Introduction
Diabetic cardiomyopathy (DCM), characterized by the
presence of functional and structural abnormalities and not
coronary heart disease, hypertension or other comorbidities, is the
major cause of disability and mortality in patients with diabetes
(1). This indicates that DCM may
be a result of direct myocardial insult, distinguishing it from
structural heart disease due to vascular complications. Identifying
the mechanisms involved in DCM onset and progression is important
for the development of therapeutic strategies to protect against
diabetic heart failure. However, the pathogenesis of DCM remains to
be fully elucidated. Increasing evidence has demonstrated that
advanced glycation end products (AGEs), which are generated at an
increased rate under chronic long-term hyperglycemia conditions,
are a major contributor in the development and progression of DCM
(2–8). Clinical studies have reported that,
in diabetic tissues, AGEs accumulate to levels that are 14-fold
higher compared with normal tissues, while serum levels of AGEs are
associated with the degree of left ventricular function and
myocardial blood flow reserve (2–4).
AGEs increase the production of intracellular reactive oxygen
species (ROS) via the receptor for AGEs (RAGE), which subsequently
activates associated signaling cascades that promote cardiomyocyte
hypertrophy (5), apoptosis
(6) and myocardial fibrosis,
ultimately resulting in diastolic and systolic dysfunction
(7,8). However, the underlying mechanisms
responsible for myocardial injury triggered by AGEs remain to be
fully elucidated.
It is established that profilin-1 is an
evolutionarily conserved actin-binding protein that has an
important function in the regulation of cytoskeleton dynamics by
promoting actin polymerization (9). Profilin-1 also binds to
polyphosphoinositide-based lipids and various proteins with
poly-L-proline motifs, and serves an important role in the
regulation of membrane trafficking, the small-GTPase signaling
pathway, receptor activity and potentially transcriptional
activity, which indicates that profilin-1 may be a central hub that
controls molecular interactions (10,11).
An increasing amount of evidence has demonstrated that upregulated
expression of profilin-1 is associated with endothelial
dysfunction, vascular inflammation and remodeling under
pathological stimulations, including AGEs (12), oxidized low-density lipoprotein
(13) and angiotensin II (14,15),
and is not associated with high glucose (13). Furthermore, profilin-1 may also be
secreted into the extracellular space, where it functions as an
extracellular ligand leading to atherogenic effects and activates
various signaling pathways, including the phosphatidylinositol
3-kinase/Akt and extracellular signal-regulated kinase 1/2 pathways
(16). In addition, it has been
reported that profilin-1 is highly expressed in heart (17), and overexpression of profilin-1
promoted cardiac hypertrophy and contractile dysfunction in
profilin-1 transgenic mice (18,19),
while knockdown of profilin-1 expression in spontaneous
hypertensive rats attenuated cardiac hypertrophy and fibrosis
(18). A number of additional
studies have indicated that the abnormality of the cytoskeleton is
an ultra-early change during left ventricular hypertrophy and
fibrosis, and subsequent heart failure (20,21).
However, whether profilin-1 contributes to AGE-induced cardiac
injury, and the underlying mechanism, remains unclear.
Due to the critical role of profilin-1 in actin
dynamics, cytoskeletal reorganization and pathological vascular and
cardiac hypotrophy, we hypothesize that profilin-1 may be involved
in cardiac injury induced by AGEs, and that attenuated expression
of profilin-1 may ameliorate AGE-induced adverse effects in the
heart.
