Introduction
Heat stress is a common stressful factor that
affects many biological systems (1). In the process of heat stroke,
intestinal epithelial cells are attacked by environmental heat and
stimulated by intestinal bacteria and bacterial lipopolysaccharide
(LPS) (2). Heat stress-induced
intestinal epithelial cell injury and apoptosis contributes to
intestinal hyperpermeability. Furthermore, bacterial products from
the intestinal lumen entering into the circulatory system causes
systemic inflammatory response syndrome and multiple organ failure
(3–5). In our previous study, heat stress was
demonstrated to induce apoptosis via transcription-independent
p53-mediated mitochondrial signaling pathways in human umbilical
vein endothelial cells (6). LPS is
a major cell wall component in gram-negative bacteria that has been
demonstrated to induce apoptosis and injury in various cell types
(7). However, little is known
about the biological effects of heat stress combined with LPS on
intestinal epithelial cell apoptosis.
Reactive oxygen species (ROS) components, including
superoxide anions, hydrogen peroxide and hydroxyl radicals, are
typically generated in the mitochondria and serve as signaling
intermediates (8,9). Under physiological conditions,
generated ROS are rapidly eliminated by antioxidant enzymes,
including superoxide dismutases, catalase, glutathione peroxidases
and peroxiredoxins (8). Numerous
studies link oxidative stress with heat stress or LPS, and suggest
synergistic augmentation of cell death and increased ROS generation
in certain cells (10,11).
The mitogen activated protein kinase (MAPK) cascades
are activated by various cellular stresses and growth factors.
Extracellular signal-regulated kinase (ERK), c-Jun NH2-terminal
kinase (JNK) and p38 are members of well-characterized subfamilies
of MAPK, and these enzymes have been implicated in the increased
sensitivity to heat stress-induced cell apoptosis exhibited by
IEC-6 cells (12–14). c-Jun is the most extensively
studied protein of activator protein-1 components and has also been
linked to apoptosis. The phosphorylation of c-Jun at Ser 63 and 73
is known to increase c-Jun activity (15–18).
However, it remains unknown whether the MAPKs and c-Jun activation
serves a critical role in LPS combined with heat stress-induced
apoptosis, or if ROS has effects on MAPK and c-Jun signaling
pathways under these conditions.
The present study aimed to investigate whether
generation of reactive oxygen species (ROS) is a critical mediator
in LPS combined with heat stress-induced apoptosis, and whether
this may involve MAPK and c-Jun activation. Furthermore, whether
cell apoptosis and permeability is exacerbated by inhibition of
ERK1/2 (PD98059), JNK (SP600125), p38 (SB203580) and c-Jun (c-Jun
peptide) activation was investigated.
Materials and methods
Cell culture and treatments
IEC-6 cells and Caco-2 cells were purchased from
Shanghai Institute of Cell Biology, Chinese Academy of Sciences
(Shanghai, China). Cells were cultured in Dulbecco's modified
Eagle's medium (DMEM) supplemented with 10% (v/v) fetal bovine
serum (FBS), 100 U/ml penicillin and 100 µg/ml streptomycin
(Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA) at
37°C in a humidified atmosphere of 5% CO2 and 95% air.
Culture dishes were placed into a circulating water bath at
37±0.5°C for control, or at 42±0.5°C for heat stress for 60 min.
The culture media was replaced with fresh media and the cells were
further incubated at 37°C for 6 h.
Intracellular ROS level
Levels of intracellular ROS were assessed using a
ROS assay kit (Beyotime Institute of Biotechnology, Haimen, China).
Dichlorofluorescein diacetate (DCFH-DA; Molecular Probes; Thermo
Fisher Scientific, Inc.) enters the cells and reacts with ROS,
producing the fluorophore DCF. Briefly, cells were exposed to 1, 5
or 10 µg/ml LPS for 30 min at 37°C, exposed to 1 µg/ml LPS for 5,
15, 30, 60 or 90 min, or cells were treated with 1 µg/ml LPS
combined with heat stress (42°C) simultaneously for 60 min, the
cell culture media for each plate was then replaced with fresh
media and the cells were further incubated at 37°C for 6 h, control
cells were always incubated at 37°C. Control cells were treated
with PBS instead of LPS. Cells (3×105) were harvested,
washed with serum-free DMEM culture medium, and stained with 10 µM
DCFH-DA for 30 min at 37°C in the dark. Following this, cells were
harvested, washed and resuspended in serum-free DMEM culture medium
three times. The fluorescence intensity was determined using a flow
cytometer (FACSCanto™ II; BD Biosciences, San Jose, CA,
USA) and analyzed using FlowJo software version 9.0 (Tree Star,
Inc., Ashland, OR, USA).
Mitochondrial membrane potential
assay
The fluorescent dye
5,5′,6,6′-tetrachloro-1,1′,3,3′tetraethylbenzimidazo carbocyanine
iodide (JC-1; Molecular Probes; Thermo Fisher Scientific, Inc.) was
used to detect the mitochondrial membrane potential. IEC-6 cells
were treated with LPS (1 µg/ml), heat stress (42°C for 60 min), or
a combination of LPS and heat stress, and further incubated for 12
h at 37°C. Cells were washed three times with PBS. A JC-1 kit was
used to detect the mitochondrial cross membrane potential. Results
were observed through a fluorescence microscope.
Flow cytometric analysis of cell
apoptosis using Annexin V-fluorescein isothiocyanate
(FITC)/propidium iodide (PI) staining
Cells were pretreated with butylated hydroxyanisole
(BHA; 5 µM; Abcam, Cambridge, MA, USA), SP600125 (10 µM; Cell
Signaling Technology, Inc., Danvers, MA, USA), SB203580 (5 µM; Cell
Signaling Technology, Inc.), PD98059 (10 µM; Cell Signaling
Technology, Inc.) and c-Jun peptide (10 µM; R&D Systems, Inc.,
Minneapolis, MN, USA), the specific inhibitors of ROS, JNK, p38,
ERK and c-Jun, respectively, at 37°C for 30 min prior to
simultaneous treatment with 1 µg/ml LPS and heat stress (42°C) for
60 min, followed by a recovery period at 37°C for 6 h. Control
cells were treated with PBS instead of LPS and were always
incubated at 37°C. Cell apoptosis was analyzed using an Annexin
V-FITC apoptosis kit (Lianke Biological Engineering Co., Ltd.,
Zhejiang, China) using a flow cytometer according to the
manufacturer's protocol. Cells (1×106) cells were
collected, washed in ice-cold PBS, and resuspended in the binding
buffer containing 5 µl FITC Annexin V and 10 µl PI stain. The
suspension was mixed and incubated at room temperature for 10 min.
Samples were subsequently analyzed using a flow cytometer.
Measurement of transepithelial
electrical resistance (TEER)
Approximately 2×106 IEC-6 and Caco-2
cells per well were seeded into collagen-coated Transwell membrane
inserts separately (6.5-mm diameter inserts; 3-mm pore size;
Corning Incorporated, Corning, NY, USA) with 200 ml culture medium
added to the apical chamber and 600 ml to the basolateral chamber.
Cells were treated with LPS (1 µg/ml) for 60 min at 37°C, heat
stress (42°C for 60 min) or simultaneous treatment with a
combination of LPS (1 µg/ml) and heat stress (42°C) for 60 min. the
cells were subsequently returned to a temperature of 37°C, after 0,
2, 6 and 12 h recovery period, the electrical resistance of
confluent polarized IEC-6 and Caco-2 monolayers was measured by
TEER with an electrical resistance system (EVOM; World Precision
Instruments GmbH, Berlin, Germany) at 0, 2, 6 and 12 h, separately.
