Introduction
Pre-diabetes, referring to impaired fasting glucose
and/or impaired glucose tolerance, manifest both core
pathophysiological features of type 2 diabetes mellitus (T2DM),
that is, insulin resistance and β-cell dysfunction (1,2).
Several key insulin target tissues including muscle, fat and liver
become resistant to the actions of insulin in the progression to
pre-diabetes. Accumulated data have identified that pancreatic
β-cells are also targets of insulin action (3). Indeed, the insulin signal
transduction pathway in β-cells serves a crucial role in regulating
the synthesis, secretion of insulin and maintaining the growth,
proliferation and survival of cells (4). Furthermore, tissue-specific knockout
of the insulin receptor (IR), insulin receptor substrate (IRS)-1 or
IRS-2 in β-cells both display defects in insulin secretory
responses to glucose stimuli (5–8).
Considering the significant role of insulin on β-cells, insulin
resistance is also thought to affect the functioning of β-cells in
addition to that of the muscle, fat and liver tissues. Therefore,
defects in insulin signaling in β-cells appear to be implicated in
the pathogenesis of pre-diabetes. Phosphoinositide 3-kinase (PI3K)
is proposed to be a key signaling molecule in insulin signaling.
Insulin binds to the IR of β-cells, triggering receptor
auto-phosphorylation, receptor tyrosine kinase activation, IRS-1
phosphorylation and PI3K activation (9). The insulin-PI3 K signaling pathway
has been demonstrated to be involved in regulating insulin
secretion and insulin gene transcription (10,11).
1,2-dicarbonyl compounds, such as methylglyoxal
(MGO) and 3-deoxyglucosone (3DG), which are easily formed from
carbohydrates in caramelization and Maillard reactions in food,
have been reported to increase the risk of T2DM and its
complications (12,13). Indeed, MGO was also reported to
induce insulin resistance in cultured L6 muscle cells (14) and 3T3-L1 adipocytes (15) through impairing insulin signaling
pathways. Incubation of cultured INS-1 β-cells with MGO also
impairs insulin signaling and insulin action on glucose-induced
insulin secretion (16). Despite
advances in understanding of the pathogenesis linking MGO with
T2DM, MGO has been reported to be less toxic than 3DG in embryonic
malformation (17). Furthermore,
dietary intake of 3DG was estimated to be range from 20 to 160
mg/day, which is higher than that of MGO in the range of 5–20
mg/day, thus 3DG was proved to be the predominant 1,2-dicarbonyl
compound (18). Importantly,
although 3DG has been reported to exert deleterious effects on many
aspects similar to MGO (19–22),
the previous findings surprisingly presented that MGO and 3DG
modulated collagen expression differently (23). Such observations are the important
cause, which suggests that the role of 3DG on the development of
T2DM deserves to be further investigated. Previously, exogenous 3DG
was administrated to normal mice for two weeks and led to a
significant increase in blood glucose concentration at 30 min
during an oral glucose tolerance test (OGTT) (24). These findings strongly suggest that
exogenous 3DG may serve an important role in development of
pre-diabetes.
It has reported that the level of plasma 3DG was
elevated ~two-fold in patients with diabetes, compared with healthy
humans (25–27). Previous studies of the authors
indicated that there was an abnormal elevation of plasma 3DG in
non-diabetic seniors, and the increasing accumulation of plasma 3DG
may eventually leads to impaired glucose regulation (IGR) through
decreasing insulin sensitivity and reducing insulin secretion
(28). These findings suggest that
accumulation of endogenous 3DG may serve an important role in the
development of diabetes. This idea is further supported by the
previous finding that acute administrated 3DG to normal rats to
achieve the pathologically relevant plasma levels of 3DG that
induced glucose intolerance through impairing the insulin-PI3K
signaling pathway involving insulin resistance and impaired β-cell
function (29). In addition, the
authors indicated that 3DG induced insulin resistance in
hepatocellular carcinoma cells HepG2 through impairing the
insulin-PI3 K signaling pathway and insulin action on glucose
uptake and glycogen synthesis (30). These results are sufficient to
indicate that abnormal elevation of circulating 3DG may participate
in worsening of diabetic state and the development of diabetes,
associated with the impairment of insulin action on target tissues.
Importantly, β-cells maintain normal glucose tolerance by
increasing insulin output in the present of insulin resistance, and
only when β-cells are incapable of releasing sufficient insulin do
glucose concentrations rise (31).
This islet-centered view is also reflected by current diabetes
treatment options (32). Finally,
the dominant thought was that β-cell dysfunction was the main
pathogenesis of T2DM. Therefore, β-cell dysfunction is believed to
involve in the deleterious effects of exogenous 3DG on blood
glucose concentrations. In this case, the link between exogenous
3DG and β-cell dysfunction may be increased plasma levels of 3DG,
which remains to be clarified. The aim of the present work was to
investigate whether continuous oral administration of 3DG leads to
normal individuals developing IGR, resulting from β-cell
dysfunction induced by an increase in plasma 3DG levels.
In the present study, 3DG was administered by
gastric gavage to normal mice for two weeks to examine the plasma
3DG levels. The effects of intragastric administration of 3DG on
the fasting blood glucose and oral glucose tolerance were
investigated. In addition, the effect of 3DG on β-cell function in
mice was assessed by calculating homeostasis model assessment
(HOMA)-β and ∆Ins30–0/∆G30–0 and in cultured
pancreas islets and INS-1 cells by measuring glucose-stimulated
insulin secretion (GSIS). Furthermore, the authors investigated the
ability of 3DG to interfere with insulin-PI3K signaling in
glucose-induced INS-1 cells.
Materials and methods
Synthesis of 3DG
According to the method of Kato et al
(33), 3DG was synthesized from
glucose as previously described (30).
Determination of appropriate doses of
intragastric administration of 3DG
Previous reports have estimated an average dietary
3DG intake of about 50 mg/day, based on the 3DG content in commonly
consumed foods (18). In order to
achieve the equivocal effect of a potential 3DG intake of 50
mg/day, the authors calculated doses based on body surface area
(6.5 mg/kg for mice and 4.5 mg/kg for rats). Previously, it was
reported that intragastric administration of 5 mg/kg 3DG for two
weeks increased blood glucose level under oral glucose tolerance
tests in mice (24). Therefore,
the present study involved administration of 5, 20 and 50 mg/kg 3DG
by gastric gavage.
