Introduction
Wiskott-Aldrich syndrome (WAS) is a rare X-linked
recessive primary immunodeficiency disease recognized by symptoms
including eczema, thrombocytopenia, immune deficiency and bloody
diarrhea (1). It is also known as
eczema-thrombocytopenia-immunodeficiency syndrome, in line with
Aldrich's original description in 1954 (2). The molecular defects of WAS were
discovered in 1994 by Derry et al (3), who isolated the pathogenic gene by
positional cloning. The gene that encodes the WAS protein (WASP) is
located in the short arm of X chromosome (XP11.22–11.23) and is ~9
kb, containing 12 exons and encoding 502 amino acids.
Epidemiological studies have demonstrated that the
incidence of neonatal WAS in the developed countries is
1/1,000,000–10/1,000,000 (4), and
WAS is caused by a mutation in the WASP gene. WASP is a
hematopoietic system-specific intracellular signal transduction
molecule, which is proline rich, expressed only in hematopoietic
cell lines. It is well documented that WASP gene mutations affect
the expression of WASP, thereby resulting in disorders in the
response of non-red blood cells to external stimuli, which in turn
leads to issues in signal transduction and cytoskeleton
dysfunction. This further influences the number, size and
aggregation of platelets, hence leading to defects in lymphocytes
migration, signal transduction and immunological synapse formation
(5).
WASP has been revealed to be critical for
cell-signaling (6), actin
polymerization (7), synapse
formation (8), cell/cell
interaction (9), chemotaxis
(10), and Treg function (11). As a result, clinical manifestations
of patients with alterations of the WASP gene show a great
heterogeneity. Categorized by the mutations in the WASP gene, WAS
can be divided into typical classic WAS, X-linked thrombocytopenia
(XLT) and X-linked neutropenia (XLN). Classic WAS is characterized
by triad of thrombocytopenia/micro-platelets, recurrent infections,
and eczema. The milder XLT variant behaves predominantly as
thrombocytopenia, sometimes occurring intermittently. Patients with
congenital neutropenia but without the clinical findings
characteristic of WAS or XLT are classified as XLN (12).
Different mutations of WASP could lead to the
heterogenicity of WAS in patients. In addition to WASP, WASP
interacting proteins have been demonstrated to be involved in the
onset of WAS (13–15). Disruption of the binding of WASP to
its interacting proteins could also lead to patients with clinical
features of WAS, in whom the WASP gene sequence and mRNA levels are
normal (16–18). The binding of WASP with its
interacting proteins have been confirmed by crystallography
(19–21).
It is not difficult to diagnose WAS according to the
clinical features; however, as the clinical manifestations of
patients demonstrate a great heterogeneity, definite diagnosis is
required in many cases. Previously, Sanger sequencing of the WASP
gene has been applied for the confirmation of WAS diagnosis
(22,23), as WASP gene mutations account for
the majority of WAS cases. However, as WAS may be caused by
mutations of other WASP interacting proteins, other genes may be
involved; therefore, performing only WASP gene detection may lead
to misdiagnosis. Therefore, it is necessary to identify multiple
genes simultaneously. In the present study, next generation
sequencing was performed on a WAS patient so as to give the
definite conclusion and provide evidence for therapy based on the
genetic profile.
Patients and methods
Patient data
A 5-month old male pediatric patient with
thrombocytopenia for 3 months and rash for 2 months was admitted to
Hunan Provincial People's Hospital (Changsha, China). His mother
was a 25-year old gravida 1 para 1 healthy woman without familial
diseases. This child was identified to have thrombocytopenia in a
local hospital 35 days after birth, when he was hospitalized due to
a lung infection. Gamma globulin protein shock treatment was
ineffective and his platelet levels maintained at (30–36)x109/l. When he was 2
months old, repeated eczematous rashes appeared. His parents
confirmed the family history. The present study was approved by the
ethics committee of Hunan Provincial People's Hospital, and
informed consents were obtained from the parents of the participant
and healthy controls.
Investigation of WASP-interacting
proteins
To investigate potential proteins that may interact
with WASP, the online server STRING (http://www.string-db.org/) was applied. This server
provides a database of known and predicted protein-protein
interactions (24). The network
analysis was performed strictly according to the user
documentation. The search was initiated using the Single Protein by
Name/Identifier selection of the website.
Next-generation sequencing (NGS)
For specimen preparation, genomic DNA from 2 ml
peripheral blood was extracted from the trio of the proband
following the instruction of BloodGen Midi kit (CWBIO, Beijing,
China) and the DNA was sheared by sonication. The sheared genomic
DNA was then hybridized with NimbleGen 2.0 probe sequence capture
array (Roche Diagnostics, Basel, Switzerland). The captured DNA was
firstly applied for exonic DNA enrichment (Roche Diagnostics) and
the libraries were then tested for enrichment by quantitative
polymerase chain reaction (qPCR) using the Agilent Bioanalyzer 2100
(Agilent Technologies, Inc., Santa Clara, USA). The samples were
thereby sequenced on an Illumina Hiseq2500 system (Illumina, Inc.,
San Diego, CA, USA).