A pair of electrodes were placed at each of the apical and
basolateral chambers of three different points to evaluate
TEER.
Intestinal paracellular permeability
assay
Intestinal paracellular permeability across cell
monolayers was determined by measuring the flux of horseradish
peroxidase (HRP; type V; Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany). HRP (3.4×10−6 mol/l) was added to the medium
in the apical chamber of Transwell chambers. After pre-treatment
with LPS for 30 min and exposure to heat stress for 1 h, samples
were carefully taken from basolateral chambers and assayed for HRP
by TMB HRP Color Development Solution for ELISA (Beyotime, China) 6
h later. Enzyme activity was determined from the rate of increase
in optical density at a wavelength of 370 nm by an automatic
microplate reader (SpectraMax® M5; Molecular Devices,
LLC, Sunnyvale, CA, USA).
Western blot analysis
Protein concentration was determined by western
blotting. IEC-6 cells were exposed to LPS (1 µg/ml) for 60 min at
37°C, heat stress (42°C for 60 min) or simultaneous treatment with
a combination of LPS (1 µg/ml) and heat stress treatment (42°C) for
60 min, and the cells were returned to a temperature of 37°C for 6
h. In addition, cells were pretreated with or without BHA for 30
min at 37°C, which was followed by simultaneous treatment with LPS
(1 µg/ml) and heat stress (42°C) with incubation for 0, 15, 30 or
60 min. Control cells treated with PBS instead of LPS and were
incubated at 37°C. Cells were homogenized in
radioimmunoprecipitation assay lysis buffer with
phenylmethylsulfonyl fluoride (Sigma-Aldrich; Merck KGaA).
Following centrifugation at 14,000 × g at 4°C for 10 min, the
supernatants were used for western blot analysis. Protein
concentration was determined by a Bicinchoninic Acid Protein Assay
kit (Thermo Fisher Scientific, Inc.). Proteins (20 µg/well) were
separated by SDS-PAGE using 10% SDS polyacrylamide gels and
transferred onto polyvinylidene difluoride membranes. Membranes
were blocked with blocking solution (5% skimmed milk diluted with
PBS) at room temperature for 2 h, followed by incubation with
primary antibodies. The following rabbit primary antibodies were
used at a 1:2,000 dilution: GAPDH (cat. no. ab70699; Abcam),
phosphorylated (p)-JNK (cat. no. 4668; Cell Signaling Technology,
Inc.), p-p38 (cat. no. 4511; Cell Signaling Technology, Inc.),
p-ERK1/2 (cat. no. 4370; Cell Signaling Technology, Inc.), JNK
(cat. no. 9252; Cell Signaling Technology, Inc.), p38 (cat. no.
8690; Cell Signaling Technology, Inc.), ERK1/2 (cat. no. 4695; Cell
Signaling Technology, Inc.), c-Jun (cat. no. 9165p; Cell Signaling
Technology, Inc.), p-c-Jun (cat. no. 8222S; Cell Signaling
Technology, Inc., Danvers, MA, USA), caspase-3 (cat. no. 14220S;
Cell Signaling Technology, Inc.) and cleaved caspase-3 (cat. no.
9654S; Cell Signaling Technology, Inc.) overnight at 4°C. A
HRP-conjugated goat anti-rabbit IgG antibody served as the
secondary antibody (1:5,000; cat. no. 7074; Cell Signaling
Technology, Inc.) for incubation at room temperature for 2 h.
Proteins were visualized using an Enhanced Chemiluminescence
reagent (Pierce; Thermo Fisher Scientific, Inc.). Membranes were
exposed to light-sensitive film and quantified using Image J
software version 1.3.4.67 (National Institutes of Health, Bethesda,
MD, USA).
Statistical analysis
All data were analyzed for statistical significance
using SPSS software version 13.0 (SPSS, Chicago, IL, USA). Data are
expressed as the mean ± standard deviation from at least three
independent experiments performed in duplicate. One-way analysis of
variance was performed followed by Fisher's least significant
difference post hoc test for multiple comparisons. *P<0.05 was
considered to indicate a statistically significant difference.
Results
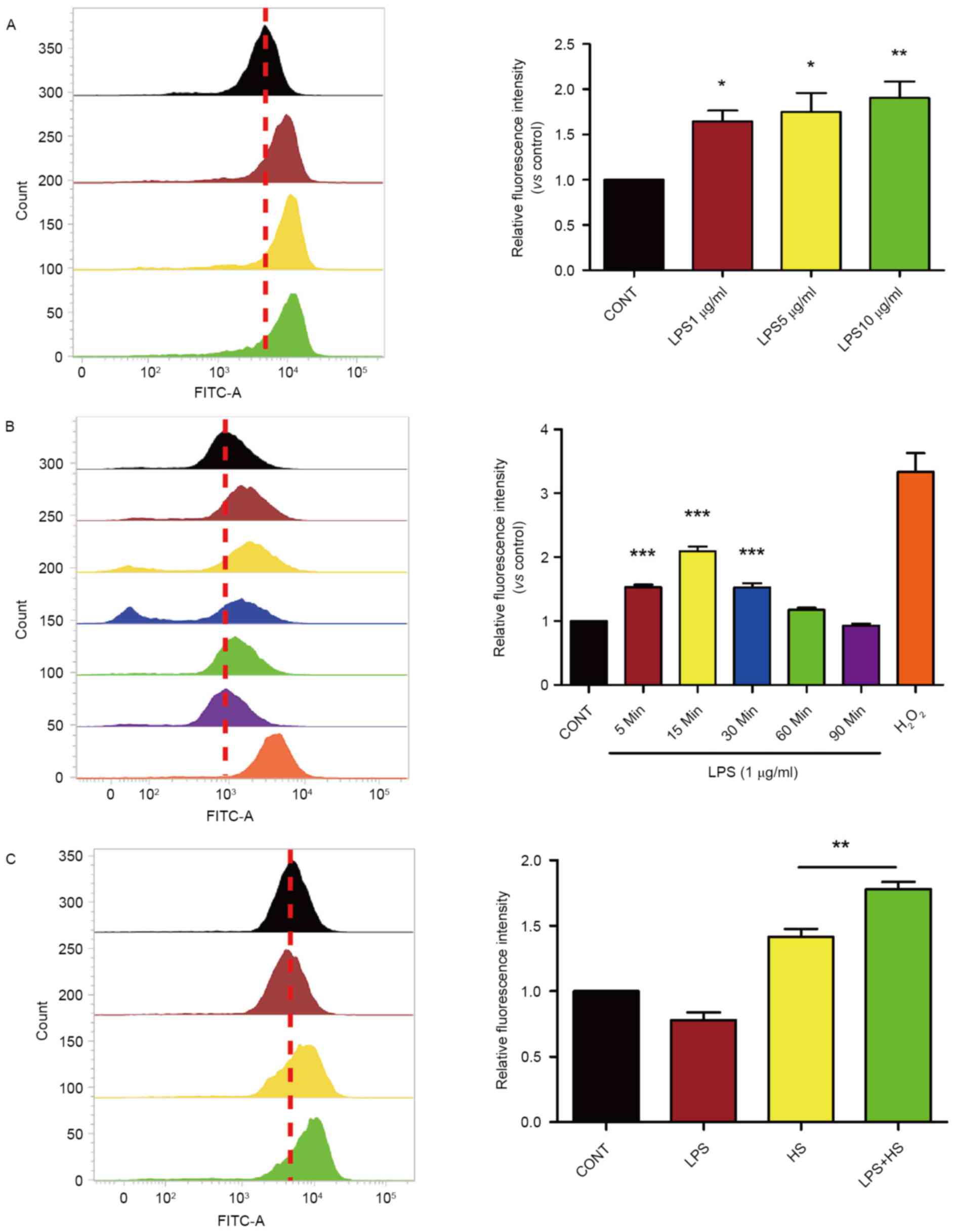
Heat stress plus LPS increases the
production of ROS and permeability in IEC-6 cells
Based on the evidence that ROS generation serves as
an important role in heat stress and LPS, the present study
examined whether heat stress associated with LPS influences ROS
accumulation in IEC-6 cells. Cells were exposed to 1, 5 or 10 µg/ml
of LPS for 30 min. Following incubation with LPS, levels of
intracellular ROS increased in a dose-dependent manner (Fig. 1A). As presented in Fig. 1B, a time-course experiment revealed
that 1 µg/ml LPS increased intracellular ROS levels in IEC-6 cells
compared with controls, with maximal induction being observed after
a 15-min incubation and returning to baseline levels at 90 min,
H2O2 was used as a positive control. To test
the combined effects of LPS treatment with heat stress on ROS
accumulation, IEC-6 were treated with 1 µg/ml LPS and exposed to
heat stress (42°C) for 60 min. At 6 h after treatment, the levels
of intracellular ROS in IEC-6 cells exposed to both LPS and heat
stress were increased compared with controls and cells exposed to
heat stress alone (Fig. 1C).