Materials and methods
Chemicals and reagents
Bovine serum albumin (BSA), D-glucose and rabbit
anti-GAPDH antibody were purchased from Sigma-Aldrich (Merck KGaA,
Darmstadt, Germany). Rabbit anti-profilin-1 and anti-Rho antibodies
were purchased from Abcam (Shanghai, China), mouse anti-RAGE
antibody was purchased from Santa Cruz Biotechnology, Inc. (Dallas,
TX, USA) and rabbit anti-p65 antibody was obtained from Bioworld
Technology, Inc. (St. Louis Park, MN, USA). Blood glucose and total
cholesterol (TC) detection kits were obtained from Nanjing
Jiancheng Bioengineering Institute (Nanjing, China). The
procollagen type III N-terminal peptide (PIIINP) ELISA kit was
purchased from Uscn Life Sciences, Inc. (Wuhan, China). TRIzol
reagent was purchased from Invitrogen (Thermo Fisher Scientific,
Inc., Waltham, MA, USA). The reverse transcription kit was obtained
from Thermo Fisher Scientific, Inc. The QuantiFast SYBR Green PCR
kit was from Qiagen GmbH (Hilden, German). The profilin-1 short
hairpin RNA (shRNA) adenovirus vectors and blank control adenovirus
vectors were designed and synthesized by Hanbio Biotechnology Co.,
Ltd. (Shanghai, China).
Preparation of AGEs
AGEs were prepared according to the protocol
described by Wu et al (22). Briefly, BSA (10 mg/ml) was
incubated in 0.2 M PBS with D-glucose (90 g/l) containing 100 U/ml
penicillin and 100 µg/ml streptomycin in the dark at 37°C for 12
weeks. After 12 weeks incubation, the preparations were dialyzed
three times for 18 h at 4°C against PBS (pH 7.4) each time to
remove free glucose, and were subsequently separated into aliquots
and stored at −20°C prior to use.
Animal protocol
Male (n=30; weight, 150±10 g; age, 1 month)
Sprague-Dawley rats were purchased from Shanghai SLAC Laboratory
Animal Co., Ltd. (Shanghai, China). The rats were raised at an
ambient temperature of 24°C with 12-h light/dark cycle and 50±5%
humidity, and free access to a standard chow diet and water. Rats
were randomly divided into the following four groups (n=6 each):
Control, rats were treated with vehicle saline; AGEs, rats were
treated with AGEs; AGEs + S, rats were treated with AGEs and
profilin-1 shRNA adenovirus vectors; and AGEs + V, rats were
treated with AGEs and blank control adenovirus vectors. For
transfection efficiency detection, the remaining 6 rats were
randomly divided into two groups (n=3 each) for 4 weeks treatment:
Ad-vector group, rats were treated with blank control adenovirus
vector; Ad-profilin-1 shRNA group, rats were treated with
adenovirus vector containing profilin-1 shRNA. AGEs (50 mg/kg/day)
was administered by tail vein injection for 8 weeks, with the same
volume of saline injected as a control. Ad-profilin-1 shRNA or
blank control vector was injected twice by tail vein at a dose of
3×109 plaque forming units with an interval of every 4
weeks, beginning with the initial AGEs injection. The experimental
procedures and protocols performed in the present study were
approved by the Medicine Animal Welfare Committee of Xiangya
Hospital, Central South University (Changsha, China), conforming
with the National Institutes of Health Guide for the Care and Use
of Laboratory Animals (NIH publication 85–23, revised 1996)
(23).
Echocardiography
The cardiac contractile function in vivo was
determined after 8 weeks of daily administration of AGEs (50
mg/kg/day) using a BL-420E Data Acquisition and Analysis system
(Chengdu TME Technology Co., Ltd., Chengdu, China). Following
anesthesia with pentobarbital sodium (60 mg/kg; intraperitoneal),
all rats were examined and, using the left ventricular long axis
view, the following parameters were measured: Left ventricular
ejection fraction (LVEF) and left ventricular fractional shortening
(LVFS).
Preparation of serum and tissue
samples
After 8 weeks of daily administration of AGEs (50
mg/kg/day), 800 µl blood samples were collected and centrifuged for
10 min at 4°C and 3,000 × g. Supernatants were stored at −80°C
until the analysis of biochemistry parameters. The whole heart was
harvested immediately following blood sample selection, the atrium
and right ventricle were removed, and left ventricle samples were
divided into the following three groups: One group was fixed with
4% paraformaldehyde at room temperature for 1 week and embedded in
paraffin. Paraffin sections were used for Masson's trichrome
staining and immunohistochemistry; the second group was homogenized
by TRIzol reagent for reverse transcription-quantitative polymerase
chain reaction (RT-qPCR) analysis; the final group was homogenized
in radioimmunoprecipitation assay lysis buffer (P0013B; Beyotime
Institute of Biotechnology, Beijing, China) and 0.1 mmol/l
phenylmethylsulfonyl fluoride (ST506; Beyotime Institute of
Biotechnology) for western blot analysis.
