Introduction
Bilirubin is the end product of heme catabolism,
which is produced primarily from the breakdown of erythrocyte
hemoglobin in the reticuloendothelial system. There are several
major steps in the hepatic clearance of bilirubin, including
hepatocytes ingesting and storing unconjugated bilirubin, the
conjugation of bilirubin to bilirubin glucuronides, the excretion
of conjugated bilirubin into bile, and hepatocytes resorbing the
conjugated bilirubin (1).
Hyperbilirubinemia is not only induced by increased bilirubin
synthesis, but it can also be caused by decreased bilirubin
clearance (2). There are several
inherited disorders, which can contribute to hyperbilirubinemia
(3), including Dubin-Johnson
syndrome (DJS), Crigler-Najjar syndrome, Gilbert syndrome and
Lucey-Driscoll syndrome. As a rare autosomal recessive disorder,
DJS is characterized by predominantly conjugated hyperbilirubinemia
without progression to end-stage liver disease (4–6).
To date, there are several genes, which have been
identified to contribute to hyperbilirubinemia, including
UGT1A1, SLCO1B1/OATP1B1, SLCO1B3/OATP1B3,
MRP2/ABCC2 and ABCG2/BCRP (1,7,8). At
present, at least 24 ABCC2/MRP2 point mutations have been
reported in DJS, a number of which are predicted to result in
truncated proteins. In the present study, the possible causative
gene was investigated in a patient with hyperbilirubinemia. The
results revealed two novel mutations (c.1303A>C/p.T435P and
c.1326G>A/p.W442X) in exon 10 of ABCC2. To the best of
our knowledge, these mutations have not been reported in previous
studies, neither have they been presented in the single nucleotide
polymorphism (dbSNP) databases (https://www.ncbi.nlm.nih.gov/projects/SNP/) and Exome
Variant Server databases (http://evs.gs.washington.edu/EVS/).