Raw image files were processed by the BclToFastq
(Illumina, Inc.) for base calling and raw data generating. The
low-quality variations were filtered out using quality score ≥20
(Q20). The sequencing reads were aligned to the NCBI human
reference genome (hg19) using the Burrows-Wheeler Alignment tool
(25). The Genome Analysis Toolkit
(26) was used to analyzed single
nucleotide polymorphism and insertion-deletion of the sequences.
The genes that encoded WASP interacting proteins predicted by
STRING were analyzed in detail.
PCR amplification
PCR amplification was performed using whole genome
DNA of the proband and a 25-year-old female healthy control. Whole
genome DNA was extracted from peripheral blood using DNAzol™ BD
Reagent (Thermo Fisher Scientific, Inc., Waltham, MA, USA).
Specific primers covering most exons of the WASP gene were designed
using Primer premier 5.0 software (Premier Biosoft International,
Palo Alto, CA, USA). DNA polymerase was purchased from Takara Bio,
Inc. (Otsu, Japan). The primer sequences and relative product
lengths are presented in Table I.
The PCR conditions were as follows: Initial denaturation at 94°C
for 5 min, followed by 40 cycles of denaturation at 94°C for 30
sec, annealing at 60°C for 30 sec, elongation at 72°C for 30 sec,
and extension at 72°C for 5 min. PCR products were applied to 1%
agarose gel electrophoresis and analyzed by image acquisition and
analysis system (Tanon Science and Technology Co., Ltd., Shanghai,
China).
 | Table I.Primers used for specific truncated
WASP gene detection for the proband and a healthy control by
polymerase chain reaction amplification. |
Table I.
Primers used for specific truncated
WASP gene detection for the proband and a healthy control by
polymerase chain reaction amplification.
| Primer | Sequence (5′-3′) | Product length
(bp) |
|---|
| WAS-1F |
TCTAAGCAGTCAAGTGGAGGAG |
930 |
| WAS-1R |
ATCTGGATGAGTCTTTGGTTCTG |
|
| WAS-2F |
GAGCCTCAACTTCCTAAGACTAGA | 1,063 |
| WAS-2R |
TCAGCCATCTACCGCCAATC |
|
| WAS-3F |
TACCTCCATGACCATCCAACA |
380 |
| WAS-3R |
CCATCCTTCCATTCACTCAGC |
|
| WAS-4F |
TTCCATAACTCCTGCCTATACTCA |
680 |
| WAS-4R |
CACTGACCAACTCCTGACTGA |
|
| WAS-5AF |
TCACTCAGTCCTTATGGGAGCACCT | 1,021 |
| WAS-5AR |
TCAAACAGATGGGGCTGATGTCACT |
|
| WAS-6F |
TTAACCAGACAGGAAGCAAT |
593 |
| WAS-6R |
CTTGAGTGAAGAGAACTGAGA |
|
qPCR amplification
qPCR amplification was performed using whole genome
DNA extracted from peripheral blood of the mother and a 25-year-old
female healthy control using DNAzol™ BD Reagent (Thermo Fisher
Scientific, Inc.). Specific fluorescence quantitative primers were
designed for the N terminal, middle and C terminal of the WASP gene
with Primer premier 5.0 software. The primer sequences and relative
product lengths are presented in Table II. The primers were synthetized by
Sangon Biotech Co., Ltd. (Shanghai, China). The PCR conditions were
as follows: Initial denaturation at 95°C for 1 min, followed by 40
cycles of denaturation at 95°C for 15 sec, annealing at 60°C for 40
sec and elongation at 68°C for 30 sec. For each sample, two
parallel reactions were performed and tyrosine-protein kinase ABL1
(ABL1) served as a reference gene. Homogenization of the PCR
products were firstly performed using the output of ABL.
Amplification efficiency was calculated according to the
comparative Cq method (27) by
drawing the standard curve of each pair of primers. The copy number
ratio of each truncated gene was calculated by comparing with that
of the control.
| Table II.Primers used for specific truncated
WASP gene detection for the mother and a healthy control by
quantitative polymerase chain reaction amplification. |
Table II.
Primers used for specific truncated
WASP gene detection for the mother and a healthy control by
quantitative polymerase chain reaction amplification.
| Primer | Sequence
(5′-3′) | Product length
(bp) |
|---|
| WAS-1QF |
AAGACCTTGTGGCTACCCCT | 144 |
| WAS-1QR |
AGCACACAGCCCCACAATGCTC |
|
| WAS-3QF |
GTCAATGAGCCAACCACCCTA | 151 |
| WAS-3QR |
TTCTTATCAGCTGGGCTAGGTC |
|
| WAS-5QF |
CTAAGCCCTCTGTGCTGATCCC | 136 |
| WAS-5QR |
GGCTCTGCTTCTCTTCTGCATCAC |
|
Statistical analysis
Statistical analysis was performed using SPSS
software version 17.0 (SPSS, Inc., Chicago, IL, USA). The rations
of the truncated WASP genes of the mother were calculated to that
of the control and then expressed as mean ± standard deviation. The
value was then compared with the estimated value of 0.5 using
single-sample Student's t-test. P<0.05 was considered to
indicate a statistically significant difference.