Animals
Male Kunming mice (6–8-weeks-old) and male
Sprague-Dawley rats (11-weeks-old) were purchased from Joinn
Laboratories Co., Ltd. (Suzhou, Jiangsu, China) and housed in a
temperature-controlled room (23°C) and 12-h light/dark cycle. All
of animal experimental procedures were conducted in compliance with
Guide for Care and Use of Laboratory Animals of the National
Institutes of Health (Bethesda, MD, USA). The study was approved by
the local ethic committee of Suzhou Hospital of Traditional Chinese
Medicine (Suzhou, Jiangsu, China). The mice and rats had free
access to a standard diet (Shuangshi Laboratory Animal Feed Science
Co., Ltd., Suzhou, Jiangsu, China) and water. The mice were
randomly distributed into four groups and each group consisted of
six mice. They were treated with water (control group), 5 mg/kg
3DG, 20 mg/kg 3DG and 50 mg/kg 3DG by gastric gavage at 9:00 am
every day for two weeks. Following two weeks, the mice were fasted
overnight and killed following completion of an OGTT. In a second
study, following overnight fasting, the rats were anaesthetized
with pentobarbital sodium (50 mg/kg) and given intravenous
injection of 3DG (5, 20 and 50 mg/kg). At 2 h, pancreas islet
tissues were isolated and subsequently cultured for the measurement
GSIS.
OGTT
The mice were fasted overnight, and then
intragastric administration of 2.5 g/kg glucose was performed
following measurement of the fasting blood glucose and fasting
insulin levels. The blood glucose was detected at 30, 60 and 120
min, and the insulin level was detected at 30 min following the
glucose load. The blood samples were obtained from caudal vein and
a glucose meter determined blood glucose. Blood samples were
obtained by enucleating the eyeballs and insulin concentration was
determined using an insulin radioimmunoassay kit (Beijing North
Institute of Biological Technology, Beijing, China). The blood
glucose concentration-time curves were plotted and the area under
curve (AUC) was calculated for each group. The β-cell function was
assessed by HOMA-β and ∆Ins30-0/∆G30-0. All indexes were calculated
using the following formulas: AUC mmol.h/l=1/2A+B+C+1/2D (A, B, C,
D indicated the blood glucose of 0, 30, 60 and 120 min
respectively, mM), HOMA-β=20xFINS/(FBG-3.5),
∆Ins30-0/∆G30-0=(Ins30-Ins0)/(G30-G0) (FBG, fasting blood glucose,
mM; FINS, fasting insulin, mIU/l).
INS-1 cell culture and GSIS study
INS-1 cells were kindly provided by School of
Biology and Basic Medical Sciences of Soochow University (Suzhou,
Jiangsu, China). The islets and INS-1 cells were cultured in
phenol-red free RPMI medium (Gibco; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) containing 11 mM glucose, 10% charcoal stripped
fetal bovine serum, 1 mM glutamine, 100 U/ml penicillin and 100
mg/ml streptomycin. For GSIS experiment, the cells were plated at a
density of 4×103 per well in a 96-well plate overnight and then
treated with and without different concentrations of 3DG (0, 80,
300 and 500 ng/ml) in RPMI1640 containing different glucose content
(5.6 or 25.5 mM). Following incubation for 12 h at 37°C in a
humidified incubator, the culture solution was collected and MTT (5
mg/ml in PBS) was added to each well and incubated for 4 h, then
the medium was totally removed. Finally, 0.1 ml buffered dimethyl
sulfoxide (Wuxi Zhanwang Chemical Reagent Co., Ltd., Wuxi, Jiangsu,
China) was added to each well. The absorbance was recorded on a
microplate reader (Spectra Max M2e, Molecular Devices, LLC,
Sunnyvale, CA, USA) at the wavelength of 490 nm. The insulin of the
culture solution was detected by radioimmunoassay.
GSIS study of pancreas islets
The rats were anaesthetized with pentobarbital
sodium (50 mg/kg) and pancreas islet tissues were isolated. These
tissues subsequently were chopped and digested by collagenase (1.5
mg/ml in RPMI1640) for 20 min at 37°C for the measurement GSIS.
Briefly, the digested pancreases were filtered and centrifuged at
3,000 × g at 4°C. The supernatant was discarded, and the pellet was
resuspended in the RPMI1640. The islet cells were plated at a
density of 4×103 per well in 96-well plates overnight and then
treated with different concentrations of 3DG (0, 80, 300 and 500
ng/ml) in RPMI1640 containing different glucose content (5.6 or
25.5 mM). Following incubation for 12 h at 37°C in a humidified
incubator, the insulin of the culture solution was detected by
radioimmunoassay. In another study, the rats were anaesthetized
with pentobarbital sodium (50 mg/kg) and given an intravenous
injection of 3DG (5, 20 and 50 mg/kg). At 2 h, pancreas islet
tissues were isolated and subsequently cultured for the measurement
GSIS, as described previously (29). The insulin of the culture solution
was detected by radioimmunoassay.
Plasma 3DG measurement
At two weeks following intragastric administration
of 3DG, the mice were sacrificed and blood sample was obtained from
enucleating the eyeballs for the measurement of 3DG contents by
high performance liquid chromatography, as described previously
(29).
Western blot analysis
INS-1 cells were grown to confluence in six-well
culture dishes and incubated with different concentrations of 3DG
(0, 80, 300 and 500 ng/ml) for 12 h under high glucose condition
(25.5 mM). After 12 h of treatment, the cells were harvested and
solubilized in IP lysine buffer containing 20 mM Tris (pH 7.5), 150
mM NaCl, 1% Triton X-100, 2 mM SDS, 25 mM β-glycerophosphate, 1 mM
EDTA, 1 mM Na3VO4 and 0.5 µg/ml leupeptin (Beyotime Institute of
Biotechnology, Nantong, Jiangsu, China). Following centrifugation
at 12,000 × g at 4°C for 20 min, the supernatants were collected
and used for Western blot analysis. The total protein
concentrations were determined using a bicinchoninic acid protein
(BCA) assay kit (cat. no. P0012; Beyotime Institute of
Biotechnology) comprising BCA kit A (cat. no. P0012-1), BCA kit B
(cat. no. P0012-2) and standard proteins (cat. no. P0012-3). The
proteins (100 µg) were loaded onto a 12% SDS-polyacrylamide gel
(Thermo Fisher Scientific, Inc.), then subjected to electrophoresis
and transferred onto polyvinylidene fluoride membranes (Merck KGaA,
Darmstadt, Germany). The membranes were blocked for 1 h in
Tris-buffered saline with 1% Tween (TBST; Beijing Solarbio Science
& Technology Co., Ltd., Beijing, China) containing 5% dry milk.