Materials and methods
Patients
In the present study, a family from Hunan province
comprising six members across three generations, which were
admitted to the Second Xiangya Hospital in October, 2015 (Changsha,
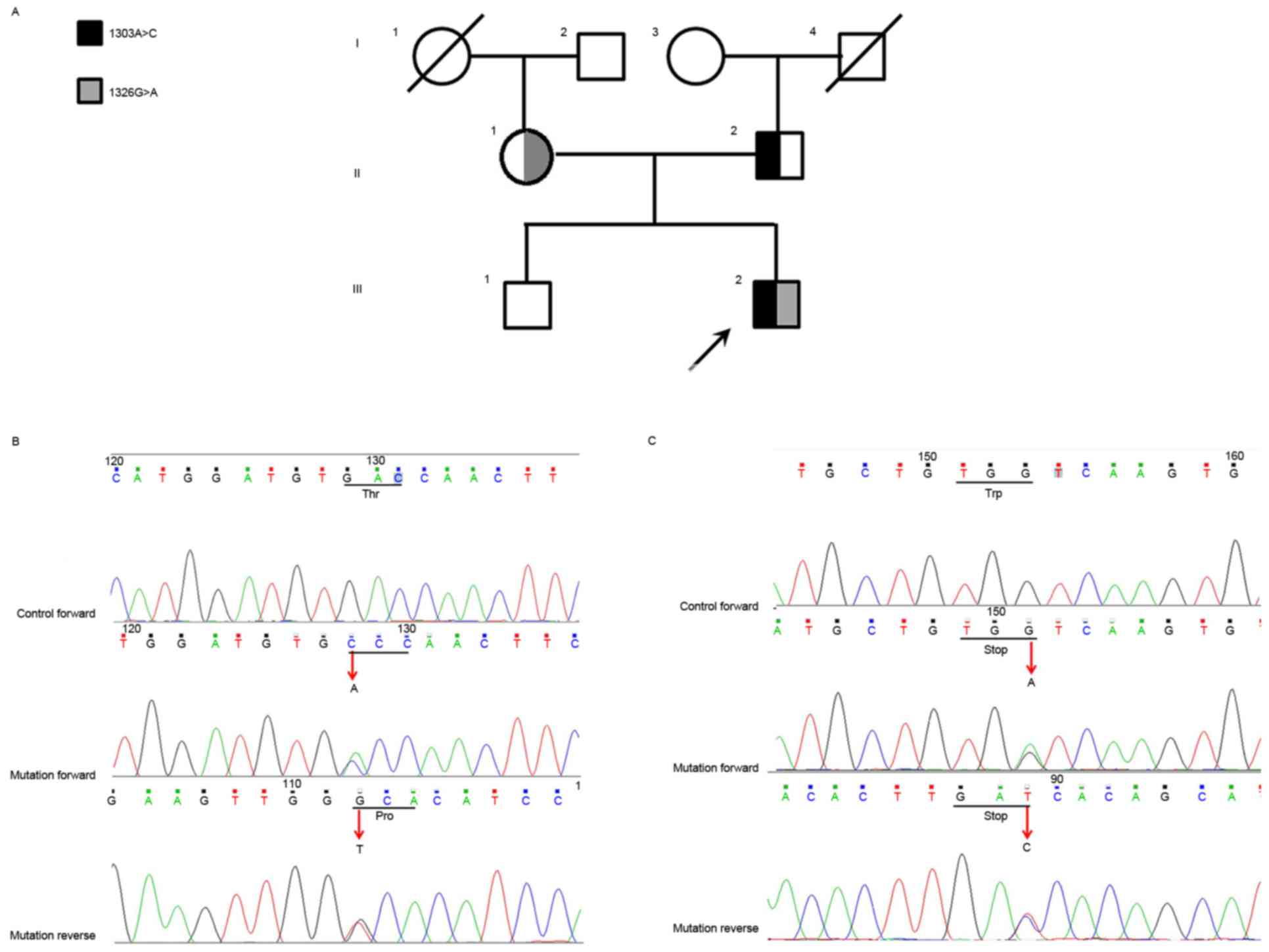
China), was included (Fig. 1A;
Table I). The proband was
diagnosed with hyperbilirubinemia (III2) with a total bilirubin
level of 30.6 µmol/l and a direct bilirubin level of 10.5 µmol/l.
No hyperbilirubinemia was present in the other family members.
Details of the family are listed in Table I. The present study was approved by
the Second Xiangya Hospital of Central South University (Changsha,
China). All subjects provided consent prior to commencement of the
study.
 | Table I.Summary of the family with
Dubin-Johnson syndrome investigated. |
Table I.
Summary of the family with
Dubin-Johnson syndrome investigated.
|
|
|
|
|
| Program
prediction |
|---|
|
|
|
|
|
|
|
|---|
| Family member | TBIL/DBIL
(µmol/l) | Age (years) | ABCC2 DNA | Protein | Polyphen-2 | SIFT | MutationTaster |
|---|
| III2 (proband) | 30.6/10.5 | 19 | 1303A>C | T435P | Possibly
damaging | Deleterious | Disease-causing |
|
|
|
| 1326G>A | W442X | Possibly
damaging | Deleterious | Disease-causing |
| I2 | 16.0/5.0 | 70 | – | – | – | – | – |
| I3 | 13.7/5.5 | 69 | – | – | – | – | – |
| II1 | 15.9/4.9 | 43 | 1326G>A | W442X | – | – | – |
| II2 | 14.9/4.9 | 44 | 1303A>C | T435P | – | – | – |
| III1 | 13.5/3.8 | 20 | – | – | – | – | – |
DNA extraction
Genomic DNA was extracted from the peripheral blood
of the proband and other family members using a DNeasy Blood &
Tissue kit (Qiagen, Inc., Valencia, CA, USA) according to the
manufacturer's protocol, on the QIAcube automated DNA extraction
robot (Qiagen, Inc.), as previously described (9).