Results
Clinical features of the patient
Following hospitalization, routine examinations of
the blood, urine and stools were performed. Physical examination
revealed scattered needlepoint to small grain-sized papules mixed
with a few pinpoint-sized bleeding points across the whole body.
Parts of the eruptions were accompanied by a little secretion, with
periauricular as the most serious. Purulent secretion was
identified in the left ear. The liver was enlarged 6 cm below the
right costal margin and the spleen could be palpated 2 cm under the
left costal margin. Blood routine examination revealed that the
white blood cell count was 14.28×109/l, the neutrophil
count was 7.42×109/l, hemoglobin was 116 g/l, the
platelet count was 26×109/l, the thrombocytocrit was
0.01%, and the platelet mean volume was 5.8 fl. Stool and urine
routine tests indicated that liver function, renal function,
myocardial enzyme, blood glucose, electrolytes, coagulation and
C-reactive protein all were roughly normal. The erythrocyte
sedimentation rate was 37 mm/h. The fecal occult blood test was
negative. Examination of immunoglobulin revealed that
immunoglobulin (Ig)A was 0.76 g/l, IgG was 9.73 g/l [after
intravenous gammaglobulin (IVIG)] and IgM was 5.88 g/l; all were
higher than healthy limits. In addition, total IgE was within the
normal range (11.36 IU/ml). Lymphocyte subsets detection
demonstrated that cluster of differentiation (CD)3+ T cells
accounted for 38.4% of lymph, CD3+ CD4 helper/inducer T cells were
32.9% of lymph, and the percentage of CD3+ CD8+
suppressor/cytotoxic T cells was 1.3%, which were a little lower
than that of normal values. The percentage of CD3-CD56+ CD16+
natural killer cells and CD3-CD19+ B cells were 16.2 and 35.9%
respectively, which were a little higher compared with normal
limits.
Respiratory virus antigen examination (influenza A,
influenza B, respiratory tract virus, adenovirus, parainfluenza
virus 1, 2 and 3) and Torch analysis were negative.
cytomegalovirus-DNA was <1.00E+03 copies and blood culture was
negative. A chest X-ray revealed scattered patchy shadow in both
lungs with a fuzzy edge, whereas no obvious abnormalities in
cardiophrenic angle were identified. A cranial plain X-ray was
normal and an abdominal ultrasound revealed hepatosplenomegaly. A
bone marrow examination revealed that proliferation of bone marrow
was active. A large number of megakaryocytes were observed, and
there was megakaryocyte mature hindrance in cell
classification.
Acute feverish and shortness of breath occurred 3
days after the child was admitted to hospital. He was transferred
to the intensive care unit for treatment as he was diagnosed severe
pneumonia, respiratory failure and sepsis. In line with the above
symptoms, the child was clinically diagnosed as typical WAS. As his
condition aggregated, his parents abandoned treatment and the child
was followed up.
WASP-interacting proteins
identification results
In order to gain insight into the proteins that
interact with WASP, the online server STRING was used. The
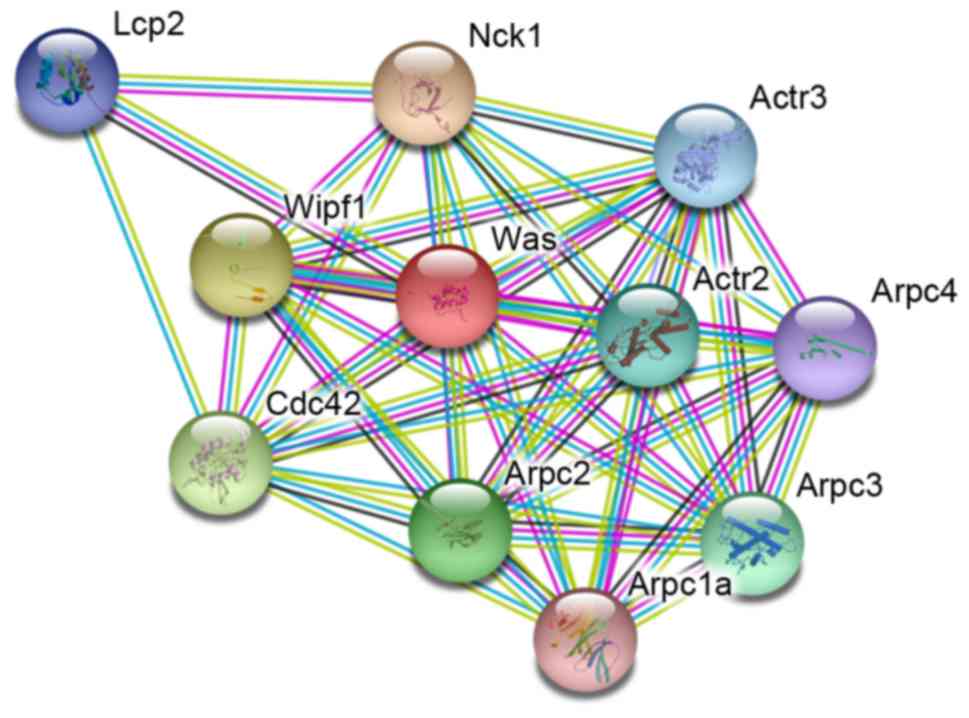
predictive result is represented in Fig. 1. Together, there were 10 proteins
that were considered to be associated with WASP directly or
indirectly. The results indicated that >one proteins are
enrolled in the interaction of WASP; NGS may reveal novel insights
into the genetic profile of WAS.