The membranes were washed with phosphate-buffered saline (PBS;
Beyotime Institute of Biotechnology) containing 0.05% Tween-20 3
times, and incubated at 4°C overnight with the following
antibodies: rabbit polyclonal anti-insulin receptor substrate 1
(IRS-1; cat. no. 2390), rabbit polyclonal anti-phospho-IRS-1
(IRS-1; cat. no. 3070), rabbit polyclonal anti-phosphoinositide
3-kinase (PI3K)-p85 (cat. no. 4292), rabbit polyclonal anti-Akt
(cat. no. 9272) and rabbit polyclonal anti-phospho-Akt (Ser473;
cat. no. 9271), all purchased from Cell Signaling Technology, Inc.
(Danvers, MA, USA; dilution, 1:1,000 each), and rabbit polyclonal
anti-glucose transporter 2 (GLUT2; cat. no. ab54460), purchased
from Abcam (Cambridge, UK; 1:500). Following washing 4 times for 5
min each in TBST, the membranes were incubated with goat
anti-rabbit secondary antibody [1:1,000; cat. no. GAR0072; Multi
Sciences (Lianke) Biotech Co., Ltd., Hangzhou, China] for 2 h and
visualized using an ECL detection kit (cat. no. PE0010; Beijing
Solarbio Science & Technology Co., Ltd.). Quantification of
protein bands was performed using ImageJ software version 1.42
(National Institutes of Health, Bethesda, MD, USA).
Statistical analysis
Results of the experimental studies are expressed as
mean ± standard deviation. Statistical significance of differences
was analyzed by the Student's t-test or one-way analysis of
variance. P<0.05 was considered to indicate a statistically
significant difference.
Results
The effects of intragastric
administration of 3DG for two weeks on plasma 3DG levels, fasting
blood glucose and oral glucose tolerance in mice
Given the evidence for the role of circulating 3DG
in the development of pre-diabetes, the author firstly investigated
whether continuous oral administration of 3DG increased plasma
levels of 3DG. At two weeks following administration, the plasma
level of 3DG was significantly increased in 3DG-treated mice
compared with the control (control, 36.22±13.51 ng/ml; 5 mg/kg 3DG,
76.67±7.98 ng/ml; P<0.05; 20 mg/kg 3DG, 84.75±6.04 ng/ml;
P<0.05; 50 mg/kg 3DG, 119.58±10.67 ng/ml; P<0.01; Fig. 1A). Then, fasting blood glucose was
measure in another two week 3DG administration experiment. The
fasting blood glucose levels of 50 mg/kg 3DG-treated mice were
higher than that of control mice (50 mg/kg 3DG vs. control,
5.45±0.603 mM vs. 4.644±0.413 mM; P<0.05; Fig. 1B). 3DG-treated mice had impaired
oral glucose tolerance in dose-dependent manner compared with
control mice (Fig. 1C; 5 mg/kg 3DG
vs. control, at 30 min, P<0.05; 20 mg/kg 3DG vs. control, at 30
and 60 min, P<0.05 and P<0.01; 50 mg/kg 3DG vs. control, at
30, 60, 90 min, P<0.05, P<0.01, P<0.05, respectively). The
AUC of OGTT results also demonstrated that the difference between
3DG-treated group with either 20 mg/kg dose or 50 mg/kg does and
control group was statistically significant (Fig. 1D; 20 mg/kg 3DG vs. control,
P<0.05; 50 mg/kg 3DG vs. control, P<0.01). These results
indicated the increased level of 3DG in the plasma may be
responsible for the 3DG-induced IGR in mice.
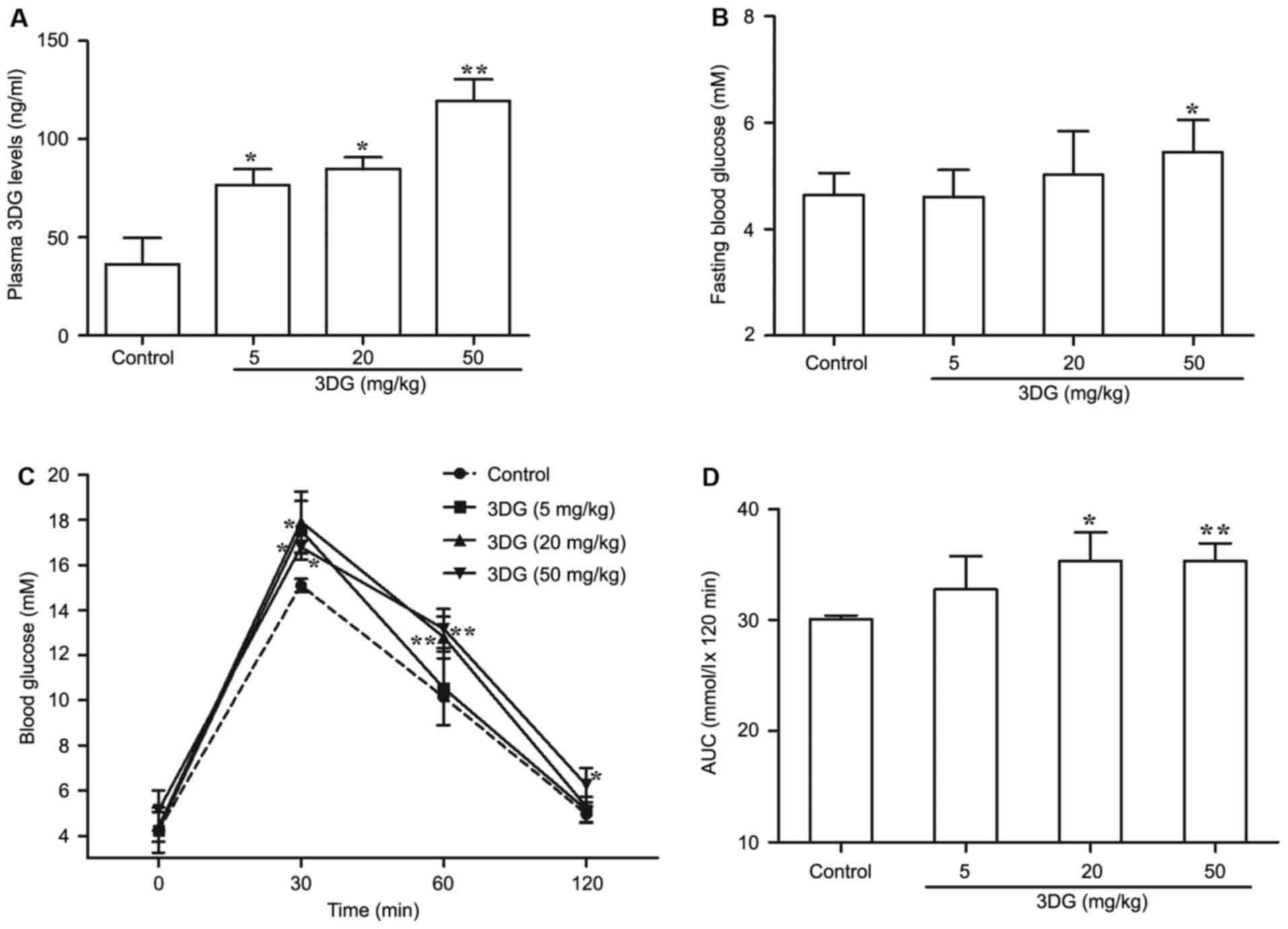 | Figure 1.The effects of intragastric
administration of 3DG for two weeks on plasma 3DG levels, fasting
blood glucose and oral glucose tolerance in mice. (A) The plasma
3DG concentrations were measured by high performance liquid
chromatography in mice two weeks following intragastric
administration of 3DG (5, 20 and 50 mg/kg). (B) Fasting blood
glucose levels were measured by a glucose meter in mice two weeks
following intragastric administration of 3DG (5, 20 and 50 mg/kg).