Measurement of metabolic parameters
and PIIINP
Metabolic parameters, including blood glucose and
TC, were detected using relevant kits. The serum levels of PIIINP
were detected using an ELISA kit (cat. no. SEA573Ra), according to
the manufacturer's instructions.
Protein preparation and western
blotting
Total protein from left ventricle tissue samples was
extracted, as described above for the preparation of serum and
tissue samples, and measured using a BCA protein assay kit.
Following heating at 95°C for 5 min, 20 µg/sample lysates were
separated by 10 or 12% SDS-PAGE and transferred to polyvinylidene
fluoride membranes. The membranes were blocked with 5% fat-free
milk in TBST buffer (0.1% Tween-20 in TBS) for 90 min at room
temperature, and subsequently incubated with primary antibodies
against profilin-1 (cat. no. ab124904, 1:1,500 dilution), p65 (cat.
no. BS1253, 1:1,000 dilution), RAGE (cat. no. sc-365154, 1:600
dilution), Rho (cat. no. ab40673, 1:1,000 dilution) and GAPDH (cat.
no. G9545, 1:5,000 dilution) at 4°C overnight. Following primary
antibody incubation, membranes were subsequently washed and
incubated with horseradish peroxidase-conjugated goat anti-mouse
(cat. no. CW0102, 1:5,000 dilution; CW Biotech, Co., Ltd., Beijing,
China) or anti-rabbit IgG (cat. no. CW0103, 1:5,000 dilution; CW
Biotech) secondary antibodies for 1 h at room temperature. Potent
ECL kit (cat. no. 70-P1425; MultiSciences Biotech Co., Ltd.,
Hangzhou, China) was used to visualize proteins. The signal was
detected and ratios of the target protein against the GAPDH control
were calculated using the Image Lab software (version 5.2.1;
Bio-Rad Laboratories, Inc., Hercules, CA, USA).
RNA isolation and RT-qPCR
Total RNA was extracted from left ventricle tissue
samples using TRIzol reagent, according to the manufacturer's
instructions. The concentration and purity of isolated RNA was
subsequently detected. cDNA was generated from 1 µg total RNA using
a high-capacity cDNA reverse transcription kit (4368814; Thermo
Fisher Scientific, Inc.) following the manufacturer's instructions.
Reverse transcription was performed as follows: 25°C for 10 min,
37°C for 2 h, 85°C for 5 min and stored at 4°C. GAPDH primers were
purchased from Sangon Biotech Co., Ltd. (Shanghai, China) and all
other primers were synthesized and purified by Sangon Biotech Co.,
Ltd., as listed in Table I. cDNA
was quantified using a QuantiFast SYBR Green PCR kit and qPCR was
performed using a 7500 Fast Real-Time PCR System (Applied
Biosystems; Thermo Fisher Scientific, Inc.). Briefly, the
amplification was performed with an initial step at 95°C for 5 min,
followed by 40 cycles of denaturation at 95°C for 10 sec, annealing
at 60°C for 30 sec and extension at 60°C for 30 sec for each target
gene. All amplification reactions for each sample were performed in
triplicate and the results were expressed as the ratio of target
genes to GAPDH mRNA using the 2−ΔΔCq method (24).
 | Table I.Sequences of reverse
transcription-quantitative polymerase chain reaction primers. |
Table I.
