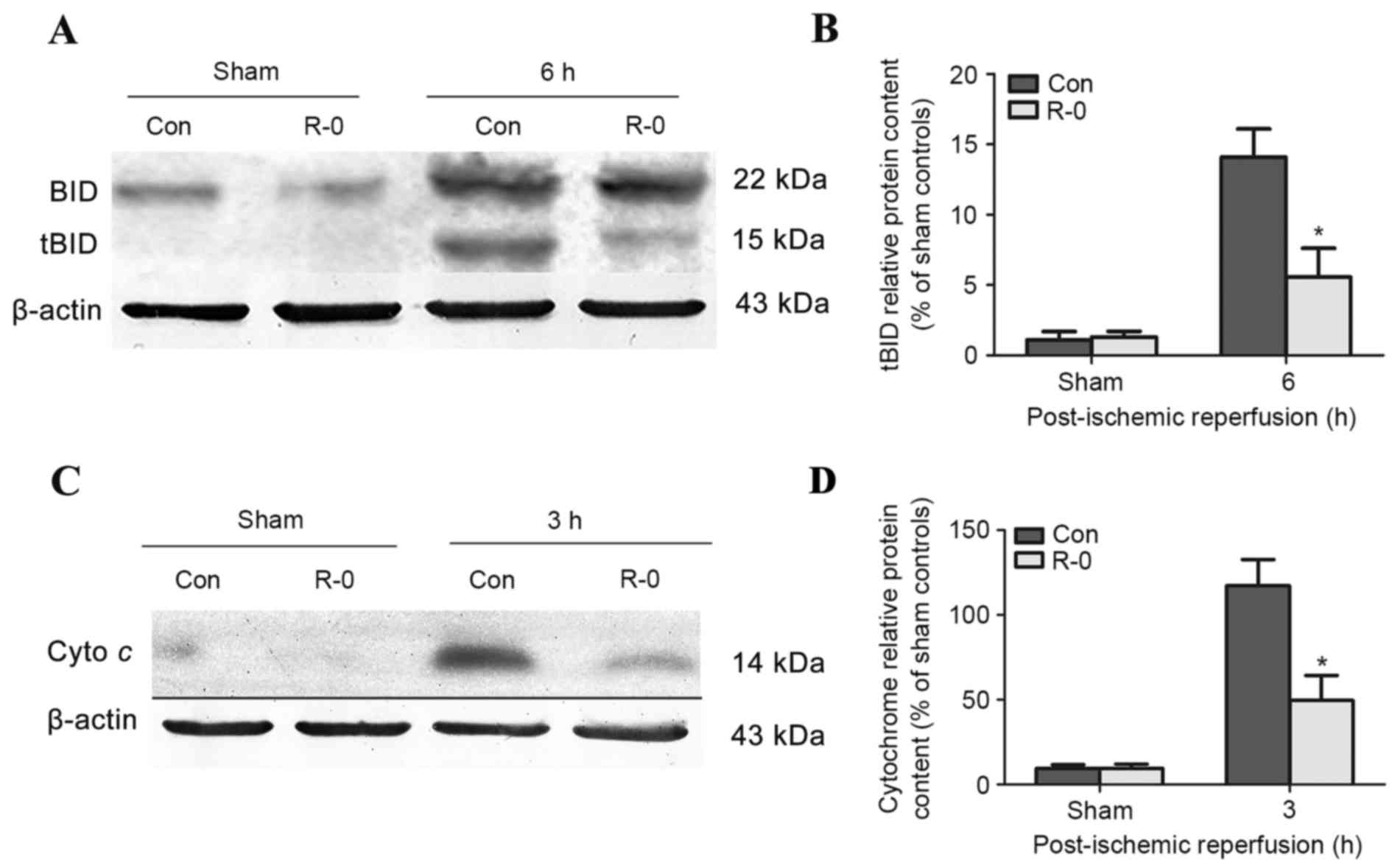
The R-0 group was selected to explore the potential
mechanism underlying the effects of RIPostC. Western blot analysis
revealed that tBID was readily detected in the control group brains
3 h following reperfusion, peaked at 6 h and was continuously
detectable through the first 12 h. The R-0 group exhibited a
similar trend, however tBID expression was significantly reduced at
each time point compared with in the control group (Fig. 4A and B). Cytochrome c
release into the cytosolic fraction was detected in the R-0 and
control groups 3 h following reperfusion and at various time points
throughout the first 24 h. However, its expression was markedly
reduced in the R-0 group compared with in the control group
(Fig. 4C and D). In the RIPostC +
sham group, the expression levels of tBID and cytochrome c
were similar to those in the sham group. The peak expression of
tBID and cytochrome c was significantly decreased in the R-0
group compared with control group (Fig. 5).
tBID level and translocation
The immunofluorescence signals of BID were very low
in the non-ischemic cortex and indicated a diffusive pattern,
consistent with its cytosolic distribution in normal cells.