Heat stress and LPS lead to the
mitochondrial membrane potential disruption in IEC-6 cells
Mitochondria may provide a permanent source of ROS
under physiological conditions. Alterations in the mitochondrial
membrane potential (ΔΨm) were assessed using JC-1. When
mitochondrial membrane potential is high, JC1 accumulates in the
mitochondrial matrix, forming JC-1 polymer aggregates which
produces red fluorescence. When the mitochondrial membrane
potential is lower, JC1 cannot accumulate in the mitochondrial
matrix, JC-1 remains a monomer, which produces green fluorescence.
As presented in Fig. 2, LPS and
heat stress resulted in the reduction of mitochondrial membrane
potential, particularly when used in combination.
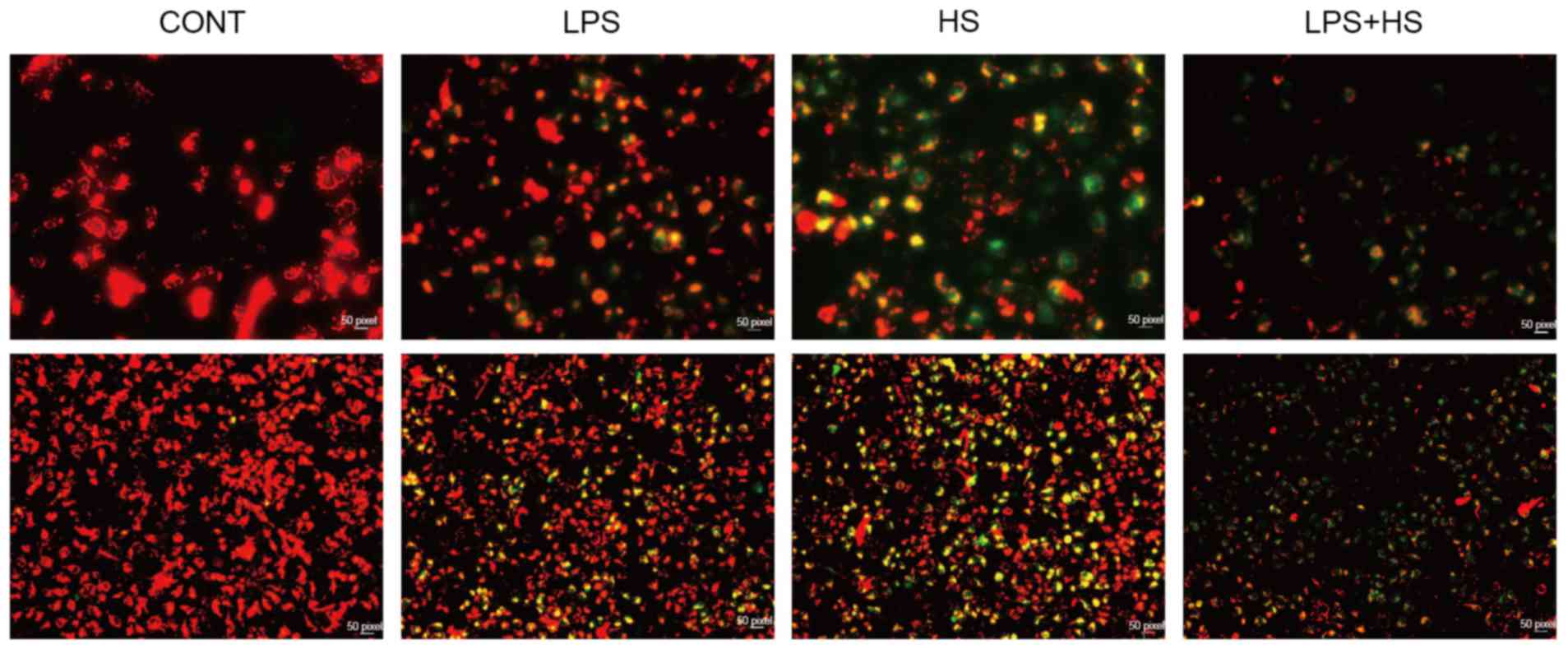 | Figure 2.Effects of heat stress and LPS on the
mitochondrial membrane potential in IEC-6 cells. IEC-6 cells were
treated with LPS (1 µg/ml), heat stress (42°C for 60 min) or a
combination of LPS and HS. The mitochondrial membrane potential was
assayed by 5,5′,6,6′-tetrachloro-1,1′,3,3′tetraethylbenzimidazo
carbocyanine iodide staining. Upper and lower images have
magnifications of ×200 and ×40, respectively. CONT, control; LPS,
lipopolysaccharide; HS, heat stress. |
Effect of heat stress and LPS on
intestinal epithelial cell apoptosis and barrier function
There were significant differences in early
apoptotic rates among the control, LPS, heat stress, and LPS
combined with heat stress groups. Early apoptotic rates in the LPS,
heat stress, and LPS combined with HS groups were significantly
higher compared with the control group. Early apoptotic rates in
the heat stress combined with LPS group were also increased
compared with the LPS and heat stress groups (Fig. 3A). These results indicated that
heat stress or LPS may increase apoptosis of IEC-6 cells, and that
the combination of these two treatments may produce a synergistic
effect.
