Introduction
Heat stroke (HS) is a life-threatening disease with
high morbidity and mortality that is characterized by extreme
hyperthermia (>40°C) with thermoregulatory failure (1). Since heatwaves are responsible for HS
injury and mortality in various parts of the world (2,3),
there is a growing concern regarding the prevention and treatment
of HS, particularly during military training in China (4,5).
Accumulating evidence suggests that endotoxins and cytokines are
implicated in the pathogenesis of HS (6). Inflammatory cytokines are highly
elevated in some HS patients and in animal models, and these levels
correlate with organ failure and a fatal outcome (7). The liver, a major organ of immunity
that produces and responds to cytokines during inflammation, is
notably damaged in HS (8). The
liver serves an important role in heat stress responses through the
production of cytokines and, particularly, in the clearance of
intestinal-derived endotoxins produced during hyperpyrexia,
including lipopolysaccharide (LPS) (9), which is associated with the
regulation of Kupffer cells (KCs). KCs are the primary source of
intrahepatic inflammatory cytokines (10) and the primary cause of increased
LPS concentrations (11).
KCs account for 80–90% of total resident macrophages
in vivo (12). KCs reside
in the liver and remove immune complexes and intestinal bacterial
products from the blood, which is essential for immunity against
various pathologies (13). KCs are
critical in the clearance of gut-derived endotoxins by
phagocytosis. Under normal conditions, KCs are involved in the
interruption of the formation of endotoxins by phagocytosis and the
simultaneous release of cytokines. Under conditions of
ischemia-reperfusion, hemorrhagic shock and hemorrhagic
pancreatitis, phagocytotic function of KCs is weakened by
endotoxemia; however, their secretory function enhances
inflammation, which results in multiple organ failure (14). A previous study has reported that
liver injury in rats with HS is associated with the increased
secretion of tumor necrosis factor (TNF)-α, interleukin (IL)-1β and
IL-6 by KCs (5). Previous evidence
suggests that the macrophage-derived macrophage inflammatory
protein-1α (MIP-1α) stimulates the secretion of TNF-α, IL-1β, and
IL-6 by peritoneal macrophages (15). The association between MIP-1α and
macrophages indicates that the expression of MIP-1α influences the
alteration in the concentration of inflammatory cytokines secreted
by KCs under heat stress.
MIP-1α is an important chemokine that activates
immunocytes, promotes their chemotaxis, regulates the synthesis of
cytokines and is involved in acute or mild inflammation (16). MIP-1α is produced by immune cells,
including macrophages, lymphocytes, neutrophils and dendritic cells
(17). As the largest group of
macrophages in the human body, KCs are the major source of MIP-1α
in the liver. In the MIP-1α knock-out rat model, the levels of
MIP-1α and the levels of cytokines, including TNF-α, IL-1β and
IL-6, secreted by KCs are reduced compared with normal rats, and
these alterations result in the downregulation of inflammatory
responses (17). A previous study
on acute inflammatory liver injury demonstrated that MIP-1α
secreted by KCs is increased, and anti-MIP-1α functions to decrease
the inflammatory responses as well as the mortality rate of the
rats (18). A previous study using
a liver ischemia-reperfusion injury model demonstrated that MIP-1α
secreted by KCs is responsible for the inflammation associated with
multiple organ failure (19).
Therefore, MIP-1α serves an important role in KC-associated
inflammatory responses.
As MIP-1α elevates inflammatory responses, it
ensures that increased levels of inflammation are more serious in
HS-induced liver injury. In order to identify the signaling
pathways that mediate MIP-1α-associated inflammation in KCs, c-Jun
N-terminal kinase (JNK) signaling was investigated, as previous
studies have demonstrated that activation of the JNK signaling
pathway contributes to the development of liver inflammation
(20). JNK, which is activated by
various stimuli, including cytokines, serves a role in cell
responses, inflammation and apoptosis (17). Previous research demonstrates that
the JNK signaling pathway is potentially activated in trauma,
stress and ischemia-reperfusion (21). A previous study have demonstrated
that JNK signaling is partially responsible for the activation of
macrophages and macrophage-induced renal injury (22). In addition, the activation of JNK
signaling stimulates the production of pro-inflammatory factors,
including TNF-α and IL-1, which lead to further exacerbation of the
inflammatory response (23). It
was hypothesized that KC-secreted MIP-1α promotes inflammation
through the JNK signaling pathway during HS. In the present study,
HS animal and cell models were constructed to detect changes in KCs
and the association with MIP-1α and the JNK signaling pathway.
Materials and methods
Experimental animals
The experiments were performed in 40 adult male
Wistar rats weighing 250–300 g and aged 10–12 weeks old, obtained
from the Animal Resource Center of Hubei Provincial Academy of
Preventive Medicine (Wuhan, China). The animals were housed at a
temperature of 25°C and were given free access to food and water in
the specific pathogen-free facility at Wuhan General Hospital of
Guangzhou Command (Wuhan, China). All of the animal studies were
approved by the Animal Ethics Committee of Wuhan General Hospital
of Guangzhou Command in accordance with the Guide for the Care and
Use of Laboratory Animals of the State Scientific and Technological
Commission of China.
The animals were randomly assigned to two groups.
The experimental group consisted of rats that were placed in a warm
blanket of the instrument for maintaining animal body temperature
(SS-20-2; Anhui Huaibei Zhenghua Biological Apparatus Co., Ltd.,
Anhui, China) at 40°C throughout the experiment and was
correspondingly named the HS group. An intraperitoneal injection of
sodium pentobarbital (50 mg/kg body weight) was administered to
anesthetize the animals.
Induction of heat stress
The rats in the experimental group were placed in a
folded heating blanket, with the temperature set to 40°C. A towel
was placed between the animals and the warm blanket to avoid burns.
The core temperature (represented by the rectal temperature) was
monitored by the PowerLab Data Acquisition and Analysis System (AD
Instruments, Shanghai, China) every 5 min. The point at which the
systolic blood pressure (represented by the caudal artery systolic
pressure) decreased from its peak value was taken as the onset of
HS. The rats were removed immediately from the heating blanket and
returned to room temperature (25°C).