However, the signal increased following reperfusion, particularly 6
h after reperfusion, suggesting mitochondrial localization.
Conversely, expression was weaker in the R-0 group compared with in
the control group. Subcellular localization in the mitochodria was
further confirmed by double-label staining with the anti-COXIV
antibody. Furthermore, tBID was activated in the neurons as
demonstrated by double-label staining with anti-NeuN antibody (a
neuronal marker) (Fig. 6).
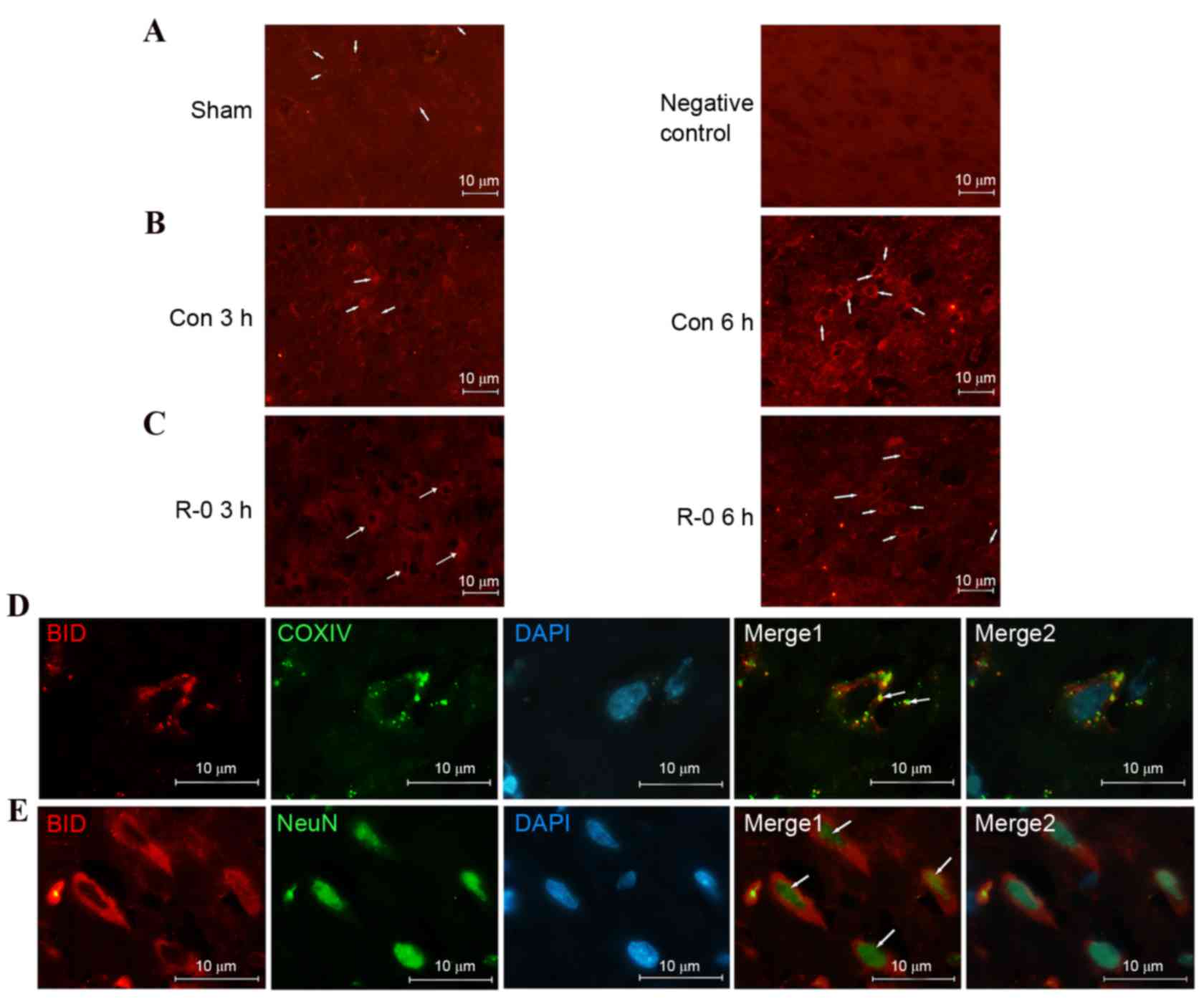 | Figure 6.Activated BID is translocated to
ischemic neurons following reperfusion. Representative
immunofluorescent images of tBID obtained from the wild-type
parietal cortex relevant to the infarct border zone. (A)
Immunofluorescence signal of tBID was weak and diffusive in the
non-ischemic cortex (left). A wild-type cortex staining control not
incubated with the primary anti-BID antibody 6 h following
reperfusion is presented on the right. (B) tBID immunofluorescence
signal was increased at 3 h (left) and 6 h (right) following
reperfusion and assumed a punctuated pattern. (C) tBID
immunofluorescence signal appeared weaker at 3 h (left) and 6 h
(right) following remote ischemic postconditioning. (A-C)
Magnification, ×400; scale bar, 10 µm. Arrows indicate tBID
signals. (D) Double staining of tBID and mitochondria COXIV in
cortical neurons from control group, 6 h following reperfusion. The
overlapping image indicated that tBID was localized in the
mitochondria (arrows mark the double-stained neurons). (E) Double
staining of tBID and NeuN in the same treated cortex as D. The
overlapping image indicated that enhanced tBID signals were
detected in neurons (as marked by the arrows). (D and E)
Magnification, ×1,000; scale bar, 10 μm. BID, B-cell lymphoma 2
homology 3 interacting-domain death agonist; tBID, truncated BID;
R-0, remote ischemic postconditioning 0 min group; COXIV,
cytochrome c oxidase IV; NeuN, neuronal nuclear antigen. |
Discussion
Reperfusion following ischemia is often associated
with induction of apoptosis-like cell death (15). The release of intramitochondrial
contents into the cytosol is considered irreversible during the
induction of apoptotic signaling (22). The present study demonstrated that
RIPostC, applied as early as possible following reperfusion, may
result in a significant neuroprotective effect in the rat MCAO
model. The protective mechanisms of RIPostC may be associated with
inhibition of the BID-mediated mitochondrial apoptotic pathway.
Hess et al (23) reported that preclinical reports of
remote limb conditioning in focal cerebral ischemia are promising;
however, the underlying mechanism remains to be elucidated. At
present, it is known that numerous mechanisms are involved: i)