(C) The blood glucose concentration-time curves were measured after
intragastric administration of glucose (2.5 g/kg) in mice treated
with 3DG (5, 20 and 50 mg/kg) for two weeks. (D) The AUCs were
calculated of the blood glucose concentration-time curves using the
following formulas: AUC mmol.h/l=1/2A+B+C+1/2D (A, B, C and D was
the blood glucose at 0, 30, 60 and 120 min, respectively, mM).
Values are presented as the mean ± standard deviation. *P<0.05,
**P<0.01 vs. control (n=6). AUC, area under the curve; 3DG,
3-deoxyglucosone. |
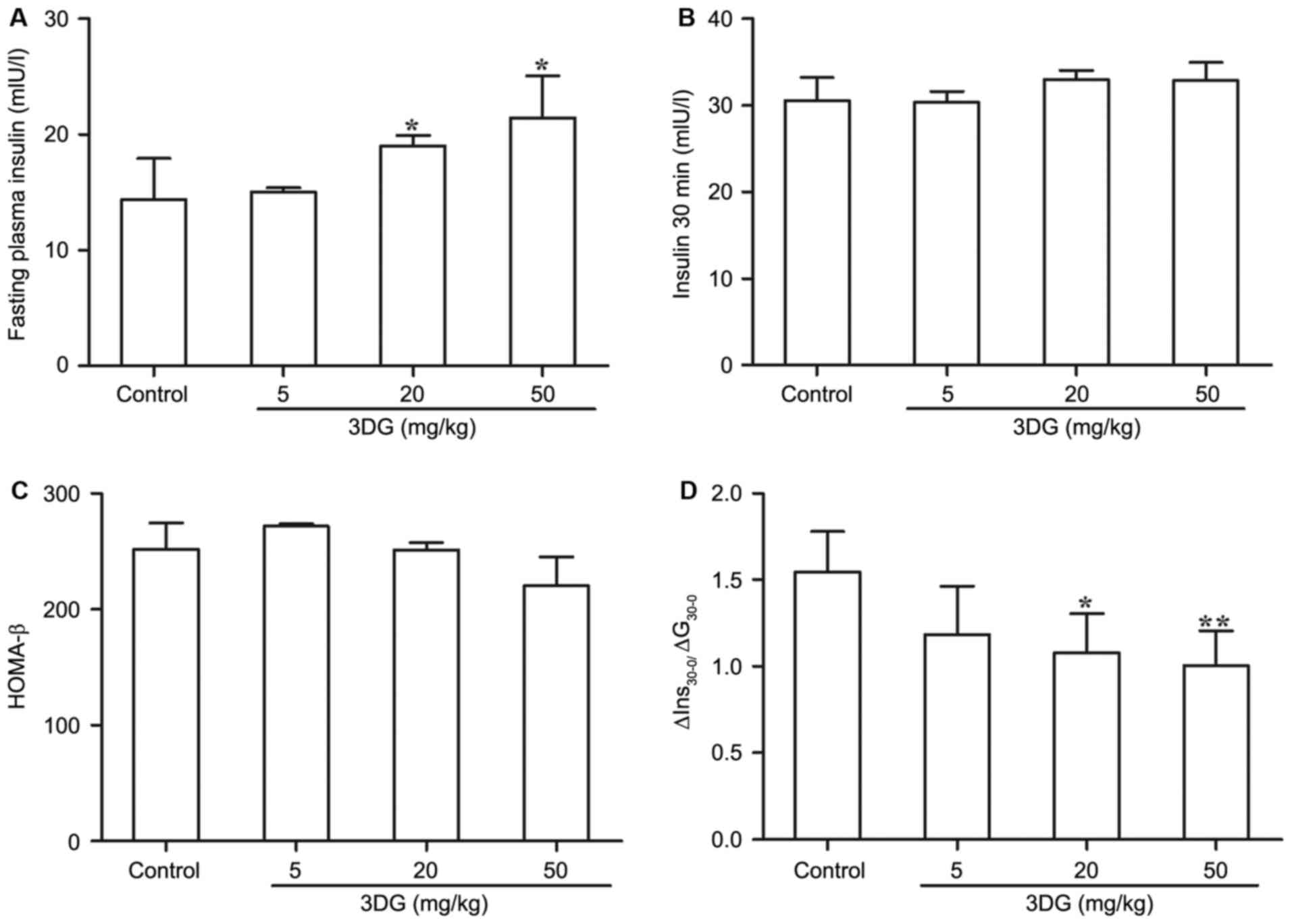
The effects of intragastric
administration of 3DG for two weeks on plasma insulin and the
insulin secretion index in mice
In consideration of the importance of β-cell
dysfunction to the pathogenesis of pre-diabetes, the authors
evaluated the potential damaging impact of exogenous 3DG on β-cell
function (Fig. 2). Under fasting
conditions, there was a noticeable increase in plasma insulin
levels between the control group (14.396±3.505 mIU/l) and
3DG-treated groups (20 mg/kg 3DG, 19.015±0.891 mIU/l, P<0.05; 50
mg/kg 3DG, 21.465±3.618 mIU/l, P<0.05; Fig. 2A). Whereas no significant
difference in insulin plasma levels was observed between
3DG-treated groups and control group 30 min following the glucose
load (Fig. 2B). In addition,
compared with the control group, 3DG-treated mice with either 20
mg/kg dose or 50 mg/kg doses exhibited a significant reduction of
∆Ins30-0/∆G30-0 (the early phase of insulin secretion) (Fig. 2D, 20 mg/kg 3DG vs. control,
1.546±0.231 vs. 1.081±0.223, P<0.05; 50 mg/kg 3DG vs. control,
1.546±0.231 vs. 1.006±0.201, P<0.01) whereas no difference in
HOMA-β was detected in 3DG-treated mice compared with control mice
(Fig. 2C). These results indicated
that administration of exogenous 3DG could have the ability to
affect β-cell function.