NGS results
To confirm the diagnosis of WAS and unravel the
underlying genetic mutations in WASP and other genes, genomic DNA
from the proband and his parents underwent NGS. A total of 8.7 G
clean data was obtained from the three samples. The average of GC
content was 42.92% and the Q20 was >95%. Quality control files
demonstrated that the data was reliable and adequate for further
analysis. Coverage and mean depth for the child, his mother and his
father were 0.24/0.6, 1/32.9 and 1/48.4, respectively. No mutation
that could support the determination of other disease besides WAS
was identified. The results indicated that the WASP gene was
deviant or even totally lost in the proband, and confirmed the
diagnosis of WAS.
PCR results indicate the whole WASP
gene is lost in the proband
To assess the deletion information in the WASP gene
in the proband, PCR was performed. As presented in Fig. 2, no bands were observed in all the
lanes (1, 3, 5, 7, 9 and 11) of PCR products from the patient,
whereas relative bands were observed in all lanes (2, 4, 6, 8, 10
and 12) from that of the healthy control. The results indicated
that the WASP gene in the proband was totally lost.
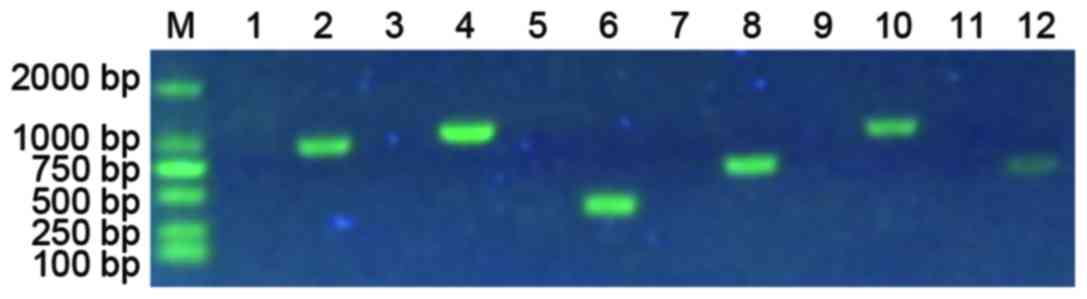 | Figure 2.PCR result indicate the whole WASP
gene is lost in the proband. Lanes 1, 3, 5, 7, 9, and 11 are PCR
products from the proband, while lanes 2, 4, 6, 8, 10, and 12 are
PCR products with the corresponding primers from a healthy control.
The lanes 2, 4, 6, 8, 10, and 12 are products of WAS-1F and WAS-1R,
WAS-2F and WAS-2R, WAS-3F and WAS-3R, WAS-4F and WAS-4R, WAS-5AF
and WAS-5AR, WAS-6F and WAS-6R primers, respectively. PCR,
polymerase chain reaction; WAS, Wiskott-Aldrich syndrome; F,
forward; R, reverse. |
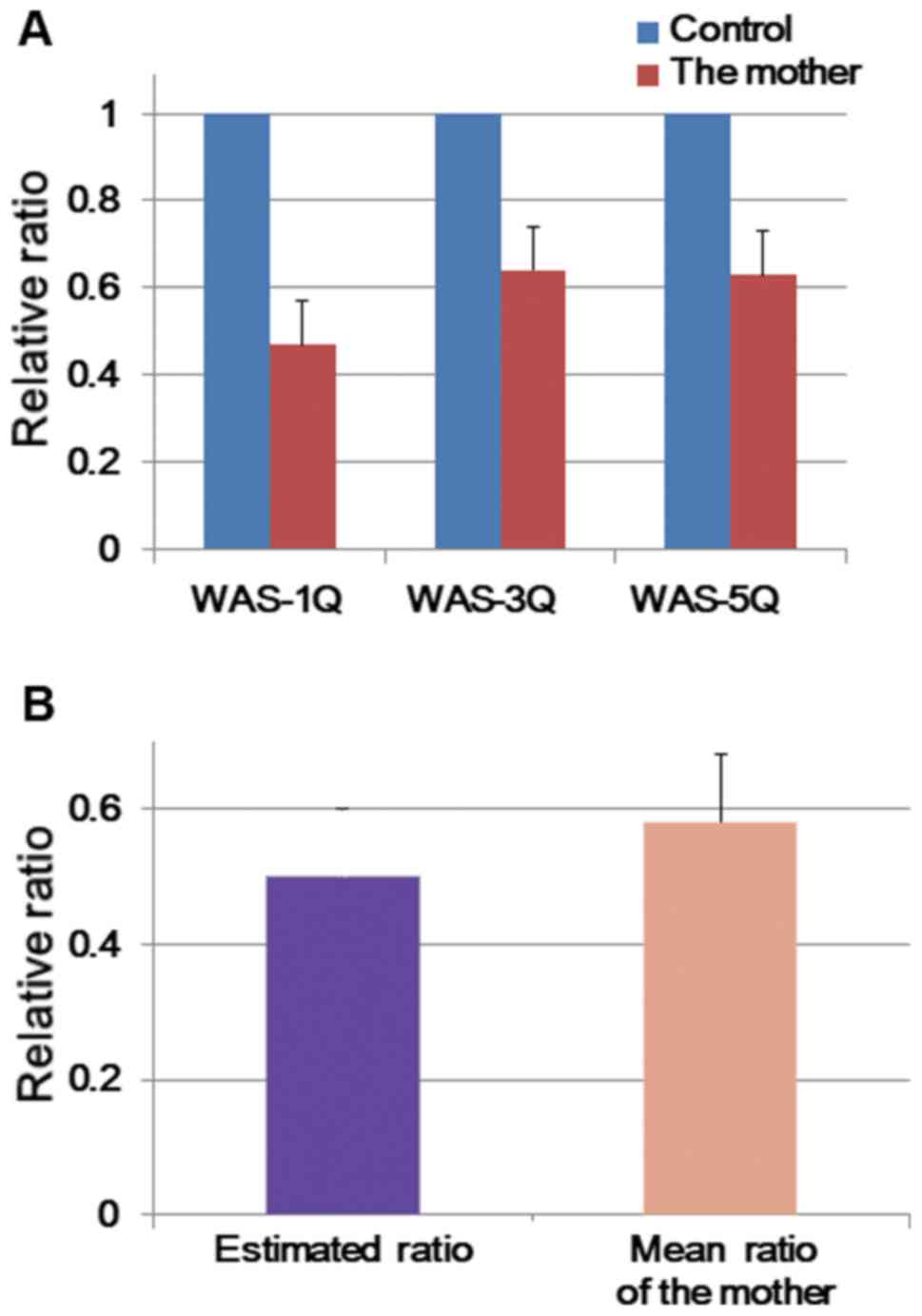
qPCR results of the mother of the
proband
As the father was healthy, the lost mutation could
only be inherited from his mother or caused de novo. To reveal the
etiology so as to guide reproduction to this family, the whole
genomic DNA of the mother underwent qPCR analysis. As presented in
Fig. 3, the ratios of the target
truncated WASP genes to that of the control all were ~0.5, with a
mean of 0.58±0.10. There were significant differences in the mean
ratio value for the mother (t=1.453, P=0.284). The results