Humoral factors acting via systemic circulation; ii) neurogenic
transmission with involvement of the autonomic nervous system; and
iii) effects on circulating immune cells (24). However, a previous study involving
the heart suggested that the pathway of protection in the target
organ involves activation of kinases to resolve the reperfusion
injury, or activation of survival factor enhancement pathways,
which ultimately converge on the mitochondria to prevent opening of
the mitochondrial permeability transition pore (25). The present study demonstrated that
infarct volume, and the number of apoptotic cells, was
significantly attenuated following administration of RIPostC;
therefore, further investigations were conducted regarding the
effects of RIPostC on the BID-mediated mitochondrial apoptotic
pathway. The results appeared to be consistent with a previous
study by Darling et al, which indicated that three cycles of
occlusion-reperfusion in the first minute are optimal, and delaying
postconditioning for even 1 min eliminates protection in the heart
(26). The differing degree of
protection between the results of the present study and other
reports may reside in the differences in the duration of the
ischemic insults and animal models. The results of the present
study indicated that humoral factors may have a temporal profile of
decay that reduces effectiveness, proportionate to the interval
between ischemic stimulus and reperfusion, confirming that timing
of the initiation of postconditioning is critical in the brain and
heart.
As a BH3 domain-containing proapoptotic Bcl-2 family
member, BID is inactive in the cytosol until exposed to various
stress stimuli. tBID has been demonstrated to be important for the
efficient recruitment of cytosolic Bax to the mitochondrial outer
membrane (27), resulting in
alterations to mitochondrial membrane permeability (7). In the present study, a western blot
analysis detected tBID and cytochrome c expression at the
early stages of reperfusion. Translocation from the cytosol to
mitochondria is considered an important activating mechanism for
tBID and the propagation of extracellular to intracellular
apoptotic signals (8), and this is
consistent with the immunofluorescence results obtained. BID was
activated and translocated to ischemic neuronal mitochondria
following reperfusion. Numerous proapoptotic members of the Bcl-2
family may induce cytochrome c release; however, tBID may
initiate release of almost all mitochondrial cytochrome c
into the cytosol at a very low concentration compared with others
(28). This may provide an
explanation as to why its expression peak appears later than that
of cytochrome c. These data suggested that BID is critical
to the early activation of the mitochondrial apoptotic pathway.