Sequences of reverse
transcription-quantitative polymerase chain reaction primers.
| Gene name | Forward primer
(5′-3′) | Reverse primer
(5′-3′) |
|---|
| Atrial natriuretic
peptide |
CTCCGATAGATCTGCCCTCTTGAA |
GGTACCGGAAGCTGTTGCAGCCTA |
| MMP-2 |
GAAAGGTGCTGACCGTATCC |
CCAGTGCCCTCCTAAGACAG |
| MMP-9 |
GCCGACTTATGTGGTCTTCC |
TGCCCGAGTGTAACCATAGC |
| Profilin-1 |
CTGATGGGCAAAGAAGGTC |
GGGAAGGGACAGATGAGGTC |
| β-myosin heavy
chain |
CCAGAAGCCTCGAAATGTC |
CTTTCTTTGCCTTGCCTTTGC |
| GAPDH |
CCATGTTCGTCATGGGTGTGAACCA |
GCCAGTAGAGGCAGGGATGATGTTC |
Histology and Immunohistochemistry
analysis
Paraformaldehyde-fixed paraffin sections were cut
from the left ventricle at a thickness of 4 µm and were stained
using a Masson's trichrome stain kit (BSBA-4079A; Zhongshan Golden
Bridge Biotechnology Co., Beijing, China), according to the
manufacturer's instructions, to evaluate ventricular remodeling.
Immunohistochemistry was performed as described previously
(14). Briefly, left ventricle
sections, at a thickness of 4 µm, were incubated overnight at 4°C
with the primary antibody against profilin-1 (1:200 dilution), and
subsequently incubated with a biotin conjugated goat anti-rabbit
immunoglobulin-G secondary antibody (cat. no. ZB-2010; Zhongshan
Golden Bridge Biotechnology Co., Beijing, China) at a 1:1,000
dilution at 37°C for 20 min. Following washing in PBS, sections
were developed using 3,3′-diaminobenzidine tetrahydrochloride
obtained from Zhongsha Golden Bridge Biotechnology Co. (cat. no.
ZLI-9018) and counterstained with 50% hematoxylin for 15 min at
room temperature. Subsequently, sections were mounted with neutral
balsam. Masson's trichrome stain and immunohistochemistry pictures
were visualized under a light microscope (magnification, ×200;
Nikon Corporation, Tokyo, Japan).