demonstrated that the WASP gene of the mother was heterozygous, and
the mutation of the proband was inherited from his mother.
Discussion
At present, >300 kinds of WAPS gene mutations
have been reported, which are distributed in the whole WAS gene,
and are more concentrated in exon 1-4s, 7 and 10 (28). Six sites are regarded as mutational
hotspots, accounting for ~25% of all mutational sites, including
three splice site mutations and three point mutations in the coding
region (29). The mutation type of
the WAS gene determines the expression of WASP, which is closely
associated with the clinical phenotype of WAS. A missense mutation
in exons 1–3 usually leads to the XLT phenotype, loss of or
truncated WASP expression of the 10th exon could induce typical
WAS, and missense mutations in GTPase binding domain (L270P, S272P
and 1294T) could lead to XLN (30).
Peng et al (31) observed that from January 1991 to
October 2013, there were 12 articles focusing on gene diagnosis of
WAS including 54 case studies. Mutations consisting of missense,
nonsense, splicing site and insertion/deletion mutations were
identified; however, no whole exon deletion was observed. In a
retrospective study containing 50 case studies from 40 pedigrees by
Catucci et al (32),
missense and fusion mutations manifested as WASP positive
expression, whereas nonsense mutations, deletion mutations and
insertional mutagenesis led to WASP-negative expression. Among the
10 cases of deletion mutations, 3 cases were large fragment
deletions, and 1 case was whole exon deletion. In the present
study, the patient had whole exon 1–12 deletion, which is rare. To
the best of our knowledge, this is the first case of whole exon
deletion reported in China, and the second case in the world.
WAS is a combined immunodeficiency disease, with
different degrees of humoral and cellular immune deficiency. As the
abnormal immune function of WAS is gradual, the immune deficiency
symptoms increase with age. Therefore, immunological examination
results vary markedly. In most cases, it is reported that serum IgM
decreases, IgG only slightly decreases or remains normal, IgA and
IgE elevate, and T and B cell proliferation is reduced in 50%
patients in vitro (4). In
the present study, T cell counts reduced whereas B lymphocyte
numbers increased, which is consistent with other studies (4,33).