Notably, BID activation and cytochrome c
release were not observed at the end of MCAO (data not shown). This
is consistent with the previous findings, which suggested that
induction of apoptosis is a delayed event (29). This delay may be attributable to
the restoration of cellular energy levels during the reperfusion
phase, which may be essential for the energy-requiring steps in the
execution of the apoptotic program (30); similarly, proapoptotic substances
produced during reperfusion may have contributed to the delay.
Notably, in response to RIPostC, neither the inhibition of
cytochrome c release nor the neuroprotection against MCAO
was complete, suggesting that the mitochondrial apoptotic pathway
may have been activated by numerous mechanisms, or other factors
were present resulting in reperfusion injury. However, BID bridges
the activation of extrinsic and intrinsic apoptotic pathways in the
early stage, due to its strategic position in the apoptotic
signaling pathway, and may therefore be considered an ideal target
for therapeutic intervention.
The present study used the bilateral femoral artery,
as it is more practical to induce limb ischemia by placing a
tourniquet on an extremity for a brief period. Therefore, RIPostC
may have translational potential as a routine procedure in
emergencies and in prehospital settings, and as a thrombolytic
treatment in acute brain ischemia. However, the long-term effects
of RIPostC were not observed, and it remains to be elucidated if
underlying comorbidities may affect the efficacy of RIPostC;
therefore, further studies are required.
In conclusion, the protective effects of RIPostC may
be due to inhibition of the BID-mediated mitochondrial apoptotic
pathway. The results of the present study suggested that RIPostC
applied at the moment of reperfusion may exert a potent
neuroprotective effect against focal cerebral ischemia/reperfusion
injury. These data may help to elucidate the complex signaling
cascades involved in the neuroprotective effects of RIPostC,
leading to its potential future use as an effective treatment for
stroke patients.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant no. 81271456).
References
|
1
|
Schaller B and Graf R: Cerebral ischemia
and reperfusion: The pathophysiologic concept as a basis for
clinical therapy. J Cereb Blood Flow Metab. 24:351–371. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Li J, Yu W, Li XT, Qi SH and Li B: The
effects of propofol on mitochondrial dysfunction following focal
cerebral ischemia-reperfusion in rats. Neuropharmacology.
77:358–368. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Li J, Han B, Ma X and Qi S: The effects of
propofol on hippocampal caspase-3 and Bcl-2 expression following
forebrain ischemia-reperfusion in rats. Brain Res. 1356:11–23.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Prasad SS, Russell M and Nowakowska M:
Neuroprotection induced in vitro by ischemic preconditioning and
postconditioning: Modulation of apoptosis and PI3K-Akt pathways. J
Mol Neurosci. 43:428–442. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhang W, Wang B, Zhou S and Qiu Y: The
effect of ischemic post-conditioning on hippocampal cell apoptosis
following global brain ischemia in rats. J Clin Neurosci.
19:570–573. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Meller R, Cameron JA, Torrey DJ, Clayton
CE, Ordonez AN, Henshall DC, Minami M, Schindler CK, Saugstad JA
and Simon RP: Rapid degradation of Bim by the ubiquitin-proteasome
pathway mediates short-term ischemic tolerance in cultured neurons.
J Biol Chem. 281:7429–7436. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang Y and Tjandra N: Structural insights
of tBid, the caspase-8-activated Bid, and its BH3 domain. J Biol
Chem. 288:35840–35851. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shamas-Din A, Bindner S, Zhu W, Zaltsman
Y, Campbell C, Gross A, Leber B, Andrews DW and Fradin C: tBid
undergoes multiple conformational changes at the membrane required
for Bax activation. J Biol Chem. 288:22111–22127. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tobaben S, Grohm J, Seiler A, Conrad M,
Plesnila N and Culmsee C: Bid-mediated mitochondrial damage is a
key mechanism in glutamate-induced oxidative stress and
AIF-dependent cell death in immortalized HT-22 hippocampal neurons.
Cell Death Differ. 18:282–292. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ding ZM, Wu B, Zhang WQ, Lu XJ, Lin YC,
Geng YJ and Miao YF: Neuroprotective effects of ischemic
preconditioning and postconditioning on global brain ischemia in
rats through the same effect on inhibition of apoptosis. Int J Mol
Sci. 13:6089–6101. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yan C, Chen J, Chen D, Minami M, Pei W,
Yin XM and Simon RP: Overexpression of the cell death suppressor
Bcl-w in ischemic brain: Implications for a neuroprotective role
via the mitochondrial pathway. J Cereb Blood Flow Metab.