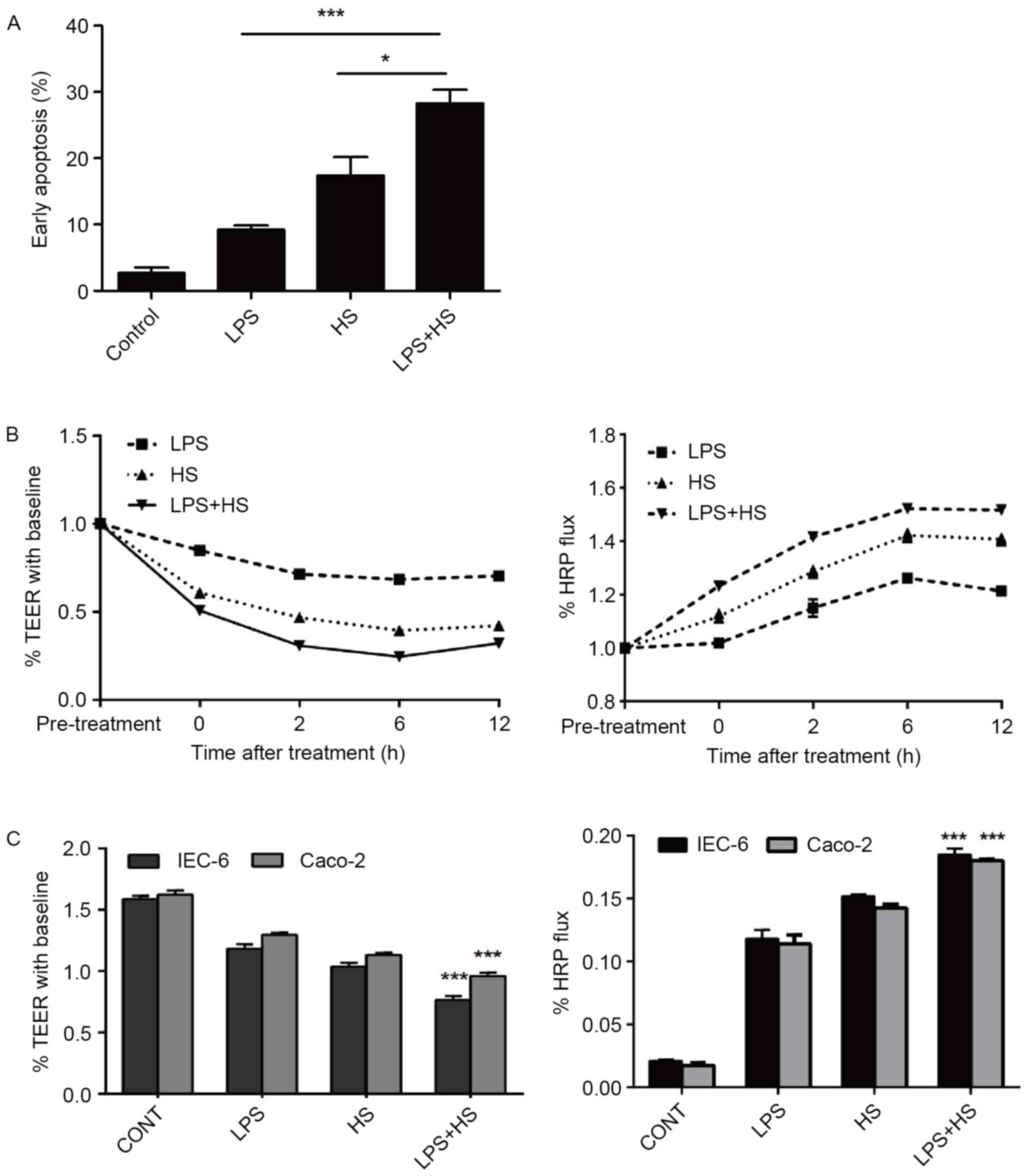 | Figure 3.Effects of heat stress, LPS, and a
combination of LPS and HS treatment on IEC-6 cell early apoptosis
and intestinal epithelial barrier function. IEC-6 cells were
treated with LPS (1 µg/ml), HS (42°C for 60 min) or a combination
of LPS and HS. (A) Quantification of flow cytometry following
Annexin V-fluorescein isothiocyanate/propidium iodide staining.
*P<0.05 and ***P<0.001. TEER and HRP permeability analysis of
epithelial barrier function of (B) IEC-6 cells after 0, 2, 6 and 12
h treatment, and (C) IEC-6 and Caco-2 cells in the different
treatment groups. Data are presented as the mean ± standard
deviation of three independent experiments. ***P<0.001 vs. HS
and LPS groups. TEER, transepithelial electrical resistance; HRP,
horseradish peroxidase; HS, heat stress; CONT, control; LPS,
lipopolysaccharide. |
The present study examined the effect of LPS and
heat stress on the effects of heat stress-induced dysfunction of
the intestinal epithelial barrier in IEC-6 and Caco-2 monolayers.
Caco-2 cells were used as a model to form a typical TJ structure
similar to the mature intestinal epithelium in vitro
(19). Epithelial barrier
integrity and paracellular permeability were determined by the
measurement of TEER and flux of HRP. As basal resistance slightly
differed in independent wells, the data are presented relative (%
TEER) to baseline (prior to heat exposure=1). The permeability for
HRP into the basolateral chambers, which was determined by the
calculated flux, was expressed as a percentage of added HRP
marker.
LPS, heat stress and LPS combined with heat stress
resulted in a reduction of TEER in time-dependent manner, and a
significant increase in paracellular permeability of HRP flux in
IEC-6 cells (Fig. 3B). The
significant reduction in TEER was accompanied by an increase in
paracellular permeability of HRP flux in the LPS combined with heat
stress group, compared with the LPS and HS groups at 6 h after
treatment (Fig. 3C). These results
indicated that LPS combined with heat stress significantly weakened
the intestinal epithelial barrier function, associated with the
reduction in TEER and the increase in HRP permeability. In
addition, IEC-6 and Caco-2 cells demonstrated the same trend.
Influence of heat stress and LPS on
MAPK activation
To determine whether p38, ERK and JNK
phosphorylation is required for apoptosis, IEC-6 cells were
stimulated with LPS combined with heat stress. As presented in
Fig. 4, heat stress alone or LPS
combined with heat stress induced activation of p38, ERK and JNK.
The phosphorylation levels of p38, ERK and JNK in the LPS combined
with heat stress groups were significantly increased compared with
the heat stress only groups. However, LPS only slightly increased
p38, ERK and JNK phosphorylation levels compared with the control
groups. A previous study indicated that LPS induces MAPK
phosphorylation at an early time point, which then rapidly
decreases to baseline levels, which coincides with ROS generation
after a 6-h recovery period from LPS, and induced ROS accumulation
was cleared (15). This may
explain the low activation level of MAPKs induced by LPS observed
in the present study.