Isolation of KCs
The liver was perfused with physiological saline
containing 0.01% EDTA through the portal vein for ~25 min until the
liver tissue was completely softened. The liver was removed,
crushed using scissors and digested into liquid, which was filtered
through a 200-mesh strainer into a 50 ml centrifuge tube. The
filtrate was washed thoroughly in RPMI-1640 (Hyclone; GE Healthcare
Life Sciences, Logan, UT, USA) at 2,500 × g for 5 min at 4°C and
suspended in 12 ml RPMI-1640. A 12 ml cell suspension was placed on
15 ml 50% Percoll solution (Pharmacia; GE Healthcare Life Sciences,
Little Chalfont, UK) and 20 ml 25% Percoll solution, and the
mixture was centrifuged at 2500 rpm for 20 min. The cells between
the layers of 25% Percoll solution and 50% Percoll were slowly
extracted and resuspended in RPMI-1640 containing 10% fetal bovine
serum (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA),
penicillin (100 U/ml) and streptomycin (100 µg/ml). The cell
suspensions were plated on 10-cm culture dishes and placed in a
humidified incubator at 37°C, containing 5% CO2. After 6
h, the cell supernatants were discarded, and the cells were washed
three times with PBS at 37°C. In total >95% of the adherent
cells were KCs, as determined by examination of cluster of
differentiation (CD)163 staining, and the cell viability >90%,
as demonstrated through trypan blue exclusion analysis. To explore
whether the JNK signaling pathway was associated with the
regulation of phagocytic and secretory functions of KCs during HS,
a JNK inhibitor (SP600125; Wuhan Myhalic Biotechnology Co., Ltd.,
Wuhan, China) was used to treat cultured KCs. KCs were cultured for
24 h, and then treated with 50 µM SP600125 for the indicated
periods of time.
Measurement of phagocytic function in
vivo and in vitro
The phagocytic activity of KCs in each group were
determined following HS termination. In vivo, the caudal
vein was injected with a 1:10 dilution of India ink in 0.9% saline
(0.25 ml/100 g). The animal was sacrificed following the India ink
injection, and the liver was processed and stained with eosin. The
consumption of carbon by KCs was observed with a microscope. LPS
concentrations were detected in the peripheral blood using a
ToxinSensor™ Chromogenic LAL Endotoxin Assay kit (GenScript
Corporation, Piscataway, NJ, USA) according to the manufacturer's
protocol. Orbital blood was collected following the injection of
diluted India ink, and the phagocytic index K value was calculated
subsequent to the absorbance being measured (24). In vitro, isolated KCs were
plated in a 96-well plate at a density of 5×106 cells/ml
and incubated overnight. pHrodo™ Escherichia coli
BioParticle-conjugated particles (Molecular Probes; Thermo Fisher
Scientific, Inc.), dissolved in RPMI-1640 at a concentration of 1
mg/ml, were added to each well. The procedure was performed using
the pHrodo™ E. coli BioParticles® Phagocytosis
kit for Flow Cytometry (Invitrogen; Thermo Fisher Scientific, Inc.)
following the manufacturer's protocol. The cells were resuspended
in 200 µl staining buffer and analyzed using an LSRII flow
cytometer (BD Biosciences, Franklin Lakes, NJ, USA). Data analysis
was conducted using the accompanying FACSDiva v8.0 and FCAP Array
v3.0 software (BD Biosciences). Interferon (IFN)-γ (RA20684), IL-1β
(RA20020), TNF-α (RA20035) and MIP-1α (RA20996) concentrations in
the medium of cultured KCs and the peripheral blood, were detected
using ELISAs (Bio-swamp Life Science Lab, Wuhan, China) in the HS
and control groups.
Silencing of MIP-1α
The small interfering RNA (siRNA) targeting rat
MIP-1α (siMIP-1α; 5′-CAGCGCCAUAUGGAGCUGAC-3′) was purchased from
Thermo Fisher Scientific, Inc. KCs were transfected with siRNAs
using Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol. Assays
were performed 48 h following transfection.
Immunofluorescence
Liver tissues were fixed in formalin and blocked in
10% normal goat serum (Wuhan Boster Biological Technology, Ltd.,
Wuhan, China) for 30 min at 37°C. The tissues were incubated with a
primary antibody against CD163 (1:100; sc-18796; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA) overnight at 4°C and
amplified with rabbit anti-goat IgG secondary antibody (1:1,000;
ab150141; Abcam, Cambridge, England) conjugated to Alexa Fluor 488
for 1 h at 37°C. For co-localization with CD163, sections were
co-stained overnight with a primary antibody against p-JNK (1:100;
sc-293136) or MIP-1α (1:100; sc-365691) (both from Santa Cruz
Biotechnology, Inc.) overnight at 4°C and amplified with goat
anti-mouse IgG secondary antibody (1:500; ab175473; Abcam)
conjugated to Alexa Fluor 568 for 1 h at 37°C. Prior to
observation, DAPI was used to label the nuclei. Images were
acquired using a fluorescence microscope (OLYMPUS BX51; Olympus
Corporation, Tokyo, Japan) with appropriate filters. The images are
presented as single-color stains of green and red to exhibit the
localization of the two markers in the cells. Merged images are
presented below the images with single-color stains. The two
markers that were co-expressed in cells at a similar location are
often observed in yellow.
Immunoblotting
Following the different treatments, the cells or
livers were harvested, washed three times with PBS and lysed in
radioimmunoprecipitation buffer [50 mM Tris-HCl (pH 7.4), 150 mM
NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM EDTA, 30
µg/ml aprotinin, 50 µg/ml leupeptin and 1 mM phenylmethane sulfonyl
fluoride] on ice for 30 min. The cell lysates were centrifuged at
12,000 × g and 4°C for 10 min, and the supernatant was collected.
Protein concentrations were measured using a Bicinchoninic Acid
Protein Assay kit (P0010S; Beyotime Institute of Biotechnology,
Shanghai, China). Protein was loaded at a concentration of 30 µg
per lane and separated by 10% SDS-PAGE. Following electrophoresis,
the proteins were transferred onto polyvinyl difluoride membranes
(Merck KGaA, Darmstadt, Germany), incubated with 5% non-fat milk
for 1 h and then incubated overnight at 4°C with the primary
antibodies. The primary antibodies were as follows: Anti-MIP-1a
antibody (1:200; sc-365691; Santa Cruz Biotechnology, Inc.) and
anti-phospho-JNK (p-JNK) antibody (1:200; sc-293136; Santa Cruz
Biotechnology, Inc.). The protein loading was normalized with
anti-β-actin antibody (1:200; sc-58673; Santa Cruz Biotechnology,
Inc.). An HRP-conjugated secondary antibody (1:5,000; BM3895;
Boster Biological Technology, Pleasanton, CA, USA) was used, and
the signals were detected using an ECL Super Signal West Pico Trial
kit (Thermo Fisher Scientific, Inc.).