However, there was difference in the humoral immunity result. Serum
IgE was normal and IgA and IgG levels were elevated, which may be
associated with intravenous Ig therapy. IgM levels increased
significantly, which may be associated with repeated intrauterine
and postnatal infection by combing with liver and spleen
enlargement. This indicates that the immune level of the patient
may be associated with age, disease course, infection and other
factors that affect disease progression. Therefore, WAS diagnosis
should not only be based on the outcome of immunodetection.
Autoimmune diseases often occur in children with
WAS, and in the United States and the European population the
incidence is as high as 40~72% (32). Autoimmune hemolytic anemia,
vasculitis, arthritis and kidney disease are the most common
symptoms. In a retrospective study, Imai et al (33) demonstrated that there is no
statistically significant difference between WASP negative and
positive groups in autoimmune disease incidence (22 vs. 26%,
P>0.05) (33). Five cases were
with IgA nephropathy and the age of onset was rather late, from 10
to 20 years old. In addition, 5 out of the 50 reviewed patients
were complicated by tumors, and they all were WASP negative. In the
present study, neither autoimmune or tumor diseases were
identified; this may be associated with the young age of the
patient and the short duration of the disease. Zhao et al
(34) suggested that the clinical
phenotype of WAS evaluation should follow the dynamic principle. In
line with their opinion, whether this patient's disease may be
associated autoimmune diseases and tumors requires further
follow-up observation.
Therapeutic methods of WAS should be planned
according to clinical severity, duration, mutations of the WASP
gene and WASP expression. Therefore, it is necessary to identify
the etiology on genetic profile. Routinely, Sanger sequencing is
performed to detect mutational situation in the WASP gene. However,
as WAS may be caused by WASP-interacting proteins, it is essential
to identify the mutations in these proteins simultaneously so as
give systemic identification. In view of this, the present study
performed whole exome sequencing for this patient.
Before sequencing, the STRING online database was
used to identify WASP-interacting proteins, and the result
demonstrated that 10 proteins are included. Among the 10 proteins,
actin-related protein 2/3 complex subunit (Arpc)1a, Arpc2, Arpc3,
Arpc4, Actr2 and Actr3 are associated with the Arp2/3 protein
complex, which has been implicated in the control of actin
polymerization in cells and are conserved through evolution
(35–38). In addition, they have been
demonstrated to interact with Cortactin (35). The protein WAS/WASL-interacting
protein family member 1 (Wipf1) is reported to interact with WASP,
Cortactin, and cytoplasmic protein NCK1 (13,14).
NCK1 is linked to glucose tolerance and insulin signaling within
certain tissues, and serves important roles in insulin signaling
and the c-Jun N-terminal kinase signaling pathway (39). Cdc42 is involved in diverse
cellular functions, including cell morphology, migration,
endocytosis and cell cycle progression (40). The functions of these proteins are
associated with that of WASP (6,9,10),
indicating that it is necessary for detecting the status of these
genes. The sequencing results indicated that there were no abnormal
WASP-interacting protein coding genes, but only whole loss of the
WASP gene. No novel mutations in WASP-interacting protein coding
genes were identified, indicating that NGS was indispensable for
WAS diagnosis.
Supportive treatment and antibiotic prophylaxis are
required for typical WAS; however, it is necessary to perform IVIG
and platelet transfusion and splenectomy. As for typical WAS,
hematopoietic stem cell transplantation is the most effective
method for early stage of onset. Without hematopoietic stem cell
transplantation, WAS patients will eventually die because of
infections, bleeding, malignant tumors and other complications
(41). As the patient in the
present study has severe WAS, hematopoietic stem cell
transplantation is the first choice for the treatment; however, his
parents terminated treatment because of economic reasons. To date,
this patient is followed up. As the genetic profile had been
perfectly revealed, the result serve a significant role for family
planning guidance for the parents.
In conclusion, this proband has a typical clinical
phenotype of WAS; however, at Sanger sequencing was insufficient to
determine the pathogenesis, therefore NGS was performed. NGS
indicated whole exon deletion of the WASP gene, and the result was
further confirmed by Sanger sequencing. Although once reported,
whole exon deletion of this gene is very rare. The one reported
case was from Japan and this case is in China, which may indicate
that whole exon deletion is prone to occur in Asians. WAS is a rare
immunodeficiency disease; due to the diversity of WASP gene
mutations, the clinical manifestations are very different, and
therapeutic methods of WAS should be planned according to clinical
severity. The traditional diagnostic approach for WAS is Sanger
sequencing, which is flawed as WASP mutation types are varied and
other genes in addition to WASP could also induce WAS. Therefore,
it is necessary to perform NGS for pathogenic identification and
subsequent family planning guidance. The present study aimed to
identify mutations in other genes beside the WASP gene to
demonstrate the powerful functionality of NGS both for genotyping
and for treatment scheme planning; however, this study identified
only whole exon deletion. However, in line with the gene deletion
type, it is presumed that the prognosis will be very poor, thereby
hematopoietic stem cells transplantation is the first choice for
treatment, which highlights the importance of NGS as a diagnostic
tool.