Statistical analysis
Data are presented as the mean ± standard deviation.
Statistical analysis was performed using SPSS 17.0 software (SPSS,
Inc., Chicago, IL, USA) either by unpaired student's t-test for two
groups or one-way analysis of variance followed by Newman-Keuls
post-hoc test for multiple groups. P<0.05 was considered to
indicate a statistically significant difference.
Results
Characteristics of the exogenous AGE
infusion rat model
As presented in Table
II, no significant differences were observed in the body
weight, blood glucose and TC of different groups of rats following
8 weeks of treatment.
| Table II.Biochemical characteristics of rats
following various treatments. |
Table II.
Biochemical characteristics of rats
following various treatments.
|
| Control | AGEs | AGEs + S | AGEs + V |
|---|
| Body weight, g | 552.2±1.7 | 554.5±4.6 | 554.3±3.3 | 553.3±2.7 |
| Glucose,
mmol/l |
3.9±0.6 |
4.4±0.5 |
4.0±0.6 |
4.1±0.6 |
| Total cholesterol,
mmol/l |
3.1±0.2 |
3.6±0.8 |
3.2±0.5 |
3.3±0.5 |
Chronic injection of AGEs causes
cardiac contractile dysfunction, hypertrophy and fibrosis, and
increases the expression of profilin-1
To determine whether chronic injection of AGEs has
an effect on cardiac injury, the present study focused on the
primary characteristics of DCM, including cardiac function,
fibrosis and hypertrophy. Compared with the control group,
echocardiography demonstrated that left ventricle systolic
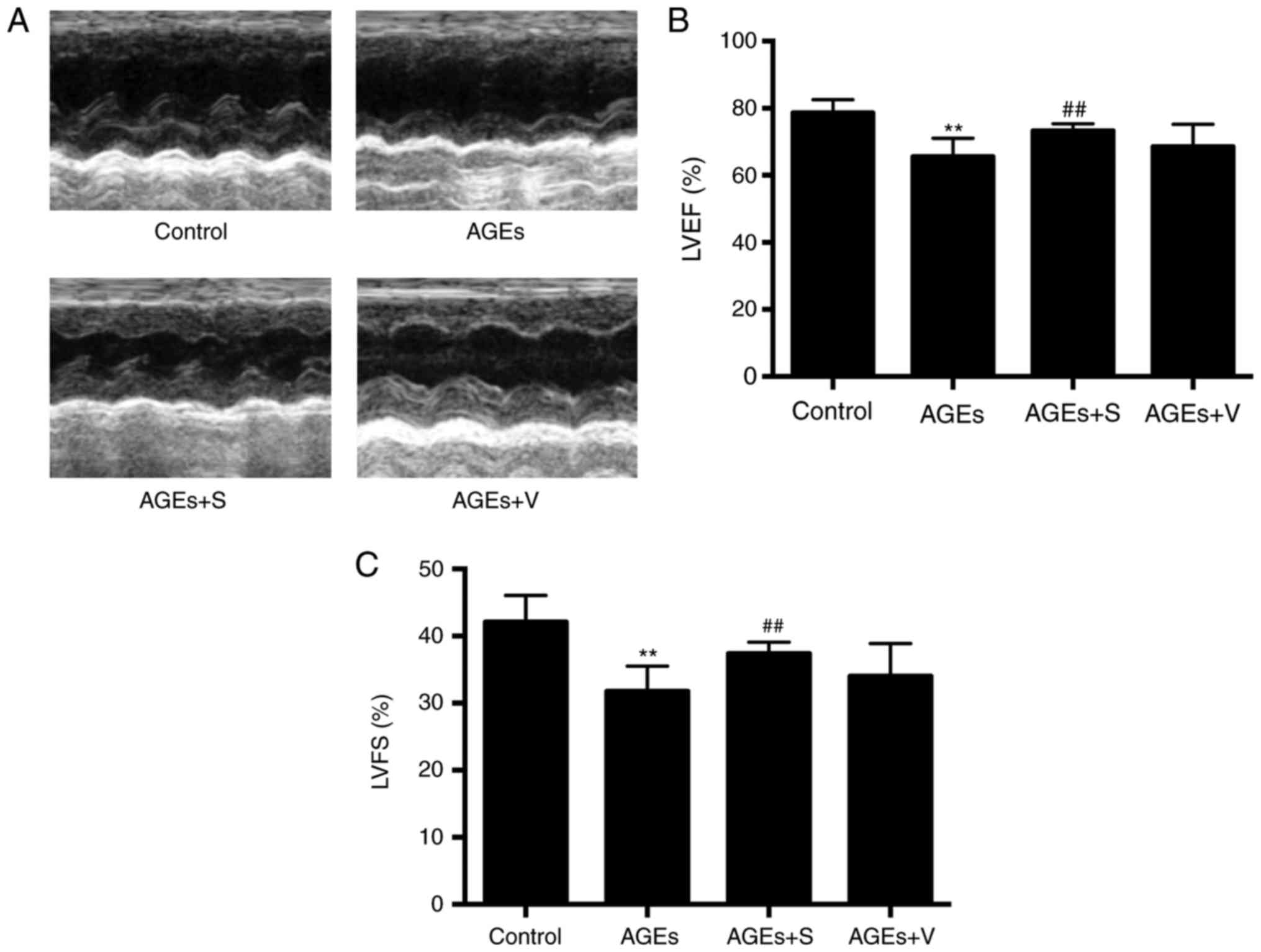
function, including LVEF and LVFS, was significantly decreased in
the AGEs group (Fig. 1). In
addition, the AGEs group developed cardiac fibrosis, as evidenced
by markedly increased collagen deposition in the heart tissue
sections, particularly around vessels, indicated by blue color in
Masson's trichrome staining (Fig.