Statistical analysis
Results are expressed as the mean ± standard
deviation. All experimental data were analyzed using one-way
analysis of variance followed by the least significant difference
t-test. P<0.05 was considered to indicate a statistically
significant difference. All analyses were performed using SPSS
software (version 19.0; IBM SPSS, Armonk, NY, USA).
Results
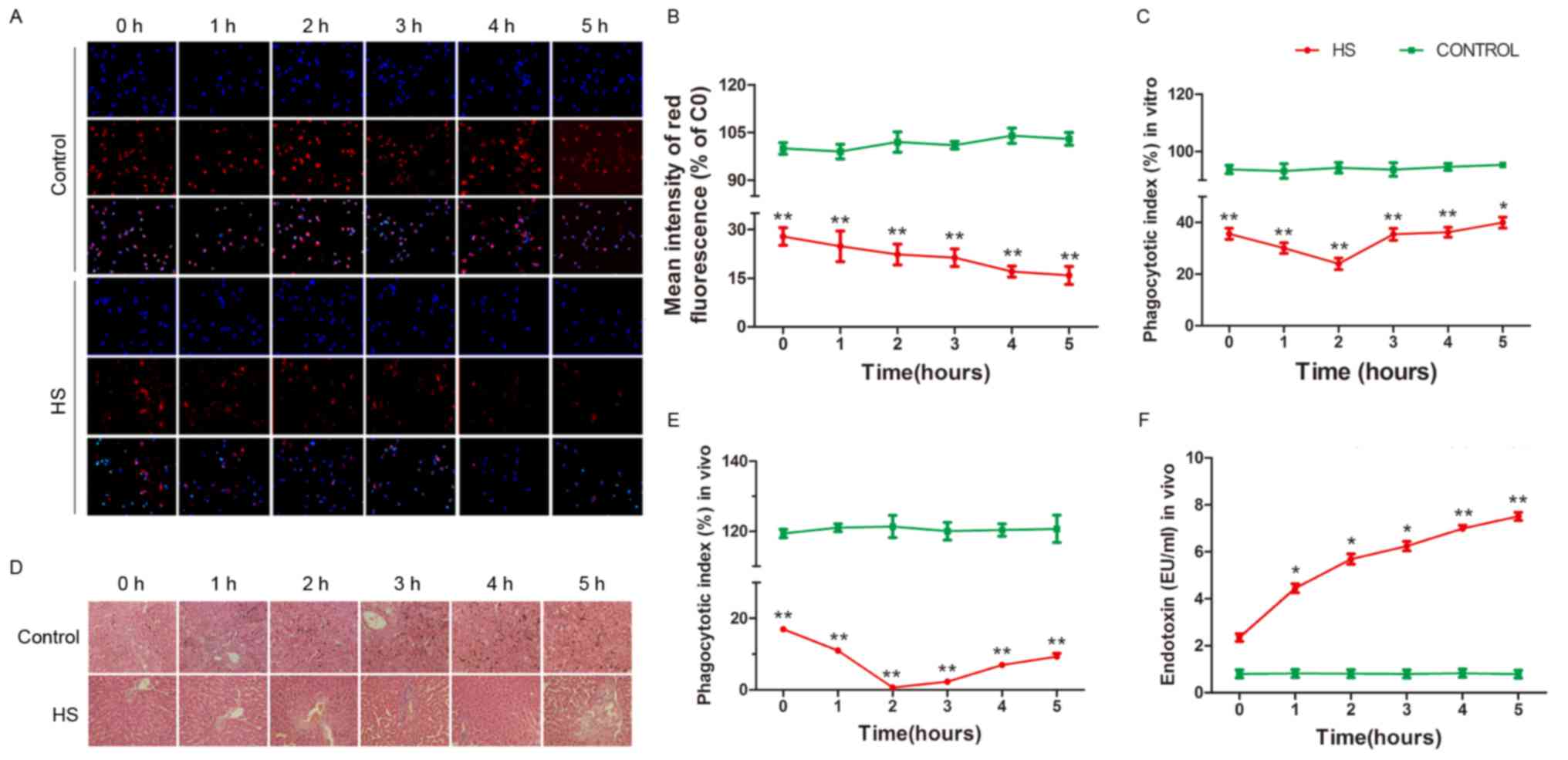
HS inhibits the phagocytic activity of
KCs
To determine whether HS is responsible for changes
in the phagocytic function of KCs, latex beads and a pHrodo E.
coli BioParticles Phagocytosis kit for Flow Cytometry was used
to detect the phagocytic activity of cultured rat KCs in each
group. The phagocytic activity of KCs was significantly decreased
in the HS group compared with the control group, as presented in
Fig. 1A-C (P<0.05).
In order to estimate the phagocytic function of KCs
in vivo and further confirm that increased temperatures
affect the phagocytic function of KCs, the livers of each group
were stained with eosin, and the consumption of carbon by the KCs
was observed microscopically subsequent to the caudal vein being
injected with diluted India ink (Fig.
1D). As presented in Fig. 1E,
the phagocytic activity of KCs exhibited a trend similar to that
observed in vitro (P<0.05). KCs help protect the
intestines from attack by bacteria and endotoxins via phagocytosis
and removal from the blood. In order to further analyze the
phagocytic function of KCs in vivo, LPS concentrations were
detected in the peripheral blood using a ToxinSensor™ Chromogenic
LAL Endotoxin Assay kit. As presented in Fig. 1F, the LPS concentration was
increased at HS onset and continued to increase to a high level
compared with that in the control group (P<0.05), indicating
that KCs failed to eliminate endotoxins from the blood. The results
of the present study suggest that the phagocytic activity of KCs is
reduced in HS conditions in vitro and in vivo.
HS activates the secretory function of
KCs
In order to determine whether HS is responsible for
altering the secretory function of KCs, the concentrations of
IFN-γ, IL-1β, TNF-α and MIP-1α in the medium of the cultured KCs
were measured in each group. As presented in Fig. 2A-D, the concentrations of IFN-γ,
IL-1β, TNF-α and MIP-1α, respectively, increased during the time
course (P<0.05). The present results demonstrate that high
temperatures strongly promote the secretion of inflammatory factors
from KCs.