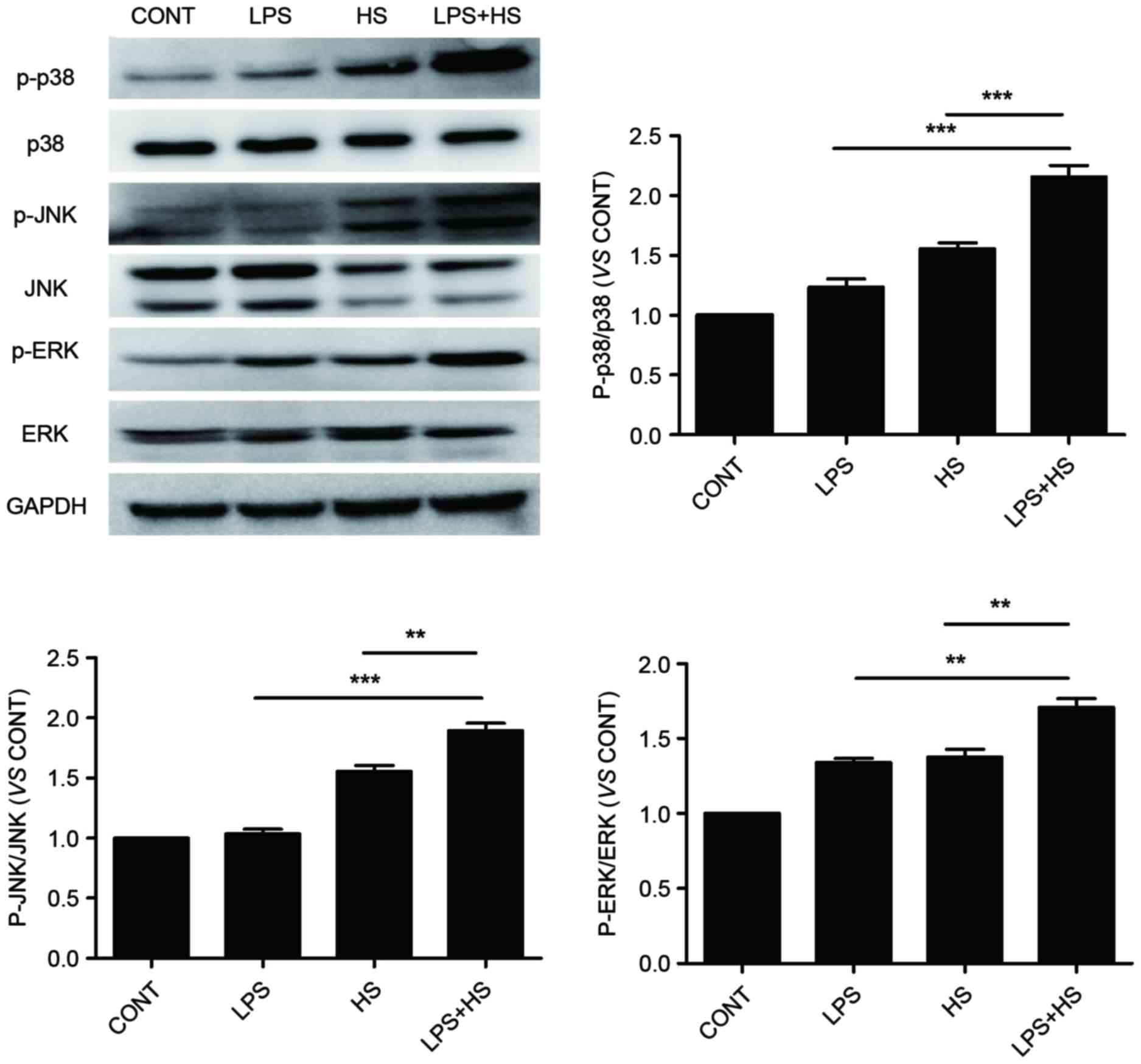 | Figure 4.Effects of HS, LPS, or a combination
of LPS and HS treatment on p38, ERK and JNK phosphorylation in
IEC-6 cells. IEC-6 cells were treated with LPS (1 µg/ml), HS (42°C
for 60 min) or a combination of LPS and HS. Protein expression
levels of p-p38, p38, p-JNK and JNK were detected by western
blotting. GAPDH served as an internal control. Data are presented
as the mean ± standard deviation of three independent experiments.
**P<0.01 and ***P<0.001. LPS, lipopolysaccharide; HS, heat
stress; JNK, c-Jun N-terminal kinase; ERK, extracellular
signal-regulated kinase; p, phosphorylated; CONT, control. |
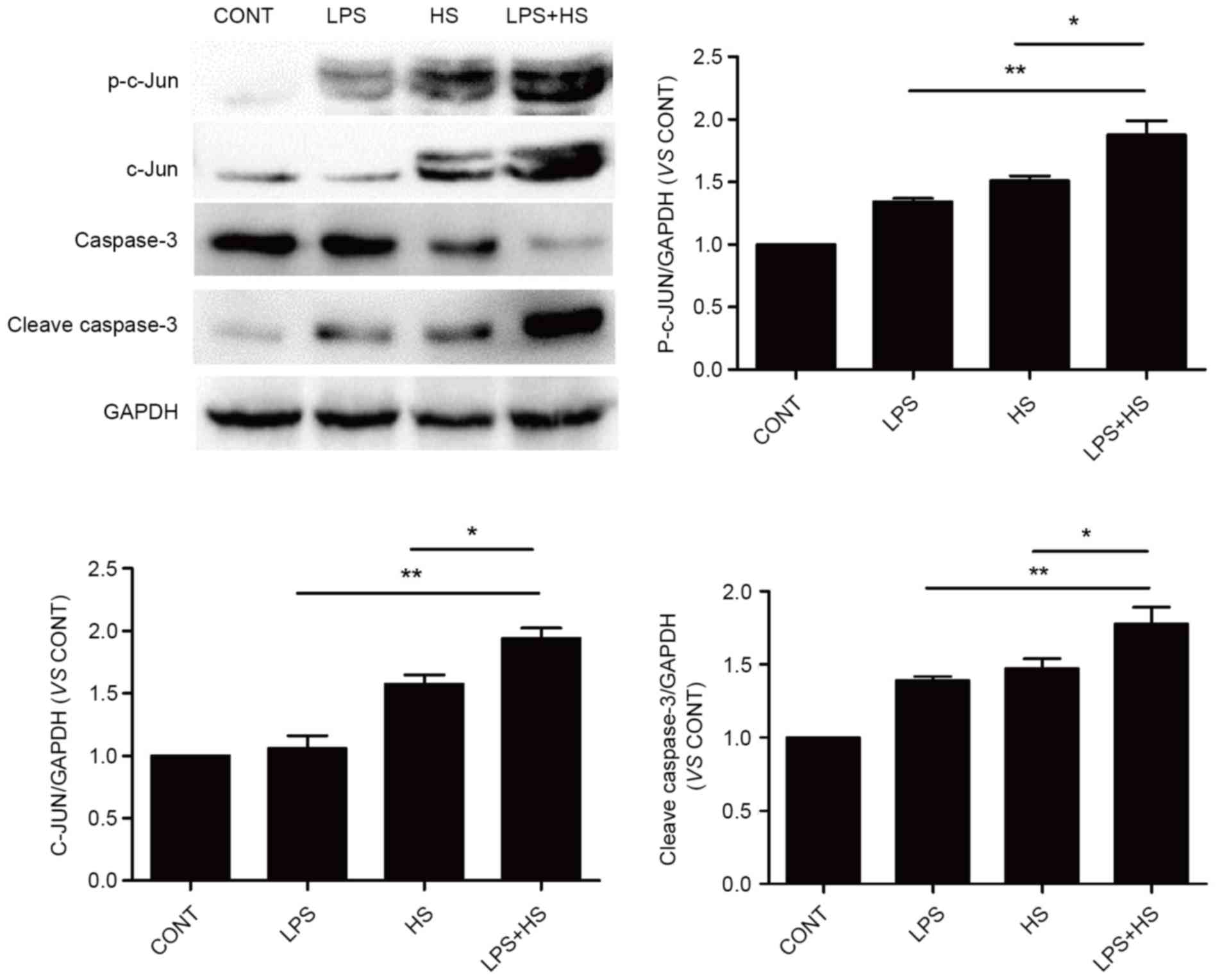
Influence of heat stress and LPS on
c-Jun and caspase-3 activation
To examine the effects of heat stress and LPS on
c-Jun activation and expression, IEC-6 cells were exposed to
simultaneous treatment with a combination of LPS (1 µg/ml) and heat
stress (42°C) for 60 min, or LPS and heat stress only, the cells
were returned to a temperature of 37°C for 6 h. As presented in
Fig. 5, heat stress and LPS
combined with heat stress caused an increase in the phosphorylation
and expression levels of c-Jun. The phosphorylation and expression
levels of c-Jun in the LPS combined with heat stress groups was
significantly increased compared with the heat stress groups. LPS
only slightly increased c-Jun phosphorylation, without affecting
its expression. Cleaved caspase-3 expression levels, which
typically indicates apoptosis, were detected in the LPS, heat
stress and LPS combined with heat stress groups. Cleaved caspase-3
expression in LPS combined with HS groups was significantly
increased compared with the heat stress and LPS groups.