Acknowledgements
The present study was supported by the Planned
Projects of Hunan Provincial Science and Technology Department
(grant no. 2013FJ6028) and the Scientific Fund of Healthy and
Family Planning Commission of Hunan Province (grant no.
C2013-023).
References
|
1
|
Lemahieu V, Gastier JM and Francke U:
Novel mutations in the Wiskott-Aldrich syndrome protein gene and
their effects on transcriptional, translational and clinical
phenotypes. Hum Mutat. 14:54–66. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Aldrich RA, Steinberg AG and Campbell DC:
Pedigree demonstrating a sex-linked recessive condition
characterized by draining ears, eczematoid dermatitis and bloody
diarrhea. Pediatrics. 13:133–139. 1954.PubMed/NCBI
|
|
3
|
Derry JM, Ochs HD and Francke U: Isolation
of a novel gene mutated in Wiskott-Aldrich syndrome. Cell.
78:635–644. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ochs HD and Thrasher AJ: The
Wiskott-Aldrich syndrome. J Allergy Clin Immunol. 117:725–739.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Blundell MP, Worth A, Bouma G and Thrasher
AJ: The Wiskott-Aldrich syndrome: The actin cytoskeleton and immune
cell function. Dis Markers. 29:157–175. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Westerberg LS, Dahlberg C, Baptista M,
Moran CJ, Detre C, Keszei M, Eston MA, Alt FW, Terhorst C,
Notarangelo LD and Snapper SB: Wiskott-Aldrich syndrome protein
(WASP) and N-WASP are critical for peripheral B-cell development
and function. Blood. 119:3966–3974. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Adriani M, Aoki J, Horai R, Thornton AM,
Konno A, Kirby M, Anderson SM, Siegel RM, Candotti F and
Schwartzberg PL: Impaired in vitro regulatory T cell function
associated with Wiskott-Aldrich syndrome. Clin Immunol. 124:41–48.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wada T, Schurman SH, Garabedian EK, Yachie
A and Candotti F: Analysis of T-cell repertoire diversity in
Wiskott-Aldrich syndrome. Blood. 106:3895–3897. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Maillard MH, Cotta-de-Almeida V, Takeshima
F, Nguyen DD, Michetti P, Nagler C, Bhan AK and Snapper SB: The
Wiskott-Aldrich syndrome protein is required for the function of
CD4(+) CD25(+) Foxp3(+) regulatory T cells. J Exp Med. 204:381–391.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yarar D, To W, Abo A and Welch MD: The
Wiskott-Aldrich syndrome protein directs actin-based motility by
stimulating actin nucleation with the Arp2/3 complex. Curr Biol.
9:555–558. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Davis BR and Candotti F: Revertant somatic
mosaicism in the Wiskott-Aldrich syndrome. Immunol Res. 44:127–131.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ochs HD: Mutations of the Wiskott-Aldrich
syndrome protein affect protein expression and dictate the clinical
phenotypes. Immunol Res. 44:84–88. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Luthi JN, Gandhi MJ and Drachman JG:
X-linked thrombocytopenia caused by a mutation in the
Wiskott-Aldrich syndrome (WAS) gene that disrupts interaction with
the WAS protein (WASP)-interacting protein (WIP). Exp Hematol.
31:150–158. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bender M, Stritt S, Nurden P, van Eeuwijk
JM, Zieger B, Kentouche K, Schulze H, Morbach H, Stegner D, Heinze
KG, et al: Megakaryocyte-specific Profilin1-deficiency alters
microtubule stability and causes a Wiskott-Aldrich syndrome-like
platelet defect. Nat Commun. 5:47462014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sasahara Y: WASP-WIP complex in the
molecular pathogenesis of Wiskott-Aldrich syndrome. Pediatr Int.
58:4–7. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lanzi G, Moratto D, Vairo D, Masneri S,
Delmonte O, Paganini T, Parolini S, Tabellini G, Mazza C, Savoldi
G, et al: A novel primary human immunodeficiency due to deficiency
in the WASP-interacting protein WIP. J Exp Med. 209:29–34. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Fried S, Matalon O, Noy E and Barda-Saad
M: WIP: More than a WASp-interacting protein. J Leukoc Biol.
96:713–727. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ramesh N, Massaad MJ, Kumar L, Koduru S,
Sasahara Y, Anton I, Bhasin M, Libermann T and Geha R: Binding of
the WASP/N-WASP-interacting protein WIP to actin regulates focal
adhesion assembly and adhesion. Mol Cell Biol. 34:2600–2610. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
St-Jean M, Izard T and Sygusch J: A
hydrophobic pocket in the active site of glycolytic aldolase
mediates interactions with Wiskott-Aldrich syndrome protein. J Biol
Chem. 282:14309–14315. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ti SC, Jurgenson CT, Nolen BJ and Pollard
TD: Structural and biochemical characterization of two binding
sites for nucleation-promoting factor WASp-VCA on Arp2/3 complex.