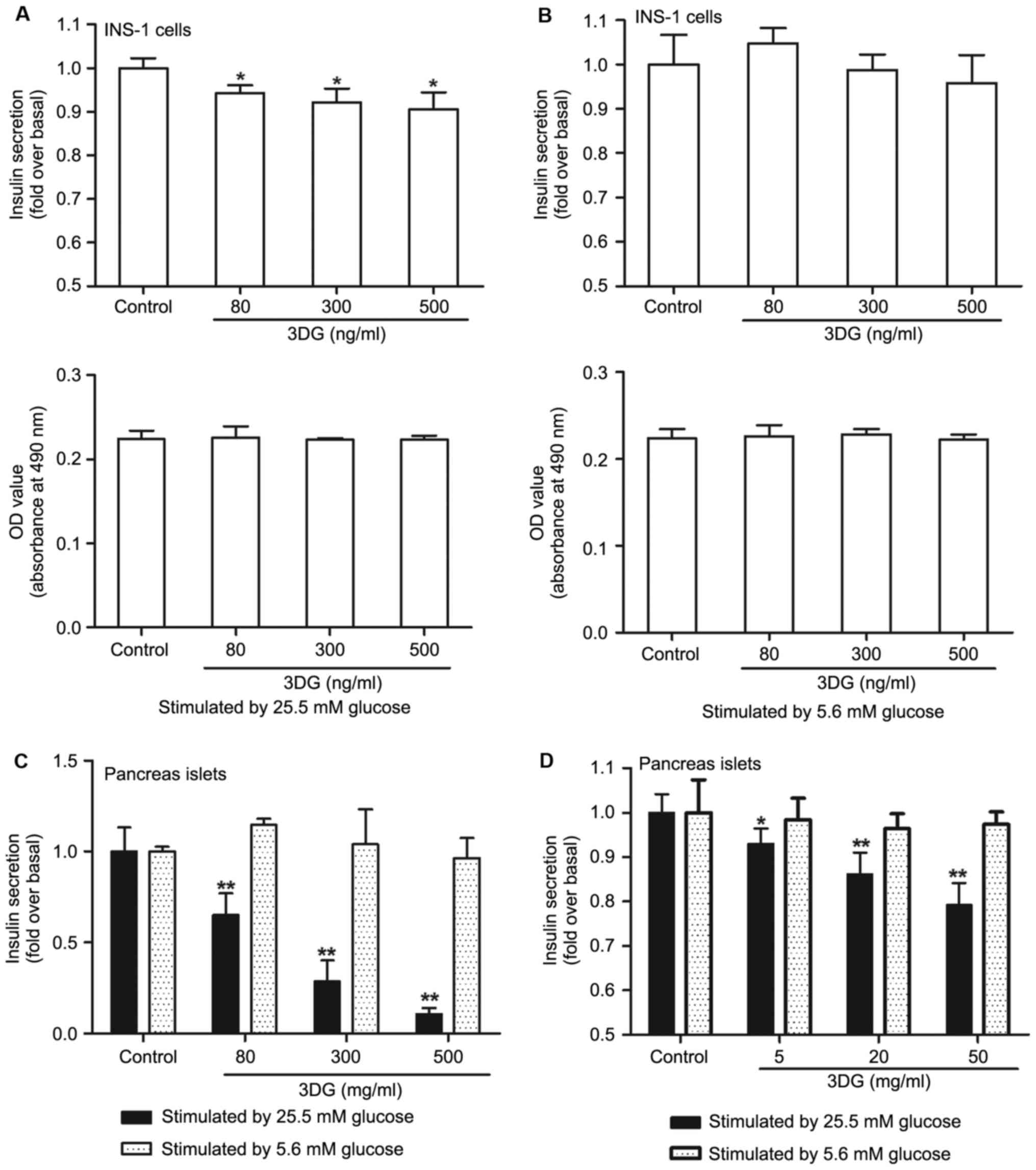
The effect of 3DG treatment on GSIS in
cultured islets and INS-1 cells
To further investigate the effect of 3DG on β-cell
function, the GSIS was measured in cultured pancreas islets and
INS-1 cells (Fig. 3). Compared
with the control group, GSIS in cultured pancreas islets and INS-1
cells treatment with 3DG was decreased following exposure to high
glucose (25.5 mM; Fig. 3A and C).
Furthermore, under the conditions tested 3DG at concentrations of
80, 300 and 500 ng/ml failed to alter INS-1 viability.
Additionally, as demonstrated in Fig.
3D, high GSIS was lower in cultured pancreas islets from the
acute intravenous injection of 3DG-treated rats than that of
control rats (5 mg/kg 3DG vs. control, 0.93-fold, P<0.05; 20
mg/kg 3DG vs. control, 0.86-fold, P<0.01; 50 mg/kg 3DG vs.
control, 0.79-fold, P<0.01). However, there was no significant
difference among the different groups under low glucose condition
(5.6 mM; Fig. 3B, C and D).
Finally, 3DG is demonstrated to exert deleterious effects on
insulin secretion under high glucose condition.