Contribution of ROS, MAPKs and c-Jun
to LPS combined with heat stress-induced cell apoptosis and
intestinal epithelial barrier disruption in IEC-6 cells
To examine whether accumulated ROS levels, or MAPK
and c-Jun activation are involved in LPS and heat stress-induced
cell apoptosis and intestinal epithelial barrier disruption, IEC-6
cells were stimulated with LPS combined with heat stress in the
presence or absence of inhibitors for ROS, MAPKs or c-Jun. As
presented in Fig. 6A, treatment
with BHA significantly decreased early apoptosis rates.
Furthermore, while IEC-6 cells pretreated with PD98059, a specific
inhibitor of ERK, did not exhibit an increase in cell survival,
cells that were pretreated with specific inhibitors of JNK, p38 and
c-Jun (SP600125, SB203580 and c-Jun peptide, respectively)
exhibited a significant increase in cell survival (Fig. 6B). Epithelial barrier integrity and
paracellular permeability were determined by the measurement of
TEER and flux of HRP, respectively. The results of intestinal
epithelial barrier integrity and permeability were consistent with
apoptosis, as presented in Fig. 6C and
D.
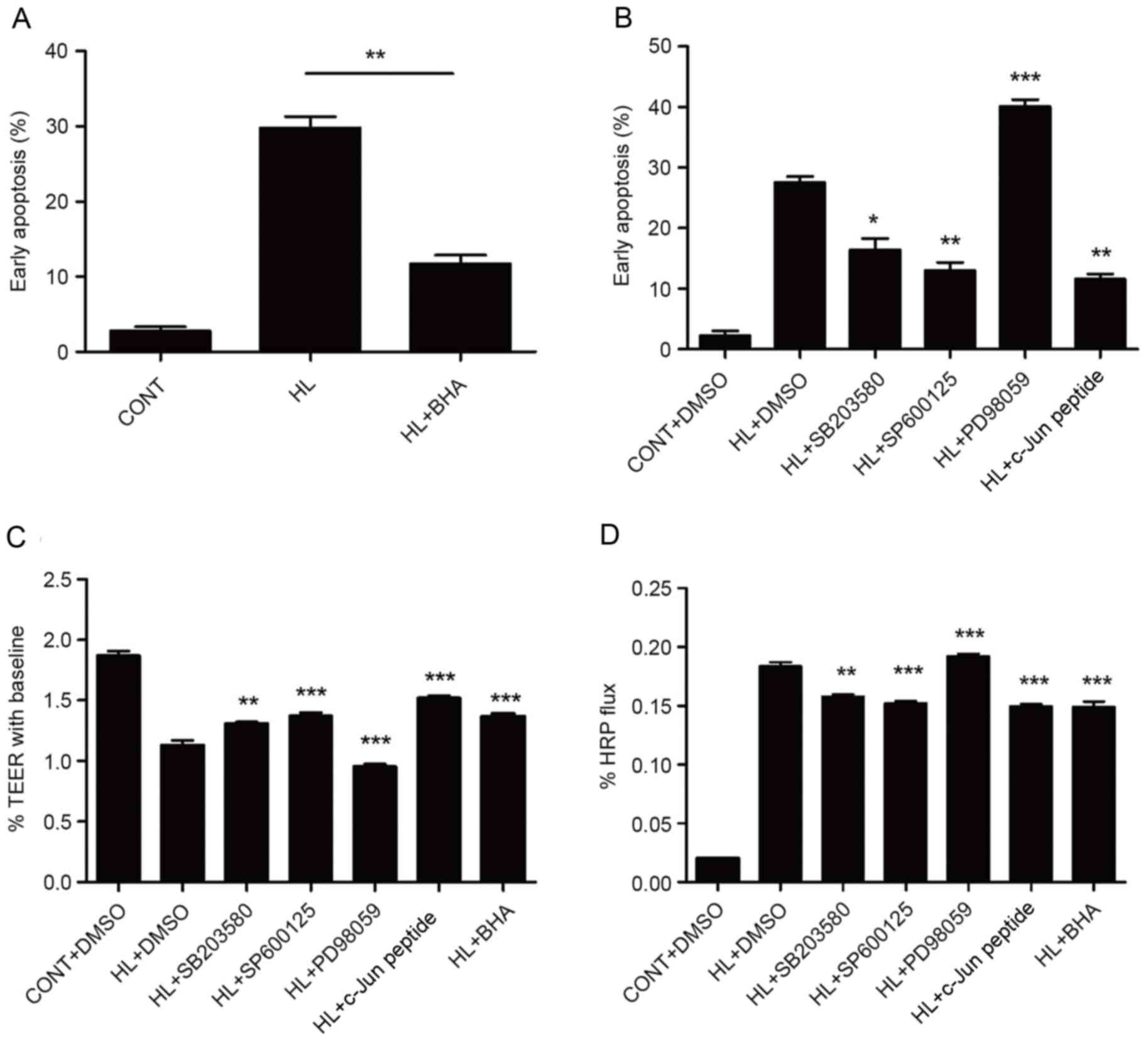 | Figure 6.ROS, MAPKs and c-Jun are involved in
LPS combined with heat stress-induced IEC-6 cell apoptosis and
intestinal epithelial barrier disruption. Early apoptosis rates of
cells that were pretreated with (A) BHA and (B) DMSO, SB203580,
SP600125, PD98059 or c-Jun peptide, prior to HL treatment followed
by a recovery period for 6 h, as assessed by flow cytometry.
Intestinal epithelial barrier function analysis following treatment
with inhibitors, as detected by (C) TEER and (D) HRP permeability.
Data are presented as the mean ± standard deviation of three
independent experiments. **P<0.01 in A; *P<0.05, **P<0.01
and ***P<0.001 vs. HL + DMSO group. CONT, control; LPS,
lipopolysaccharide; HL, combined treatment with LPS and heat
stress; DMSO, dimethyl sulfoxide; BHA, butylated hydroxyanisole;
TEER, transepithelial electrical resistance; HRP, horseradish
peroxidase. |
ROS mediates activation of MAPKs and
c-Jun
As presented in Fig.
7A, LPS combined with heat stress rapidly induced MAPK
phosphorylation, which was maintained at high levels for >60
min. When pretreated with BHA, the phosphorylation level of p38 and
ERK were inhibited slightly compared with LPS combined with heat
stress without BHA pretreatment. However, the phosphorylation of
JNK was not inhibited. To examine the effects of LPS combined with
heat stress on c-Jun activation and expression, cells were exposed
to heat stress and LPS for 0, 15, 30 or 60 min. LPS combined with
heat stress led to rapid induction of c-Jun phosphorylation on
serine 63 and subsequent accumulation of c-Jun protein in
time-dependent manner. When pretreated with BHA, c-Jun
phosphorylation and expression was inhibited within 60 min
(Fig. 7B). These results
demonstrated that p38, ERK and c-Jun activation are downstream
events of ROS accumulation.