20:620–630. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Longa EZ, Weinstein PR, Carlson S and
Cummins R: Reversible middle cerebral artery occlusion without
craniectomy in rats. Stroke. 20:84–91. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Garcia JH, Wagner S, Liu KF and Hu XJ:
Neurological deficit and extent of neuronal necrosis attributable
to middle cerebral artery occlusion in rats. Statistical
validation. Stroke. 26:627–635. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gao L, Ji X, Song J, Liu P, Yan F, Gong W,
Dang S and Luo Y: Puerarin protects against ischemic brain injury
in a rat model of transient focal ischemia. Neurol Res. 31:402–406.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li X, Luo P, Wang F, Yang Q, Li Y, Zhao M,
Wang S, Wang Q and Xiong L: Inhibition of N-myc
downstream-regulated gene-2 is involved in an astrocyte-specific
neuroprotection induced by sevoflurane preconditioning.
Anesthesiology. 121:549–562. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang Q, Peng Y, Chen S, Gou X, Hu B, Du J,
Lu Y and Xiong L: Pretreatment with electroacupuncture induces
rapid tolerance to focal cerebral ischemia through regulation of
endocannabinoid system. Stroke. 40:2157–2164. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang KC, Koprivica V, Kim JA, Sivasankaran
R, Guo Y, Neve RL and He Z: Oligodendrocyte-myelin glycoprotein is
a Nogo receptor ligand that inhibits neurite outgrowth. Nature.
417:941–944. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang Q, Zhang X, Ding Q, Hu B, Xie Y, Li
X, Yang Q and Xiong L: Limb remote postconditioning alleviates
cerebral reperfusion injury through reactive oxygen
species-mediated inhibition of delta protein kinase C in rats.
Anesth Analg. 113:1180–1187. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gidday JM: Cerebral preconditioning and
ischaemic tolerance. Nat Rev Neurosci. 7:437–448. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhou Y, Lekic T, Fathali N, Ostrowski RP,
Martin RD, Tang J and Zhang JH: Isoflurane posttreatment reduces
neonatal hypoxic-ischemic brain injury in rats by the
sphingosine-1-phosphate/phosphatidylinositol-3-kinase/Akt pathway.
Stroke. 41:1521–1527. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dirnagl U, Simon RP and Hallenbeck JM:
Ischemic tolerance and endogenous neuroprotection. Trends Neurosci.
26:248–254. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Peixoto PM, Teijido O, Mirzalieva O,
Dejean LM, Pavlov EV, Antonsson B and Kinnally KW: MAC inhibitors
antagonize the pro-apoptotic effects of tBid and disassemble
Bax/Bak oligomers. J Bioenerg Biomembr. 49:65–74. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hess DC, Hoda MN and Bhatia K: Remote limb
perconditioning [corrected] and postconditioning: Will it translate
into a promising treatment for acute stroke? Stroke. 44:1191–1197.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Weber C: Far from the heart: Receptor
cross-talk in remote conditioning. Nat Med. 16:760–762. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Szijártó A, Czigány Z, Turóczi Z and
Harsányi L: Remote ischemic perconditioning-a simple, low-risk
method to decrease ischemic reperfusion injury: Models, protocols
and mechanistic background. A review. J Surg Res. 178:797–806.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Darling CE, Jiang R, Maynard M, Whittaker
P, Vinten-Johansen J and Przyklenk K: Postconditioning via
stuttering reperfusion limits myocardial infarct size in rabbit
hearts: Role of ERK1/2. Am J Physiol Heart Circ Physiol.
289:H1618–H1626. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ott M, Norberg E, Zhivotovsky B and
Orrenius S: Mitochondrial targeting of tBid/Bax: A role for the TOM
complex? Cell Death Differ. 16:1075–1082. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Luo X, Budihardjo I, Zou H, Slaughter C
and Wang X: Bid, a Bcl2 interacting protein, mediates cytochrome c
release from mitochondria in response to activation of cell surface
death receptors. Cell. 94:481–490. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lipton P: Ischemic cell death in brain
neurons. Physiol Rev. 79:1431–1568. 1999.PubMed/NCBI
|
|
30
|
Leist M, Single B, Castoldi AF, Kühnle S
and Nicotera P: Intracellular adenosine triphosphate (ATP)
concentration: A switch in the decision between apoptosis and
necrosis. J Exp Med. 185:1481–1486. 1997. View Article : Google Scholar : PubMed/NCBI
|