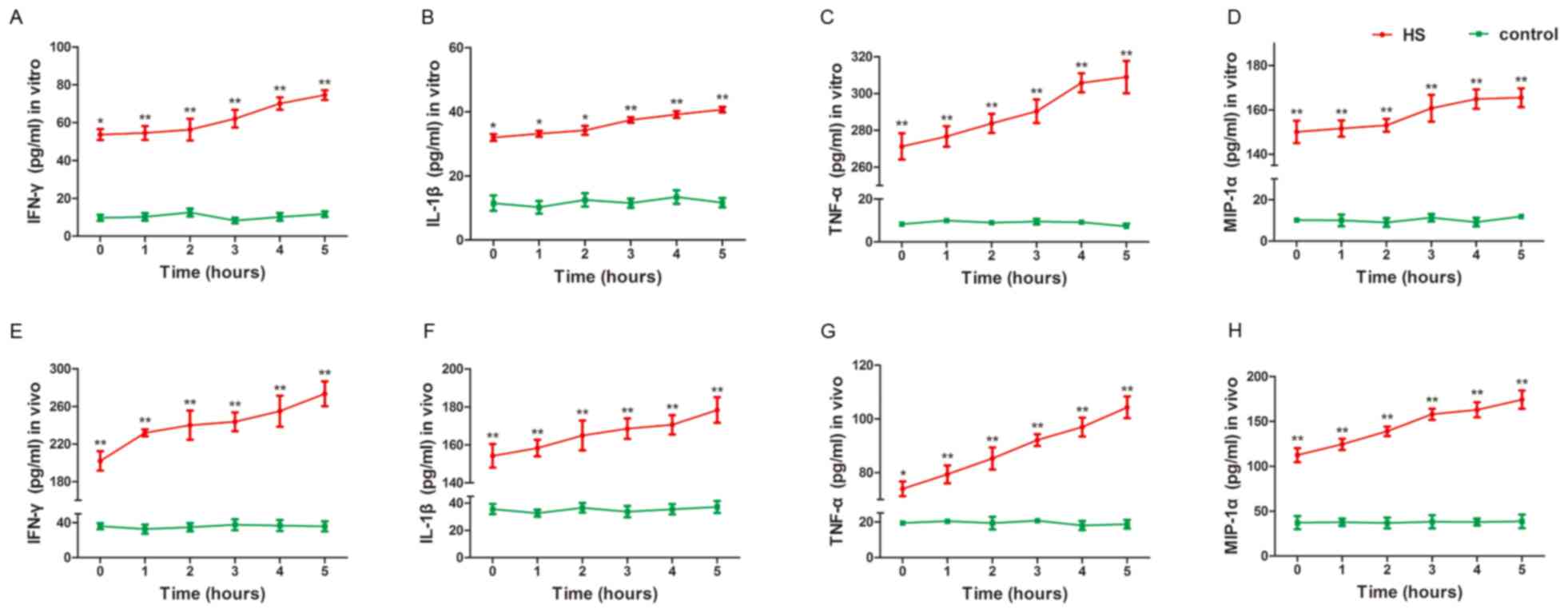 | Figure 2.HS activates the secretory function of
KCs. KCs and peripheral blood were prepared following HS for 1–5 h.
(A) IFN-γ, (B) IL-1β, (C) TNF-α and (D) MIP-1α concentrations in
the medium of cultured KCs and (E) IFN-γ, (F) IL-1β, (G) TNF-α and
(H) MIP-1α in the peripheral blood, were detected using an ELISA in
the HS and control groups. IFN-γ, IL-1β, TNF-α and MIP-1α
concentrations increased in the HS group compared with the control
group in vitro and in vivo. *P<0.05, **P<0.01
vs. control. HS, heat stroke; KCs, Kupffer cells; LPS,
lipopolysaccharide; MIP-1α, macrophage inflammatory protein 1α;
IFN, interferon; IL, interleukin; TNF, tumor necrosis factor. |
In order to estimate the secretory function of KCs
in vivo and further confirm that high temperatures influence
the secretory function of KCs, the concentrations of IFN-γ, IL-1β,
TNF-α and MIP-1α in the peripheral blood were measured in each
group. The results of the in vivo assays were similar to
those obtained in vitro. The concentrations of IFN-γ, IL-1β,
TNF-α and MIP-1α increased during the time course, as presented in
Fig. 2E-H, respectively
(P<0.05). The present results suggest that high temperatures
markedly increased the secretory functions of KCs, which promotes a
severe inflammatory response with the occurrence of HS.
Expression of MIP-1α positively
regulates the inflammatory function of KCs in HS
Since MIP-1α concentrations increased in blood and
the culture medium in the HS models, MIP-1α expression in KCs was
assessed in vitro and in vivo. MIP-1α expression in
the KCs of the rat livers increased gradually at the beginning of
HS compared with MIP-1α expression in the control group (Fig. 3A); additionally, MIP-1α protein
levels increased in the cultured liver cells and liver tissues
(Fig. 3B). By silencing MIP-1α in
the cultured KCs (Fig. 3C), the
concentrations of IFN-γ, IL-1β and TNF-α secreted by the KCs were
decreased during HS compared with the HS group (Fig. 3D).