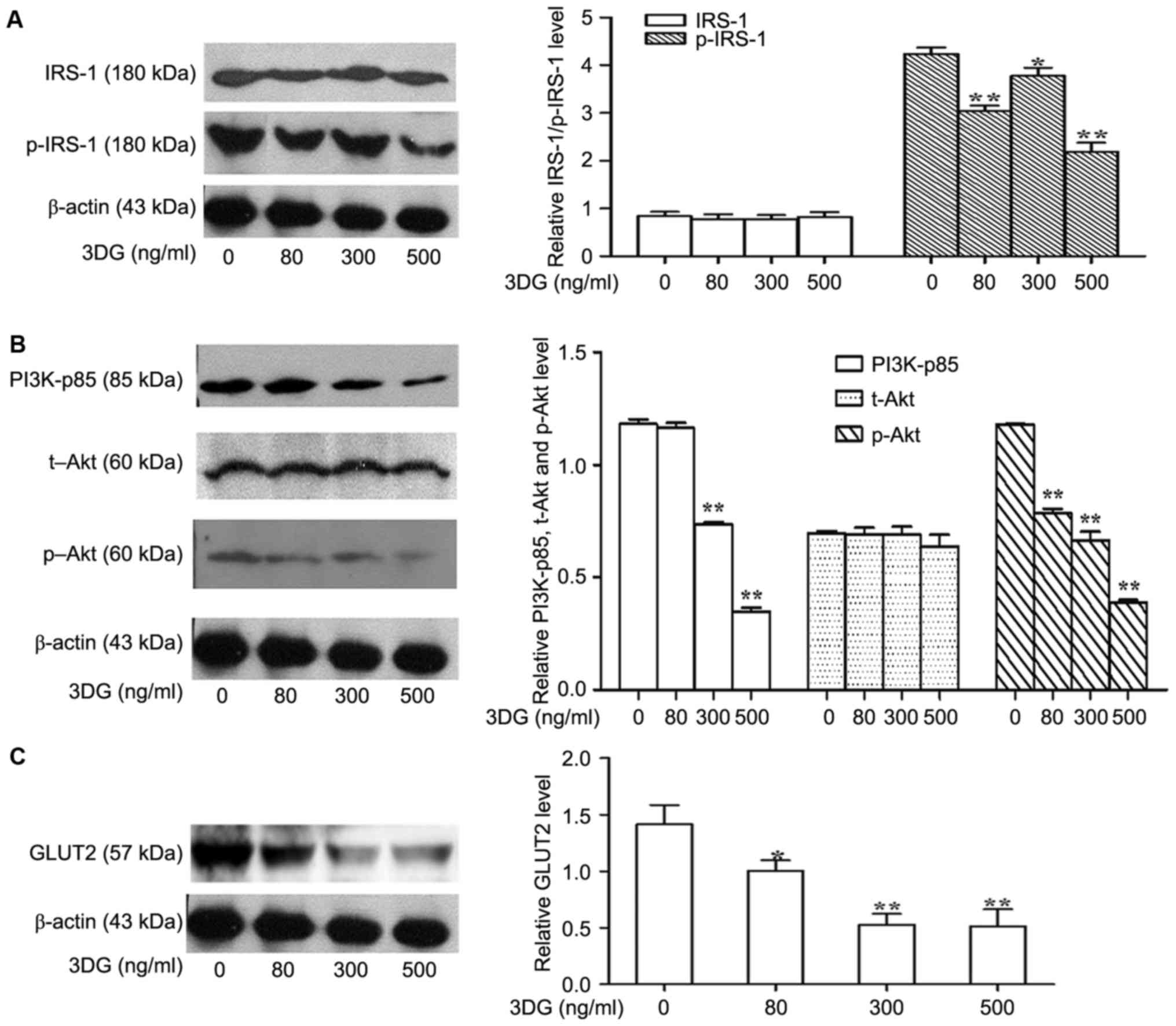
The effects of 3DG treatment on
insulin signaling in INS-1 cells
The authors further explored a potential mechanism
by which 3DG decreased insulin levels in response to a high glucose
challenge. Considering the significant role of insulin on β-cell
function, the ability of 3DG to impair insulin signaling in the
INS-1 β-cell line was investigated. Thus, the phosphorylation
levels of IRS-1 and PI3K/Akt signaling molecules involved in GSIS
were investigated in 3DG-treated cells co-incubated with high
glucose (25.5 mM) for 12 h. As presented in Fig. 4A and B, 3DG treatment with INS-1
cells led to a significant reduction of IRS-1, PI3K-p85 and Akt
phosphorylation in a dose dependent manner. These results suggested
that 3DG treatment impaired insulin secretion, at least in part,
through interfering with insulin-PI3K signaling. The idea was
further supported by the fact that 3DG treatment decreased the
expression of GLUT2 (Fig. 4C).
Discussion
The objective of the present study was to
investigate the effects of continuous intragastric administration
of a dietary composition, 3DG, on fasting blood glucose levels,
oral glucose tolerance, plasma 3DG levels and β-cell function in
normal mice. The study has confirmed that normal mice developed
impaired glucose tolerance in conjunction with slightly increased
fasting blood glucose concentrations, associated with the impaired
β-cell function. This occurred together with an obvious increase in
plasma 3DG levels following administration of 3DG that further
support for the impaired β-cell function.
Impaired glucose regulation (IGR) has been
recognized as a high risk state for type 2 diabetes mellitus
(T2DM). The rapid conversion of pre-diabetes to T2DM (annualized
conversion rate in the range of 5–10%) and the increasing
prevalence of pre-diabetes (>470 million people projected to
live with pre-diabetes worldwide by 2030) resulting in an
ever-increasing prevalence of T2DM (34). A significance of dietary factors on
understanding of the epidemic has been well recognized. The
increased intake of dietary carbohydrate or fat has been clearly
demonstrated to be important to development of pre-diabetes
(35). Aside from the known
nutrient composition, other dietary factors seem to serve an
essential part. This result clearly indicates that 3DG, the
predominant 1,2-dicarbonyl compound in foods, was administrated to
normal mice by gastric gavage for two weeks to induce the
development of IGR (Fig. 1). IGR
may be due to decreased β-cell function and/or an impairment in
insulin action on peripheral tissues. Several studies have
documented that decreased β-cell function is already present in the
IGR and gradually recognized to be the primary cause of IGR
(2,36,37).
Furthermore, at concentrations similar to those obtained from
plasma contents in 3DG-treated mice, 3DG directly decreased GSIS in
a dose-dependent manner in cultured pancreas islets and INS-1 cells
under high glucose conditions (Fig. 3A
and C), which further provide a possible explanation for the
observed β-cell dysfunction in 3DG-treated mice. In addition, a
rise in the fasting glucose concentrations within the normal range
was observed in 3DG-treated mice (Fig.
1B), is suggested to be due to a continuous fall in β-cell
function (38). A previous report
documented that the acute elevated plasma 3DG through the
intravenous injection impaired β-cell function (29). In the present study, increased
plasma 3DG levels in 3DG-treated mice were observed (Fig. 1A). These results not only further
support the role of β-cell dysfunction in IGR, but suggested that
abnormal elevation of circulating 3DG participates in inducing a
decreased β-cell function.
The function of β-cells depends on glucose uptake
and the subsequent signaling pathways that influence insulin
secretion. The uptake of glucose in pancreatic β-cells is primarily
mediated by GLUT2 that permits rapid glucose uptake regardless of
the extracellular sugar concentration, and is identified in high
levels in β-cells (39). Previous
reports have documented that GLUT2 is one of key mechanisms in
control of glucose-dependent insulin secretion (40,41).
In the present study, treatment with 3DG decreased the expression
of GLUT2 in high glucose-induced INS-1 cells (Fig. 4), which partly accounts for the
decreased GSIS. This occurs together with a significant reduction
in the phosphorylation levels of IRS-1, p85-PI3K and Akt, which
further confirmed the effect of 3DG on GLUT2 expression (Fig. 4). The finding in INS-1 cells is
consistent with those obtained in hepatocellular carcinoma cells
HepG2, in which 3DG impairs insulin signaling (30). In addition to regulating GLUT2
(42), pancreatic and duodenal
homeobox factor-1 (PDX-1) is also a key downstream molecule of the
insulin-PI3K signal pathway (43).