Proc Natl Acad Sci USA. 108:E463–E471. 2011; View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jurgenson CT and Pollard TD: Crystals of
the Arp2/3 complex in two new space groups with structural
information about actin-related protein 2 and potential WASP
binding sites. Acta Crystallogr F Struct Biol Commun. 71:1161–1168.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wada T, Itoh M, Maeba H, Toma T, Niida Y,
Saikawa Y and Yachie A: Intermittent X-linked thrombocytopenia with
a novel WAS gene mutation. Pediatr Blood Cancer. 61:746–748. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Takimoto T, Takada H, Ishimura M, Kirino
M, Hata K, Ohara O, Morio T and Hara T: Wiskott-Aldrich syndrome in
a girl caused by heterozygous WASP mutation and extremely skewed
X-chromosome inactivation: A novel association with maternal
uniparental isodisomy 6. Neonatology. 107:185–190. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Szklarczyk D, Franceschini A, Wyder S,
Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos
A, Tsafou KP, et al: STRING v10: Protein-protein interaction
networks, integrated over the tree of life. Nucleic Acids Res.
43(Database Issue): D447–D452. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li H and Durbin R: Fast and accurate short
read alignment with Burrows-Wheeler transform. Bioinformatics.
25:1754–1760. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wassner AJ, Cohen LE, Hechter E and Dauber
A: Isolated central hypothyroidism in young siblings as a
manifestation of PROP1 deficiency: Clinical impact of whole exome
sequencing. Horm Res Paediatr. 79:379–386. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Thrasher AJ: New insights into the biology
of Wiskott-Aldrich syndrome (WAS). Hematology Am Soc Hematol Educ
Program. 1–138. 2009.
|
|
29
|
Jin Y, Mazza C, Christie JR, Giliani S,
Fiorini M, Mella P, Gandellini F, Stewart DM, Zhu Q, Nelson DL, et
al: Mutations of the Wiskott-Aldrich syndrome protein (WASP):
Hotspots, effect on transcription and translation, and
phenotype/genotype correlation. Blood. 104:4010–4019. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ancliff PJ, Blundell MP, Cory GO, Calle Y,
Worth A, Kempski H, Burns S, Jones GE, Sinclair J, Kinnon C, et al:
Two novel activating mutations in the Wiskott-Aldrich syndrome
protein result in congenital neutropenia. Blood. 108:2182–2189.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Peng F, Nong G, Jiang M, Liu X, Liu H and
Li Y: Clinical features, genotype and phenotype analysis and
literature review of Wiskott-Aldrich syndrome. J Applied Clin
Pediatr. 29:675–679. 2014.
|
|
32
|
Catucci M, Castiello MC, Pala F,
Bosticardo M and Villa A: Autoimmunity in wiskott-Aldrich syndrome:
An unsolved enigma. Front Immunol. 3:2092012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Imai K, Morio T, Zhu Y, Jin Y, Itoh S,
Kajiwara M, Yata J, Mizutani S, Ochs HD and Nonoyama S: Clinical
course of patients with WASP gene mutations. Blood. 103:456–464.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhao Q, Jiang L, Yu J, Xiao J and Zhao X:
Genotype and phenotype correlation of Wiskott-Aldrich syndrome: A
report based on 24 Chinese patients. J Third Mil Med Univ.
33:1404–1407. 2011.
|
|
35
|
Welch MD, DePace AH, Verma S, Iwamatsu A
and Mitchison TJ: The human Arp2/3 complex is composed of
evolutionarily conserved subunits and is localized to cellular
regions of dynamic actin filament assembly. J Cell Biol.
138:375–384. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wu M, Katta A, Gadde MK, Liu H, Kakarla
SK, Fannin J, Paturi S, Arvapalli RK, Rice KM, Wang Y and Blough
ER: Aging-associated dysfunction of Akt/protein kinase B:
S-nitrosylation and acetaminophen intervention. PLoS One.
4:e64302009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Weed SA, Karginov AV, Schafer DA, Weaver
AM, Kinley AW, Cooper JA and Parsons JT: Cortactin localization to
sites of actin assembly in lamellipodia requires interactions with
F-actin and the Arp2/3 complex. J Cell Biol. 151:29–40. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Di Ciano C, Nie Z, Szászi K, Lewis A,
Uruno T, Zhan X, Rotstein OD, Mak A and Kapus A: Osmotic
stress-induced remodeling of the cortical cytoskeleton. Am J
Physiol Cell Physiol. 283:C850–C865. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Latreille M, Laberge MK, Bourret G, Yamani
L and Larose L: Deletion of Nck1 attenuates hepatic ER stress
signaling and improves glucose tolerance and insulin signaling in
liver of obese mice. Am J Physiol Endocrinol Metab. 300:E423–E434.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Qadir MI, Parveen A and Ali M: Cdc42: Role
in cancer management. Chem Biol Drug Des. 86:432–439. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Boztug K, Schmidt M, Schwarzer A, Banerjee
PP, Díez IA, Dewey RA, Böhm M, Nowrouzi A, Ball CR, Glimm H, et al:
Stem-cell gene therapy for the Wiskott-Aldrich syndrome. N Engl J
Med. 363:1918–1927. 2010. View Article : Google Scholar : PubMed/NCBI
|