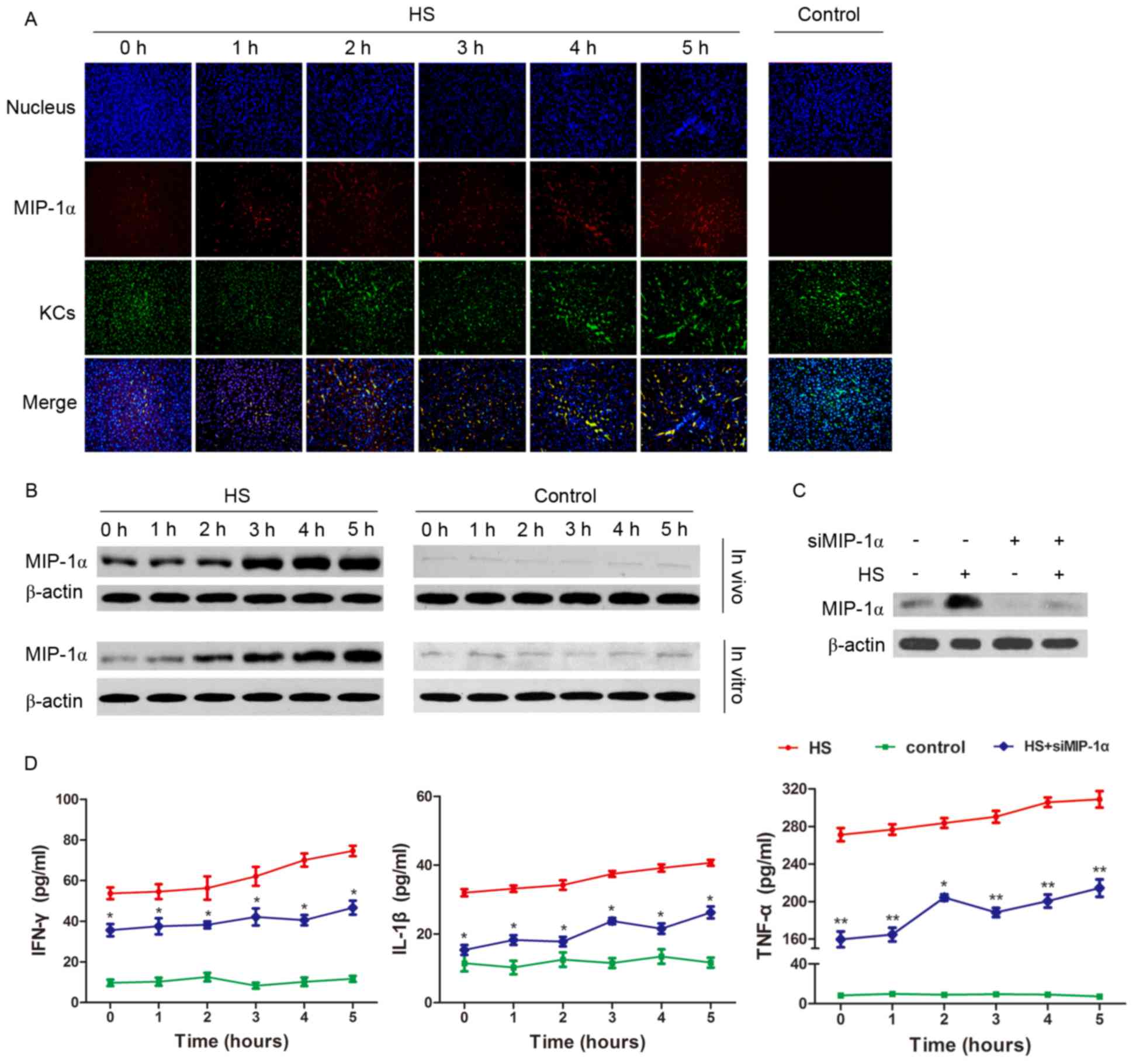 | Figure 3.Expression of MIP-1α positively
regulates KC inflammatory function in HS. (A) The livers of the
experimental animals were prepared following the induction of HS
for 1–5 h. MIP-1α (red) expression in the KCs (green) detected by
immunofluorescence revealed an increase in expression during the
time course in the HS group compared with the control group. (B)
KCs and liver extracts were prepared following the induction of HS
for 1–5 h. The amount of MIP-1α that was up-regulated in
vitro and in vivo was investigated by western blot
analysis. (C) MIP-1α expression was reduced using specifically
targeted siRNAs in cultured KCs transfected using
Lipofectamine® 2000. (D) IFN-γ, IL-1β, and TNF-α
concentration in the medium of cultured KCs decreased in the
MIP-1α-silenced HS group compared with the HS group detected by
ELISA. *P<0.05, **P<0.01 vs. HS group. HS, heat stroke; KCs,
Kupffer cells; MIP-1α, macrophage inflammatory protein 1α; IFN,
interferon; IL, interleukin; TNF, tumor necrosis factor; siRNA,
short interfering RNA. |
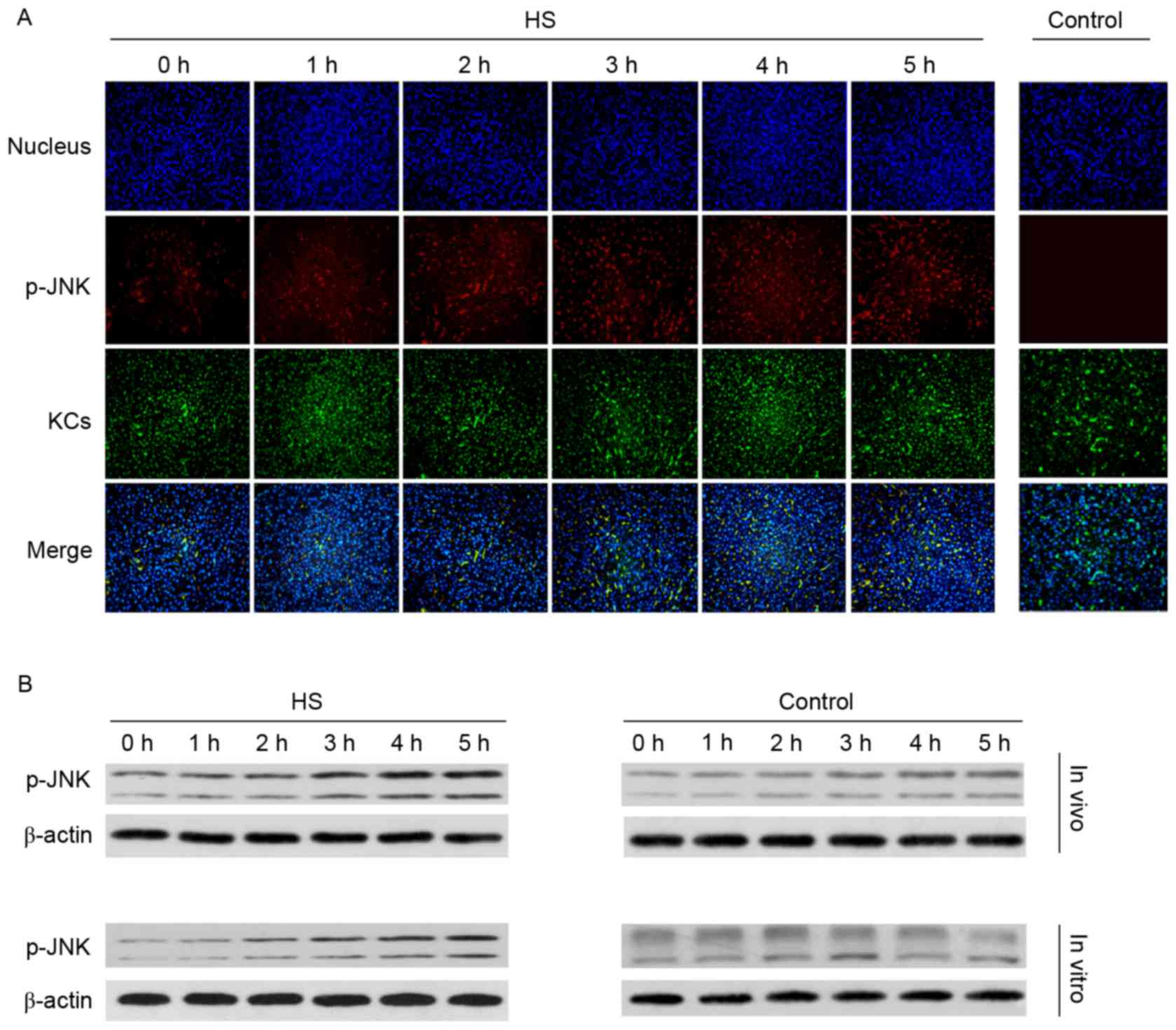
Phosphorylation of JNK increases
during HS
The JNK signaling pathway may be activated in
trauma, stress, and ischemia-reperfusion. In order to determine
whether the JNK signaling pathway is activated by KCs in the HS
model, p-JNK was assessed. As presented in Fig. 4A, the red fluorescence demonstrates
that JNK phosphorylation in KCs and other liver cells increased
during the time course following HS compared with the control
group. As presented in Fig. 4B and
C, p-JNK protein levels increased during the time course
following HS compared with the control group. The present results
indicate that JNK is activated during HS.