Mutation sequencing
Through the use of polymerase chain reaction (PCR),
several genes were amplified, including UGT1A1 (Refseq:
NM_000463), ABCC2 (Refseq: NM_000392) and OATP1B1
(Refseq: NM_0,06446). PCR was performed using 25 µl reaction
volumes, containing 0.3 mM dNTPs, 1X PCR buffer (10 mM Tris-HCl pH
9.0, 50 mM KCl, 0.1% Triton X-100 and 0.01% w/v gelatin), 2.0 mM
MgCl2, 0.5 µM of each primer (forward and reverse), 1.5
U of Taq polymerase (Thermo Fisher Scientific, Inc., Waltham, MA,
USA), and 50 ng of genomic DNA. Thermocycling conditions were as
follows: Initial denaturation at 95°C for 5 min, followed by 35
cycles of amplification consisting of denaturation at 95°C for 30
sec, annealing at 55–61°C for 30 sec and extension at 72°C for 1
min. A final extension step was performed at 72°C for 7 min. The
sequences of the PCR products were obtained using the ABI 3100
genetic analyzer (Applied Biosystems; Thermo Fisher Scientific,
Inc.). Results were compared with normal control samples, as
defined in our previous study (9).
Bioinformatics sequence analysis and
mutation prediction
In several species, the multiple ABCC2 protein
sequences were aligned (version 3.6; http://www.ncbi.nlm.nih.gov). Polymorphism
Phenotyping-2 (Polyphen 2; http://genetics.bwh.harvard.edu/pph2/) (10), Sorting Intolerant From Tolerant
(SIFT; http://sift.bii.astar.edu.sg/)
(11) and MutationTaster
(www.mutationtaster.org) (12) were used to predict the effects of
these sequence variants on the function of the protein.
Results
The present study reported on a patient with
hyperbilirubinemia with a total bilirubin level of 30.6 µmol/l and
direct bilirubin of 10.5 µmol/l, whereas the reference standard
values are 5.1–17.1 and 0–6.0 µmol/l, respectively. The possibility
of the induction of hyperbilirubinemia by known genes was
investigated. Using Sanger sequencing, a missense mutation
(c.1303A>C/p.T435P) and a nonsense mutation
(c.1326G>A/p.W442X) in ABCC2 were identified and co-segregated
with the affected members. (Fig. 1B
and C). The allelic segregation analysis revealed that the
missense mutation was carried by the mother, whereas the nonsense
mutation was inherited from the father. These newly identified
missense mutations c.1303A>C and c.1326G>A were not found in
a cohort of 200 controls, as described in our previous study
(9). In addition, these two
mutations were not present in the dbSNP and Exome Variant Server
databases. In humans, macaques, cats, mice and zebrafish, the amino
acid sequences of ABCC2 were found to be aligned, which revealed
that the affected amino acids were evolutionarily conserved
(Fig. 2A and B). Three programs
were used for analyzing the protein functions of ABCC2; polyphen2,
SIFT and Mutation Taster, predicted that the two variants were
likely to be damaging, deleterious and disease-causing,
respectively. The consistent findings of the detrimental effects of
the variants by all these bioinformatics programs suggested that
these mutations are important in the function of ABCC2.