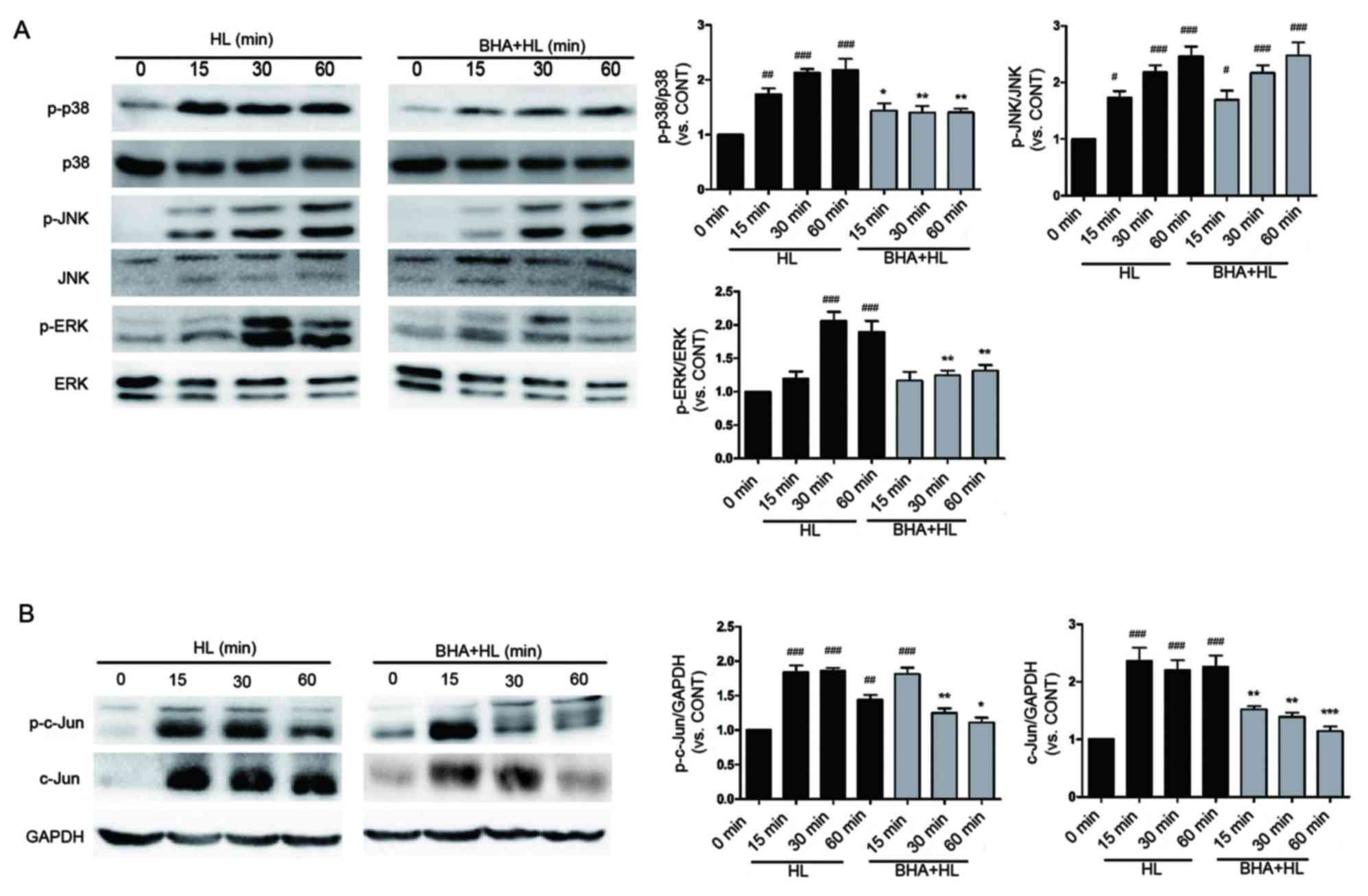 | Figure 7.BHA inhibits p38, ERK and c-Jun
activation following LPS combined with heat stress treatment in
IEC-6 cells. Representative western blot images of protein
expression levels of (A) p-p38, p-JNK and p-ERK, and (B) p-c-Jun
and c-Jun in IEC-6 cells following pretreatment with BHA prior to
HL treatment. Total p38, total JNK, total ERK and GAPDH served as
internal controls. Each band is representative of three
experiments. *P<0.05, **P<0.01 and ***P<0.001 vs. HL group
at same time point, #P<0.05, ##P<0.01
and ###P<0.001 vs. 0 min group. LPS,
lipopolysaccharide; HL, combined treatment with LPS and heat
stress; BHA, butylated hydroxyanisole; JNK, c-Jun N-terminal
kinase; ERK, extracellular signal-regulated kinase; p,
phosphorylated; CONT, 0 min control group. |
Discussion
It has previously been reported that endotoxin
levels in the blood increase in heatstroke patients at a mean
rectal temperature of 42.1°C (19,20).
Intestinal permeability to endotoxin or LPS from the gut entering
the circulation increases in heat-stressed animals (4,21).
On the contrary, anti-LPS antibodies protect against the transition
from heat stress to heatstroke (1). This suggests that LPS and heat stress
may serve an important role in the pathogenesis of heatstroke. The
present study mimicked the micro-environment of intestinal
epithelial cells under severe heat stress conditions to investigate
cell apoptosis and its potential underlying mechanisms.
In the present study, LPS and heat stress induced
production of ROS, mitochondrial membrane potential disruption and
cell apoptosis which eventually led to increased intestinal
permeability and reduced epithelial resistance in IEC-6 cells.
Furthermore, ROS production contributed to activation of p38, ERK
and c-Jun. In previous studies, intracellular ROS production was
increased by heat stress in endothelial cells (6), and IEC-6 cells by LPS in macrophages
(5). Previous studies have
suggested that environmental heat stress may stimulate production
of ROS, which contributes to intestinal barrier dysfunction and
cell apoptosis (22). As
predicted, heat stress or LPS stimulation induced early apoptosis
in IEC-6 cells, a synergistic effect produced when heat stress and
LPS stimulation were combined. A similar profile was obtained when
the corresponding cell lysates were analyzed for cleaved caspase-3
and activation of MAPKs and c-Jun. These results indicated that the
heat stroke environment may induce multiple types of cell damage
mediated by different molecules and signaling pathways.
Studies have demonstrated that the level of ROS is
associated with cell apoptosis. The present study revealed that
cell apoptosis may be inhibited by using the ROS scavenger, BHA,
which is consistent with a previous study (23). Cell apoptosis was assessed using
the inhibitors of JNK, p38, ERK and c-Jun by flow cytometry.
Activation of JNK, p38 and c-Jun serve pro-apoptotic roles, whereas
ERK is resistant to apoptosis. It is hypothesized that ERK is
important for cell survival, while JNK and p38 have been
characterized as stress-responsive factors, thus serving important
roles in apoptosis (12–16). In addition, the phosphorylation of
c-Jun is required for apoptosis following survival signal
withdrawal (24–27). These conclusions were obtained in
the present study. Therefore, the combination of LPS and heat
stress may induce both pro- and anti-apoptotic signaling pathways
via a ROS-MAPK/c-Jun signaling pathway in IEC-6 cells.
Previous studies have demonstrated that ROS mediates
MAPK and c-Jun activation in various cellular stress conditions and
cell types (4,26,28).
To investigate the molecular mechanism underlying MAPK and c-Jun
activation, cells were pretreated with an antioxidant. The results
indicated that the phosphorylation levels of p38, ERK and c-Jun are
suppressed. However, JNK phosphorylation levels did not alter
significantly. These results demonstrated that p38, ERK and c-Jun
activation is a downstream event of ROS accumulation.
In conclusion, the results reported in the present
study suggested that combined LPS and heat stress contributed to
IEC-6 cell apoptosis and intestinal epithelial barrier disruption,
and that more than one single mechanism was involved. The present
study demonstrated a critical role of ROS modulating LPS combined
with heat stress-induced MAPK and c-Jun activation in IEC-6 cells.
Elucidation of the mechanism by which ROS regulates activation of
MAPKs and c-Jun may facilitate understanding of pathological
conditions involving ROS, such as heat stroke.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant no. 81471839) and the
project team of the Natural Science Foundation of Guangdong
Province (grant no. s2013030013217).