JNK signaling pathway is involved in
MIP-1α-mediated regulation of KC function in HS
In order to explore whether the JNK signaling
pathway is associated with the regulation of KC phagocytic and
secretory function during HS, the JNK inhibitor sp600125 was used
in the experiments with cultured KCs. The latex beads and pHrodo
E. coli BioParticles Phagocytosis kit for Flow Cytometry
analysis demonstrated that JNK inhibition improved the phagocytic
activity of the KCs (Fig. 5A-C),
whereas the detection of the inflammatory factors in the culture
medium revealed that KC secretory function was reduced compared
with the HS group (Fig. 5D-F).
Secreted MIP-1α levels in the KC medium were decreased via JNK
inhibition. In addition, MIP-1α levels in KC total protein
extractions were reduced compared with the HS group. The present
results suggest that JNK is involved in the regulation of KC
function.
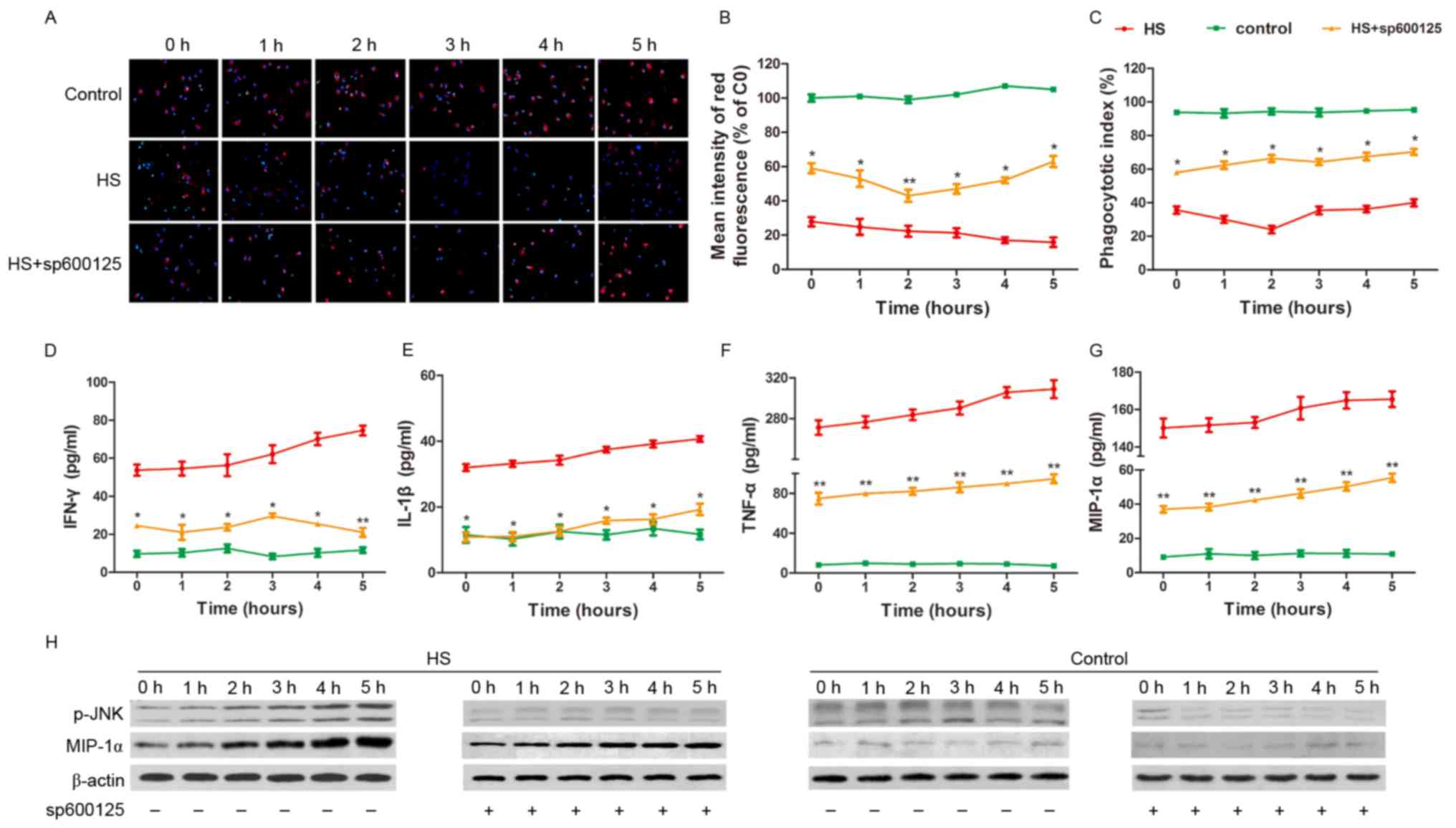 | Figure 5.JNK signaling pathway is involved in
MIP-1α-mediated regulation of KC function in HS. (A) Latex beads
(red) were applied to measure the phagocytic function of KCs, and
blue fluorescence represents the nucleus. (B) With the sp600125
treatment, the red fluorescence intensity in the KCs significantly
increased following HS compared with the cells without sp600125
treatment. (C) The phagocytic index increased following sp600125
treatment compared with HS cells without the sp600125 treatment.