Discussion
The present study presented a case of
hyperbilirubinemia associated with a compound heterozygous mutation
(c.1303A>C/p.T435P and c.1326G>A/p.W442X) in exon 10 of
ABCC2. A previous study found that mutations in ABCC2
may cause DJS, and this syndrome was characterized by biphasic,
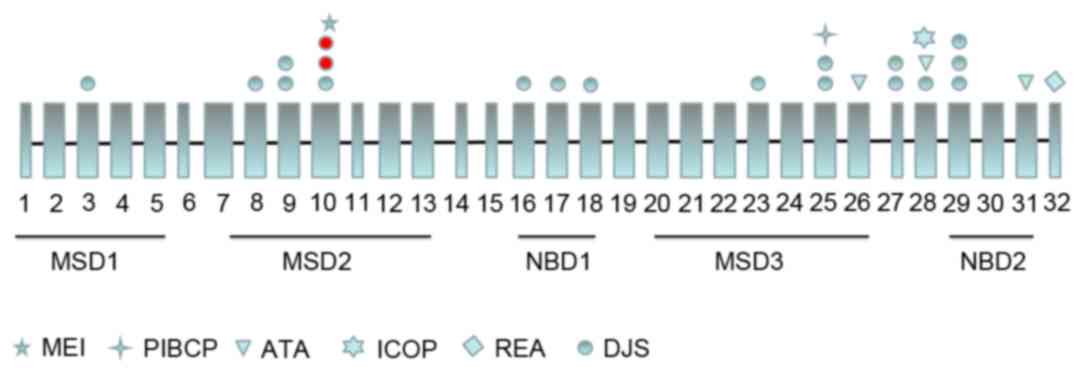
predominantly conjugated, hyperbilirubinemia. At present, ~17 point
mutations of ABCC2 have been reported in patients with DJS
(Fig. 3). The outcome of the
molecular genetic investigations performed in the present study was
consistent with and confirmed the clinical diagnosis of DJS.
Under normal conditions, hepatocytes take up
unconjugated bilirubin by transporters of the organic
anion-transporting polypeptide family, followed by conjugation with
glucuronic acid and ATP-dependent transport into bile. This efflux
across the canalicular membrane is mediated by ABCC2/MRP2, which
has a high affinity and efficiency for monoglucuronosyl and
bisglucuronosyl bilirubin into bile. Therefore, mutations in ABCC2
may lead to DJS (13,14).
In the present study, the missense and nonsense
mutations were located in a conserved membrane-spanning domain
(MSD), namely MSD2, of the ABCC2 protein (15). The nonsense mutation can also lead
to the absence of the complete nucleotide-binding domain (NBD)-1,
MSD3 and NBD2. Mutations in MSD2 may affect the subcellular
localization of ABCC2, and the truncated mutation may cause the
functional defect of ABCC2. These two point mutations were present
in patients, and this compound heterozygous mutation was associated
with DJS recessive hereditary mode.
Among the compound heterozygous mutations in DJS, 13
cases have been reported, including that identified in the present
study (5,16). At present, a total of 24
MRP2/ABCC2 point mutations have been reported, 17 of which
are associated with DJS (17–19)
(Fig. 3). In the last 25 years,
different types of viral vectors have been used in clinical trials
for the treatment of a variety of monogenetic disorders. It has
been suggested that this technique may be used to treat hereditary
hyperbilirubinemia (20,21), however, further investigation and
improvements are required (22).
In conclusion, the present study identified an
ABCC2 compound heterozygous mutation (c.1303A>C/p.T435P
and c.1326G>A/p.W442X) in a patient with DJS. To the best of our
knowledge, this may be the first report of these two mutations
worldwide. The results of the present study offer further support
for the significant involvement of ABCC2 in DJS. The results
also expand on the spectrum of ABCC2 mutations, and
contribute to the genetic diagnosis and counseling of families with
DJS.
Acknowledgements
The authors would like to thank the State Key
Laboratory of Medical Genetics of China for their technical
assistance. This study was supported by the National Natural
Science Foundation of China (grant nos. 81370394 and 81400831).
References
|
1
|
Keppler D: The roles of MRP2, MRP3,
OATP1B1, and OATP1B3 in conjugated hyperbilirubinemia. Drug Metab
Dispos. 42:561–565. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Memon N, Weinberger BI, Hegyi T and
Aleksunes LM: Inherited disorders of bilirubin clearance. Pediatr
Res. 79:378–386. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Radlovic N: Hereditary
hyperbilirubinemias. Srp Arh Celok Lek. 142:257–260. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sticova E, Elleder M, Hulkova H, Luksan O,
Sauer M, Wunschova-Moudra I, Novotny J and Jirsa M: Dubin-Johnson
syndrome coinciding with colon cancer and atherosclerosis. World J
Gastroenterol. 19:946–950. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Uchiumi T, Tanamachi H, Kuchiwaki K,
Kajita M, Matsumoto S, Yagi M, Kanki T and Kang D: Mutation and
functional analysis of ABCC2/multidrug resistance protein 2 in a
Japanese patient with Dubin-Johnson syndrome. Hepatol Res.