References
|
1
|
Wang X, Yuan B, Dong W, Yang B, Yang Y,
Lin X and Gong G: Humid heat exposure induced oxidative stress and
apoptosis in cardiomyocytes through the angiotensin II signaling
pathway. Heart Vessels. 30:396–405. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gathiram P, Wells MT, Brock-Utne JG and
Gaffin SL: Antilipopolysaccharide improves survival in primates
subjected to heat stroke. Circ Shock. 23:157–164. 1987.PubMed/NCBI
|
|
3
|
Yang PC, He SH and Zheng PY: Investigation
into the signal transduction pathway via which heat stress impairs
intestinal epithelial barrier function. J Gastroenterol Hepatol.
22:1823–1831. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gathiram P, Wells MT, Raidoo D, Brock-Utne
JG and Gaffin SL: Portal and systemic plasma lipopolysaccharide
concentrations in heat-stressed primates. Circ Shock. 25:223–230.
1988.PubMed/NCBI
|
|
5
|
Xiao G, Tang L, Yuan F, Zhu W, Zhang S,
Liu Z, Geng Y, Qiu X, Zhang Y and Su L: Eicosapentaenoic acid
enhances heat stress-impaired intestinal epithelial barrier
function in Caco-2 cells. PLoS One. 8:e735712013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gu ZT, Wang H, Li L, Liu YS, Deng XB, Huo
SF, Yuan FF, Liu ZF, Tong HS and Su L: Heat stress induces
apoptosis through transcription-independent p53-mediated
mitochondrial pathways in human umbilical vein endothelial cell.
Sci Rep. 4:44692014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yokochi T, Morikawa A, Kato Y, Sugiyama T
and Koide N: Apoptotic cell death in response to LPS. Prog Clin
Biol Res. 397:235–242. 1998.PubMed/NCBI
|
|
8
|
Indo HP, Yen HC, Nakanishi I, Matsumoto K,
Tamura M, Nagano Y, Matsui H, Gusev O, Cornette R, Okuda T, et al:
A mitochondrial superoxide theory for oxidative stress diseases and
aging. J Clin Biochem Nutr. 56:1–7. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fiers W, Beyaert R, Declercq W and
Vandenabeele P: More than one way to die: Apoptosis, necrosis and
reactive oxygen damage. Oncogene. 18:7719–7730. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lee JA, Song HY, Ju SM, Lee SJ, Kwon HJ,
Eum WS, Jang SH, Choi SY and Park JS: Differential regulation of
inducible nitric oxide synthase and cyclooxygenase-2 expression by
superoxide dismutase in lipopolysaccharide stimulated RAW 264.7
cells. Exp Mol Med. 41:629–637. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Skibba JL, Powers RH, Stadnicka A,
Cullinane DW, Almagro UA and Kalbfleisch JH: Oxidative stress as a
precursor to the irreversible hepatocellular injury caused by
hyperthermia. Int J Hyperthermia. 7:749–761. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Burdon RH, Gill VM and Rice-Evans C:
Oxidative stress and heat shock protein induction in human cells.
Free Radic Res Commun. 3:129–139. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kim EK and Choi EJ: Compromised MAPK
signaling in human diseases: An update. Arch Toxicol. 89:867–882.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lee SI, Min KS, Bae WJ, Lee YM, Lee SY,
Lee ES and Kim EC: Role of SIRT1 in heat stress- and
lipopolysaccharide-induced immune and defense gene expression in
human dental pulp cells. J Endod. 37:1525–1530. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sakon S, Xue X, Takekawa M, Sasazuki T,
Okazaki T, Kojima Y, Piao JH, Yagita H, Okumura K, Doi T and Nakano
H: NF-kappaB inhibits TNF-induced accumulation of ROS that mediate
prolonged MAPK activation and necrotic cell death. EMBO J.
22:3898–3909. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Guo X, Chen S, Zhang Z, Dobrovolsky VN,
Dial SL, Guo L and Mei N: Reactive oxygen species and c-Jun
N-terminal kinases contribute to TEMPO-induced apoptosis in L5178Y
cells. Chem Biol Interact. 235:27–36. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bai L, Mao R, Wang J, Ding L, Jiang S, Gao
C, Kang H, Chen X, Sun X and Xu J: ERK1/2 promoted proliferation
and inhibited apoptosis of human cervical cancer cells and
regulated the expression of c-Fos and c-Jun proteins. Med Oncol.
32:572015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Su SH, Wu YF, Lin Q, Yu F and Hai J:
Cannabinoid receptor agonist WIN55,212-2 and fatty acid amide
hydrolase inhibitor URB597 suppress chronic cerebral
hypoperfusion-induced neuronal apoptosis by inhibiting c-Jun
N-terminal kinase signaling. Neuroscience. 301:563–575. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sambuy Y, De Angelis I, Ranaldi G, Scarino
ML, Stammati A and Zucco F: The Caco-2 cell line as a model of the
intestinal barrier: Influence of cell and culture-related factors
on Caco-2 cell functional characteristics. Cell Biol Toxicol.
21:1–26. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Dörfel MJ and Huber O: Modulation of tight
junction structure and function by kinases and phosphatases
targeting occludin. J Biomed Biotechnol. 2012:8073562012.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kimura Y, Shiozaki H, Hirao M, Maeno Y,
Doki Y, Inoue M, Monden T, Ando-Akatsuka Y, Furuse M, Tsukita S and
Monden M: Expression of occludin, tight-junction-associated
protein, in human digestive tract. Am J Pathol. 151:45–54.
1997.PubMed/NCBI
|
|
22
|
Shapiro Y, Alkan M, Epstein Y, Newman F
and Magazanik A: Increase in rat intestinal permeability to
endotoxin during hyperthermia. Eur J Appl Physiol Occup Physiol.
55:410–412. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hall DM, Buettner GR, Oberley LW, Xu L,
Matthes RD and Gisolfi CV: Mechanisms of circulatory and intestinal
barrier dysfunction during whole body hyperthermia. Am J Physiol
Heart Circ Physiol. 280:H509–H521. 2001.PubMed/NCBI
|
|
24
|
Liu L, Fu J, Li T, Cui R, Ling J, Yu X, Ji
H and Zhang Y: NG, a novel PABA/NO-based oleanolic acid derivative,
induces human hepatoma cell apoptosis via a ROS/MAPK-dependent
mitochondrial pathway. Eur J Pharmacol. 691:61–68. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhu J, Zhang J, Huang H, Li J, Yu Y, Jin
H, Li Y, Deng X, Gao J, Zhao Q and Huang C: Crucial Role of c-Jun
phosphorylation at Ser63/73 mediated by PHLPP protein degradation
in the cheliensisin a inhibition of cell transformation. Cancer
Prev Res (Phila). 7:1270–1281. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Herdman ML, Marcelo A, Huang Y, Niles RM,
Dhar S and Kiningham KK: Thimerosal induces apoptosis in a
neuroblastoma model via the cJun N-terminal kinase pathway. Toxicol
Sci. 92:246–253. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Watson A, Eilers A, Lallemand D, Kyriakis
J, Rubin LL and Ham J: Phosphorylation of c-Jun is necessary for
apoptosis induced by survival signal withdrawal in cerebellar
granule neurons. J Neurosci. 18:751–762. 1998.PubMed/NCBI
|
|
28
|
Qin Z, Robichaud P, He T, Fisher GJ,
Voorhees JJ and Quan T: Oxidant exposure induces cysteine-rich
protein 61 (CCN1) via c-Jun/AP-1 to reduce collagen expression in
human dermal fibroblasts. PLoS One. 9:e1154022014. View Article : Google Scholar : PubMed/NCBI
|