(D-G) IFN-γ, IL-1β, TNF-α and MIP-1α concentrations in the medium
of cultured KCs decreased following sp600125 treatment measured by
ELISA, compared with HS cells without sp600125 treatment. (H)
Following HS, MIP-1α levels decreased in vitro subsequent to
sp600125 pretreatment, as detected by western blot analysis,
compared with cells not exposed to pretreatment. *P<0.05,
**P<0.01 vs. HS group. MIP-1α, macrophage inflammatory protein
1α; IFN, interferon; IL, interleukin; TNF, tumor necrosis factor;
KCs, Kupffer cells; JNK, c-Jun-N-terminal kinase; HS, heat
stroke. |
Discussion
In the present study, it was demonstrated that the
phagocytic activity of KCs is reduced and the secretory function of
KCs is increased in vitro and in vivo following HS.
Additionally, the JNK signaling pathway is potentially responsible
for MIP-1α-mediated regulation of KC function in the HS cellular
model.
KCs are resident macrophages that serve important
roles in liver processes, including regeneration, fibrogenesis,
inflammation and necrosis (11).
The inflammatory cytokines produced by activated KCs are involved
in the pathogenesis of various types of liver injury, including
viral hepatitis (25), alcoholic
liver disease (26) and
ischemia-reperfusion injury (27).
Previous studies have demonstrated that the secretory function of
KCs changes in several conditions, including hyperpyrexia, and that
the inhibition of cytokine secretion by KCs leads to the reduction
of liver injury (5). In the
present study, an acute increase in the secretion of inflammatory
factors by KCs subsequent to HS being observed, indicating that HS
activates the KC-mediated inflammation response.
KCs eliminate and decompose various exogenous and
endogenous substances, including gut-derived endotoxins in the
liver, which is an essential regulatory function. LPS, which is
located on the outer wall of gram-negative bacteria, is
predominantly eliminated by KCs in the liver (28). KC activation by LPS causes the
release of IL-1, IL-6, TNF-α and other inflammatory cytokines and
triggers additional inflammatory processes (29). Elevated levels of endotoxin (LPS)
delivered to the liver via portal blood may cause liver injury
(30). In the present study, a
decrease in the phagocytic activity of KCs following HS was noted,
and increased LPS levels indicated that the ability of KCs to
decompose LPS was weakened.
As the phagocytic activity of KCs decreases while
the secretory function increases during HS to promote inflammatory
responses, and since MIP-1α is a pro-inflammatory factor in
macrophages, MIP-1α was the focus of the present study. MIP-1α is a
member of the cysteine chemokine subfamily and is involved in the
regulation of cell-mediated immunity (31). MIP-1α attracts and stimulates
basophils to secrete pro-inflammatory substances (32). Previous studies have demonstrated
MIP-1α protein and mRNA overexpression in KCs following liver
ischemia-reperfusion injury (19).
As little is known regarding MIP-1α expression in heat-stressed
conditions, a model of HS was established in order to investigate
alterations in MIP-1α expression in KCs, and it was identified that
MIP-1α expression increased gradually following HS onset. However,
MIP-1α silencing in KCs resulted in reduced levels of inflammatory
cytokine secretion by KCs subsequent to HS compared with the HS
group.
In order to investigate the mechanism of
MIP-1α-associated translation of the production of pro-inflammatory
cytokines by KCs in HS, the stress-associated nuclear import
pathways, including JNK signaling, were examined. The JNK signaling
pathway is reported to be associated with the development of liver
inflammation and mammalian TNF-α signaling (20,33).
JNK serves a major role in cell responses, inflammation, apoptosis
and heat stress, particularly in KCs (16,34,35).
However, an association between the function of KCs and JNK has not
been established. Consistent with these observations, a
time-dependent activation of JNK phosphorylation was demonstrated
1–5 h following HS. Following inhibition of JNK phosphorylation by
sp600125 in KCs in vitro and in vivo, decreased
amounts of inflammatory cytokines and MIP-1α were produced from
activated KCs, whereas increased phagocytic activation of KCs was
detected following HS compared with that in the HS group without
inhibition.
In the present study, all of the experiments were
undertaken with animal and cell models, which provide a powerful
tool for HS studies. However, the results obtained with the models
may not be consistent with those obtained from studies in humans,
therefore further clinical studies are required.
In conclusion, the present study indicates that KC
function alters in HS-induced injury. Furthermore, it emphasizes
that HS enriches the expression of MIP-1α in KCs and the
phosphorylation of JNK, and that JNK signaling is required for the
HS response.
Acknowledgements
The present study was supported by the China
Postdoctoral Science Foundation (grant no. 2015M572816).
References
|
1
|
Widodo D: Heat stroke. Acta Med Indones.
37:39–42. 2005.PubMed/NCBI
|
|
2
|
Keatinge WR: Death in heat waves. BMJ.
327:512–513. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Misset B, De Jonghe B, Bastuji-Garin S,
Gattolliat O, Boughrara E, Annane D, Hausfater P, Garrouste-Orgeas
M and Carlet J: Mortality of patients with heatstroke admitted to
intensive care units during the 2003 heat wave in France: A
national multiple-center risk-factor study. Crit Care Med.
34:1087–1092. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Li L, Gu Z, Liu Z and Su L: The effect of
reactive oxygen species regulation of expression of Bcl-2 and Bax
in apoptosis of human umbilical vein endothelial cell induced by
heat stress. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue. 26:458–463.