It has been reported that decreased PDX-1 expression results in a
reduction in insulin mRNA expression and impairment of glucose
stimulated insulin secretion (44). Additionally, the insulin-PI3K
signal pathway participates in modulating intracellular
Ca2+ flux that directly affects the rate of exocytosis
of insulin (10). Therefore, 3DG
decreased β-cell function, at least in part, through interfering
with the insulin-PI3K signaling pathway. On the basis of these
results, it is not known how 3DG impairs insulin signaling, but we
can just speculate about the mechanistic insight. 1) 3DG was
reported to cause the direct chemical modification of lysine and
arginine residues in proteins by binding (45), which may lead to a decrease in the
expression of the corresponding proteins. 2) A number of
observations indicate that dicarbonyl compounds, including 3DG and
MGO, impair intracellular signaling via the production of reactive
oxygen species (20). 3) The
ability of 3DG to impair insulin signaling may be indirectly
mediated by producing advanced glycation end products.
Although a single oral administration study has
indicated that the absorption rate of 3DG from foodstuffs is very
slow (33), it is unknown whether
continuous oral administration of 3DG causes an increased plasma
3DG levels in normal individuals. In presented experiments, the
level of plasma 3DG was elevated ~two-fold in the range of 76–119
ng/ml following 2-week administration of 3DG in mice that were
already at the pre-diabetic stage. The elevated 3DG levels in
plasma are primarily attributed to the intake of exogenous 3DG. The
current study suggested a potential action pathway linking
exogenous 3DG and development of pre-diabetes though an increase in
the plasma 3DG levels.
In summary, it is the first demonstration that
exogenous 3DG at two weeks following intragastric administration
led normal mice to develop IGR resulting from β-cell dysfunction.
In addition, the present study demonstrated that increased plasma
3DG levels in the 3DG-treated mice participates in inducing β-cell
dysfunction, at least in part, through impairing with insulin-PI3K
signaling, which provides the link between exogenous 3DG and the
development of IGR. The study suggests a pathogenic mechanism
linking between dietary and β-cell dysfunction with the cumulative
toxicity of 3DG given daily, thus 3DG may be a novel target for
protection of β-cell function and treatment and prevention of
IGR.
Acknowledgments
The present work was supported by the research funds
from Suzhou Science and Technology Department (grant nos.
SYS201423, and SYSD2015122), and the research fund from the Suzhou
Youth Science and Education Project (grant no. KJXW2014027).
References
|
1
|
Giuseppe D, Muhammad AG and Defronzo RA:
What are the pharmacotherapy options for treating prediabetes?
Expert Opin Pharmaco. 15:2003–2018. 2014. View Article : Google Scholar
|
|
2
|
Abdul-Ghani MA, Tripathy D and DeFronzo
RA: Contributions of beta-cell dysfunction and insulin resistance
to the pathogenesis of impaired glucose tolerance and impaired
fasting glucose. Diabetes Care. 29:1130–1139. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rhodes CJ, White MF, Leahy JL and Kahn SE:
Direct autocrine action of insulin on β-cells: Does it make
physiological sense? Diabetes. 62:2157–2163. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Leibiger IB, Leibiger B and Berggren PO:
Insulin signaling in the pancreatic beta-cell. Annu Rev Nutr.
28:233–251. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kulkarni RN, Brüning JC, Winnay JN, Postic
C, Magnuson MA and Kahn CR: Tissue-specific knockout of the insulin
receptor in pancreatic beta cells creates an insulin secretory
defect similar to that in type 2 diabetes. Cell. 96:329–339. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kulkarni RN: Receptors for insulin and
insulin-like growth factor-1 and insulin receptor substrate-1
mediate pathways that regulate islet function. Biochem Soc Trans.
30:317–322. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kulkarni RN, Winnay JN, Daniels M, Brüning
JC, Flier SN, Hanahan D and Kahn CR: Altered function of insulin
receptor substrate-1-deficient mouse islets and cultured beta-cell
lines. J Clin Invest. 104:R69–R75. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Withers DJ, Gutierrez JS, Towery H, Burks
DJ, Ren JM, Previs S, Zhang Y, Bernal D, Pons S, Shulman GI, et al:
Disruption of IRS-2 causes type 2 diabetes in mice. Nature.
391:900–904. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rothenberg PL, Willison LD, Simon J and
Wolf BA: Glucose-induced insulin receptor tyrosine phosphorylation
in insulin-secreting beta-cells. Diabetes. 44:802–809. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Aspinwall CA, Qian WJ, Roper MG, Kulkarni
RN, Kahn CR and Kennedy RT: Roles of insulin receptor substrate-1,
phosphatidylinositol 3-kinase, and release of intracellular
Ca2+ stores in insulin-stimulated insulin secretion in
beta -cells. J Biol Chem. 275:22331–22338. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
da Silva Xavier G, Varadi A, Ainscow EK
and Rutter GA: Regulation of gene expression by glucose in
pancreatic beta -cells (MIN6) via insulin secretion and activation
of phosphatidylinositol 3′-kinase. J Biol Chem. 275:36269–36277.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Maessen DE, Hanssen NM, Scheijen JL, van
der Kallen CJ, van Greevenbroek MM, Stehouwer CD and Schalkwijk CG:
Post-glucose load plasma α-dicarbonyl concentrations are increased
in individuals with impaired glucose metabolism and type 2
diabetes: The CODAM study. Diabetes Care. 38:913–920. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dhar A, Desai KM and Wu L: Alagebrium
attenuates acute methylglyoxal-induced glucose intolerance in
Sprague-Dawley rats. Br J Pharmacol. 159:166–175. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Riboulet-Chavey A, Pierron A, Durand I,
Murdaca J, Giudicelli J and Van Obberghen E: Methylglyoxal impairs
the insulin signaling pathways independently of the formation of
intracellular reactive oxygen species. Diabetes. 55:1289–1299.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jia X and Wu L: Accumulation of endogenous
methylglyoxal impaired insulin signaling in adipose tissue of
fructose-fed rats. Mol Cell Biochem. 306:133–139. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fiory F, Lombardi A, Miele C, Giudicelli
J, Beguinot F and Van Obberghen E: Methylglyoxal impairs insulin
signalling and insulin action on glucose-induced insulin secretion
in the pancreatic beta cell line INS-1E. Diabetologia.