43:569–575. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li P, Wang Y, Zhang J, Geng M and Li Z:
Dubin-Johnson syndrome with multiple liver cavernous hemangiomas:
Report of a familial case. Int J Clin Exp Pathol. 6:2636–2639.
2013.PubMed/NCBI
|
|
7
|
Chen ZS and Tiwari AK: Multidrug
resistance proteins (MRPs/ABCCs) in cancer chemotherapy and genetic
diseases. FEBS J. 278:3226–3245. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sticova E and Jirsa M: New insights in
bilirubin metabolism and their clinical implications. World J
Gastroenterol. 19:6398–6407. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Xiang R, Fan LL, Huang H, Cao BB, Li XP,
Peng DQ and Xia K: A novel mutation of GATA4 (K319E) is responsible
for familial atrial septal defect and pulmonary valve stenosis.
Gene. 534:320–323. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sunyaev S, Ramensky V and Bork P: Towards
a structural basis of human non-synonymous single nucleotide
polymorphisms. Trends Genet. 16:198–200. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ng PC and Henikoff S: SIFT: Predicting
amino acid changes that affect protein function. Nucleic Acids Res.
31:3812–3814. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Schwarz JM, Rödelsperger C, Schuelke M and
Seelow D: MutationTaster evaluates disease-causing potential of
sequence alterations. Nat Methods. 7:575–576. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fujiwara R, Maruo Y, Chen S and Tukey RH:
Role of extrahepatic UDP-glucuronosyltransferase 1A1: Advances in
understanding breast milk-induced neonatal hyperbilirubinemia.
Toxicol Appl Pharmacol. 289:124–132. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Christensen RD and Yaish HM: Hemolytic
disorders causing severe neonatal hyperbilirubinemia. Clin
Perinatol. 42:515–527. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Emi Y, Yasuda Y and Sakaguchi M: A
cis-acting five-amino-acid motif controls targeting of ABCC2 to the
apical plasma membrane domain. J Cell Sci. 125:3133–3143. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Okada H, Kusaka T, Fuke N, Kunikata J,
Kondo S, Iwase T, Nan W, Hirota T, Ieiri I and Itoh S: Neonatal
Dubin-Johnson syndrome: Novel compound heterozygous mutation in the
ABCC2 gene. Pediatr Int. 56:e62–e64. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Devgun MS, El-Nujumi AM, O'Dowd GJ, Barbu
V and Poupon R: Novel mutations in the Dubin-Johnson syndrome gene
ABCC2/MRP2 and associated biochemical changes. Ann Clin Biochem.
49:609–612. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lee JH, Chen HL, Chen HL, Ni YH, Hsu HY
and Chang MH: Neonatal Dubin-Johnson syndrome: Long-term follow-up
and MRP2 mutations study. Pediatr Res. 59:584–589. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kruer MC, Jepperson T, Dutta S, Steiner
RD, Cottenie E, Sanford L, Merkens M, Russman BS, Blasco PA, Fan G,
et al: Mutations in gamma adducin are associated with inherited
cerebral palsy. Ann Neurol. 74:805–814. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kaufmann KB, Büning H, Galy A, Schambach A
and Grez M: Gene therapy on the move. EMBO Mol Med. 5:1642–1661.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
van Dijk R, Beuers U and Bosma PJ: Gene
replacement therapy for genetic hepatocellular jaundice. Clin Rev
Allergy Immunol. 48:243–253. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Stapelbroek JM, van Erpecum KJ, Klomp LW
and Houwen RH: Liver disease associated with canalicular transport
defects: Current and future therapies. J Hepatol. 52:258–271. 2010.
View Article : Google Scholar : PubMed/NCBI
|