2014.(In Chinese). PubMed/NCBI
|
|
5
|
Chen Y, Tong H, Zhang X, Tang L, Pan Z,
Liu Z, Duan P and Su L: Xuebijing injection alleviates liver injury
by inhibiting secretory function of Kupffer cells in heat stroke
rats. J Tradit Chin Med. 33:243–249. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lu KC, Wang JY, Lin SH, Chu P and Lin YF:
Role of circulating cytokines and chemokines in exertional
heatstroke. Crit Care Med. 32:399–403. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rodriguez-Fernandez M, Grosman B,
Yuraszeck TM, Helwig BG, Leon LR and Doyle FJ III: Modeling the
intra- and extracellular cytokine signaling pathway under heat
stroke in the liver. PLoS One. 8:e733932013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hall DM, Xu L, Drake VJ, Oberley LW,
Oberley TD, Moseley PL and Kregel KC: Aging reduces adaptive
capacity and stress protein expression in the liver after heat
stress. J Appl Physiol (1985). 89:749–759. 2000.PubMed/NCBI
|
|
9
|
Jirillo E, Caccavo D, Magrone T,
Piccigallo E, Amati L, Lembo A, Kalis C and Gumenscheimer M: The
role of the liver in the response to LPS: Experimental and clinical
findings. J Endotoxin Res. 8:319–327. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fukuda T, Mogami A, Tanaka H, Yoshikawa T,
Hisadome M and Komatsu H: Y-40138, a multiple cytokine production
modulator, protects against D-galactosamine and
lipopolysaccharide-induced hepatitis. Life Sci. 79:822–827. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kolios G, Valatas V and Kouroumalis E:
Role of Kupffer cells in the pathogenesis of liver disease. World J
Gastroenterol. 12:7413–7420. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu LM, Liang DY, Ye CG, Tu WJ and Zhu T:
The UII/UT system mediates upregulation of proinflammatory
cytokines through p38 MAPK and NF-kappaB pathways in LPS-stimulated
Kupffer cells. PLoS One. 10:e01213832015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Vollmar B and Menger MD: The hepatic
microcirculation: Mechanistic contributions and therapeutic targets
in liver injury and repair. Physiol Rev. 89:1269–1339. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kono H, Fujii H, Asakawa M, Yamamoto M,
Maki A, Matsuda M, Rusyn I and Matsumoto Y: Functional
heterogeneity of the kupffer cell population is involved in the
mechanism of gadolinium chloride in rats administered endotoxin. J
Surg Res. 106:179–187. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fahey TJ III, Tracey KJ, Tekamp-Olson P,
Cousens LS, Jones WG, Shires GT, Cerami A and Sherry B: Macrophage
inflammatory protein 1 modulates macrophage function. J Immunol.
148:2764–2769. 1992.PubMed/NCBI
|
|
16
|
Giribaldi G, Prato M, Ulliers D, Gallo V,
Schwarzer E, Akide-Ndunge OB, Valente E, Saviozzi S, Calogero RA
and Arese P: Involvement of inflammatory chemokines in survival of
human monocytes fed with malarial pigment. Infect Immun.
78:4912–4921. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hsieh CH, Frink M, Hsieh YC, Kan WH, Hsu
JT, Schwacha MG, Choudhry MA and Chaudry IH: The role of MIP-1
alpha in the development of systemic inflammatory response and
organ injury following trauma hemorrhage. J Immunol. 181:2806–2812.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Nishi T, Maier CM, Hayashi T, Saito A and
Chan PH: Superoxide dismutase 1 overexpression reduces MCP-1 and
MIP-1 alpha expression after transient focal cerebral ischemia. J
Cereb Blood Flow Metab. 25:1312–1324. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ma W, Wang ZR, Shi L and Yuan Y:
Expression of macrophage inflammatory protein-1alpha in Kupffer
cells following liver ischemia or reperfusion injury in rats. World
J Gastroenterol. 12:3854–3858. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Seki E, Brenner DA and Karin M: A liver
full of JNK: Signaling in regulation of cell function and disease
pathogenesis, and clinical approaches. Gastroenterology.
143:307–320. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mukhopadhyay P, Rajesh M, Horváth B,
Bátkai S, Park O, Tanchian G, Gao RY, Patel V, Wink DA, Liaudet L,
et al: Cannabidiol protects against hepatic ischemia/reperfusion
injury by attenuating inflammatory signaling and response,
oxidative/nitrative stress, and cell death. Free Radic Biol Med.
50:1368–1381. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ikezumi Y, Hurst L, Atkins RC and
Nikolic-Paterson DJ: Macrophage-mediated renal injury is dependent
on signaling via the JNK pathway. J Am Soc Nephrol. 15:1775–1784.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chan ED, Winston BW, Jarpe MB, Wynes MW
and Riches DW: Preferential activation of the p46 isoform of
JNK/SAPK in mouse macrophages by TNF alpha. Proc Natl Acad Sci USA.
94:13169–13174. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu JW: The effect of endotoxin tolerance
on the Galn/LPS induced MAPK and STAT signal transduction. Shanxi
Medical University; 2006
|
|
25
|
Adams DH and Hubscher SG: Systemic viral
infections and collateral damage in the liver. Am J Pathol.
168:1057–1059. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gregory SH and Wing EJ: Neutrophil-Kupffer
cell interaction: A critical component of host defenses to systemic
bacterial infections. J Leukoc Biol. 72:239–248. 2002.PubMed/NCBI
|
|
27
|
Su GL: Lipopolysaccharides in liver
injury: Molecular mechanisms of Kupffer cell activation. Am J
Physiol Gastrointest Liver Physiol. 283:G256–G265. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Diesen DL and Kuo PC: Nitric oxide and
redox regulation in the liver: Part II. Redox biology in pathologic
hepatocytes and implications for intervention. J Surg Res.
167:96–112. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tukov FF, Luyendyk JP, Ganey PE and Roth
RA: The role of tumor necrosis factor alpha in
lipopolysaccharide/ranitidine-induced inflammatory liver injury.
Toxicol Sci. 100:267–280. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Thakur V, Pritchard MT, McMullen MR, Wang
Q and Nagy LE: Chronic ethanol feeding increases activation of
NADPH oxidase by lipopolysaccharide in rat Kupffer cells: Role of
increased reactive oxygen in LPS-stimulated ERK1/2 activation and
TNF-alpha production. J Leukoc Biol. 79:1348–1356. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Matsukawa A, Hogaboam CM, Lukacs NW and
Kunkel SL: Chemokines and innate immunity. Rev Immunogenet.
2:339–358. 2000.PubMed/NCBI
|
|
32
|
Kaplan AP, Kuna P and Reddigari SR:
Chemokines and the allergic response. Exp Dermatol. 4:260–265.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Baud V and Karin M: Signal transduction by
tumor necrosis factor and its relatives. Trends Cell Biol.
11:372–377. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Shen J, Sakaida I, Uchida K, Terai S and
Okita K: Leptin enhances TNF-alpha production via p38 and JNK MAPK
in LPS-stimulated Kupffer cells. Life Sci. 77:1502–1515. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gonda RL, Garlena RA and Stronach B:
Drosophila heat shock response requires the JNK pathway and
phosphorylation of mixed lineage kinase at a conserved
serine-proline motif. PLoS One. 7:e423692012. View Article : Google Scholar : PubMed/NCBI
|