54:2941–2952. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Eriksson UJ, Wentzel P, Minhas HS and
Thornalley PJ: Teratogenicity of 3-deoxyglucosone and diabetic
embryopathy. Diabetes. 47:1960–1966. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Degen J, Hellwig M and Henle T:
1,2-dicarbonyl compounds in commonly consumed foods. J Agric Food
Chem. 60:7071–7079. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Okado A, Kawasaki Y, Hasuike Y, Takahashi
M, Teshima T, Fujii J and Taniguchi N: Induction of apoptotic cell
death by methylglyoxal and 3-deoxyglucosone in macrophage-derived
cell lines. Biochem Biophys Res Commun. 225:219–224. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Che W, Asahi M, Takahashi M, Kaneto H,
Okado A, Higashiyama S and Taniguchi N: Selective induction of
heparin-binding epidermal growth factor-like growth factor by
methylglyoxal and 3-deoxyglucosone in rat aortic smooth muscle
cells. The involvement of reactive oxygen species formation and a
possible implication for atherogenesis in diabetes. J Biol Chem.
272:18453–18459. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kalapos MP: Methylglyoxal in living
organisms: Chemistry, biochemistry, toxicology and biological
implications. Toxicol Lett. 110:145–175. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Niwa T: 3-Deoxyglucosone: Metabolism,
analysis, biological activity, and clinical implication. J
Chromatogr B Biomed Sci Appl. 731:23–36. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sassi-Gaha S, Loughlin DT, Kappler F,
Schwartz ML, Su B, Tobia AM and Artlett CM: Two dicarbonyl
compounds, 3-deoxyglucosone and methylglyoxal, differentially
modulate dermal fibroblasts. Matrix Biol. 29:127–134. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang Q, Jiang GR and Zhang LR: Effects of
3-deoxyglucosone on blood glucose of normal mice. Chinese J
Diabetes. 18:220–222. 2010.
|
|
25
|
Lal S, Kappler F, Walker M, Orchard TJ,
Beisswenger PJ, Szwergold BS and Brown TR: Quantitation of
3-deoxyglucosone levels in human plasma. Arch Biochem Biophys.
342:254–260. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hamada Y, Nakamura J, Fujisawa H, Yago H,
Nakashima E, Koh N and Hotta N: Effects of glycemic control on
plasma 3-deoxyglucosone levels in NIDDM patients. Diabetes Care.
20:1466–1469. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Beisswenger PJ, Howell SK, O'Dell RM, Wood
ME, Touchette AD and Szwergold BS: alpha-Dicarbonyls increase in
the postprandial period and reflect the degree of hyperglycemia.
Diabetes Care. 24:726–732. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jiang G, Zhang L, Ji Q, Wang F, Xu H,
Huang F and Wang C: Accumulation of plasma 3-deoxyglucosone
impaired glucose regulation in Chinese seniors: Implication for
senile diabetes? Diabetes Metab Syndr. 6:140–145. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Liang G, Song X, Xu H, Wang F, Zhang L,
Zhou L and Jiang G: 3-Deoxyglucosone induced acute glucose
intolerance in sprague-dawley rats: Involvement of insulin
resistance and impaired β-cell function. Exp Clin Endocrinol
Diabetes. 124:431–436. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liang G, Wang F, Song X, Zhang L, Qian Z
and Jiang G: 3-Deoxyglucosone induces insulin resistance by
impairing insulin signaling in HepG2 cells. Mol Med Rep.
13:4506–4512. 2016.PubMed/NCBI
|
|
31
|
Kahn SE, Cooper ME and Del Prato S:
Pathophysiology and treatment of type 2 diabetes: Perspectives on
the past, present, and future. Lancet. 383:1068–1083. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Schwartz MW, Seeley RJ, Tschöp MH, Woods
SC, Morton GJ, Myers MG and D'Alessio D: Cooperation between brain
and islet in glucose homeostasis and diabetes. Nature. 503:59–66.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kato H, van Chuyen N, Shinoda T, Sekiya F
and Hayase F: Metabolism of 3-deoxyglucosone, an intermediate
compound in the Maillard reaction, administered orally or
intravenously to rats. Biochim Biophys Acta. 1035:71–76. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tabák AG, Herder C, Rathmann W, Brunner EJ
and Kivimäki M: Prediabetes: A high-risk state for diabetes
development. Lancet. 379:2279–2290. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hu FB, Van Dam RM and Liu S: Diet and risk
of type II diabetes: The role of types of fat and carbohydrate.
Diabetologia. 44:805–817. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tabák AG, Jokela M, Akbaraly TN, Brunner
EJ, Kivimäki M and Witte DR: Trajectories of glycaemia, insulin
sensitivity, and insulin secretion before diagnosis of type 2
diabetes: An analysis from the Whitehall II study. Lancet.
373:2215–2221. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Gastaldelli A, Ferrannini E, Miyazaki Y,
Matsuda M, DeFronzo RA, et al: San Antonio metabolism study:
Beta-cell dysfunction and glucose intolerance: Results from the San
Antonio metabolism (SAM) study. Diabetologia. 47:31–39. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Stancáková A, Javorský M, Kuulasmaa T,
Haffner SM, Kuusisto J and Laakso M: Changes in insulin sensitivity
and insulin release in relation to glycemia and glucose tolerance
in 6,414 Finnish men. Diabetes. 58:1212–1221. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Thorens B, Sarkar HK, Kaback HR and Lodish
HF: Cloning and functional expression in bacteria of a novel
glucose transporter present in liver, intestine, kidney, and
beta-pancreatic islet cells. Cell. 55:281–290. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Unger RH: Diabetic hyperglycemia: Link to
impaired glucose transport in pancreatic beta cells. Science.
251:1200–1205. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Guillam MT, Hümmler E, Schaerer E, Yeh JI,
Birnbaum MJ, Beermann F, Schmidt A, Dériaz N and Thorens B: Early
diabetes and abnormal postnatal pancreatic islet development in
mice lacking Glut-2. Nat Genet. 17:327–330. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Assmann A, Ueki K, Winnay JN, Kadowaki T
and Kulkarni RN: Glucose effects on beta-cell growth and survival
require activation of insulin receptors and insulin receptor
substrate 2. Mol Cell Biol. 29:3219–3228. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kaneto H, Matsuoka TA, Miyatsuka T,
Kawamori D, Katakami N, Yamasaki Y and Matsuhisa M: PDX-1 functions
as a master factor in the pancreas. Front Biosci. 13:6406–6420.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Brissova M, Shiota M, Nicholson WE, Gannon
M, Knobel SM, Piston DW, Wright CV and Powers AC: Reduction in
pancreatic transcription factor PDX-1 impairs glucose-stimulated
insulin secretion. J Biol Chem. 13:11225–11232. 2002. View Article : Google Scholar
|
|
45
|
Sakiyama H, Takahashi M, Yamamoto T,
Teshima T, Lee SH, Miyamoto Y, Misonou Y and Taniguchi N: The
internalization and metabolism of 3-deoxyglucosone in human
umbilical vein endothelial cells. J Biochem. 139:245–253. 2006.
View Article : Google Scholar : PubMed/NCBI
|