2A). Furthermore, PIIINP expression in the serum, and MMP-2 and
−9 mRNA expression in left ventricle tissues, was significantly
increased in the AGEs group compared with the control (Fig. 2B-D). As demonstrated in Fig. 2E and F, AGEs significantly
increased the expression of atrial natriuretic peptide (ANP) and
β-myosin heavy chain (β-MHC) compared with the control, which
indicates cardiac hypertrophy. As expected, chronic delivery of
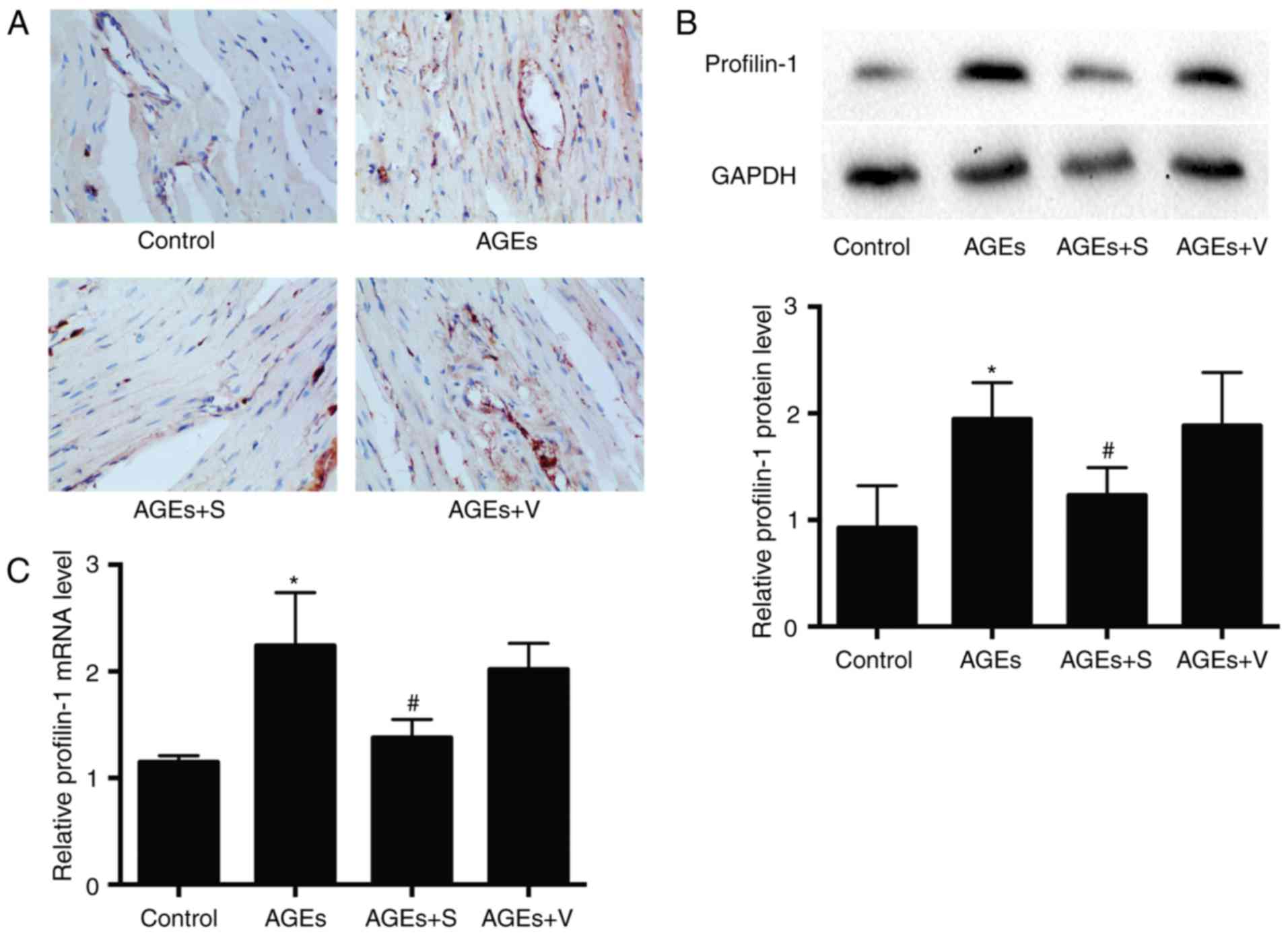
AGEs significantly increased the expression of profilin-1 compared
with the control group, as determined by immunohistochemistry,
western blot analysis and RT-qPCR (Fig. 3). No marked differences were
observed between AGEs and AGEs + V groups (Figs. 1–3).
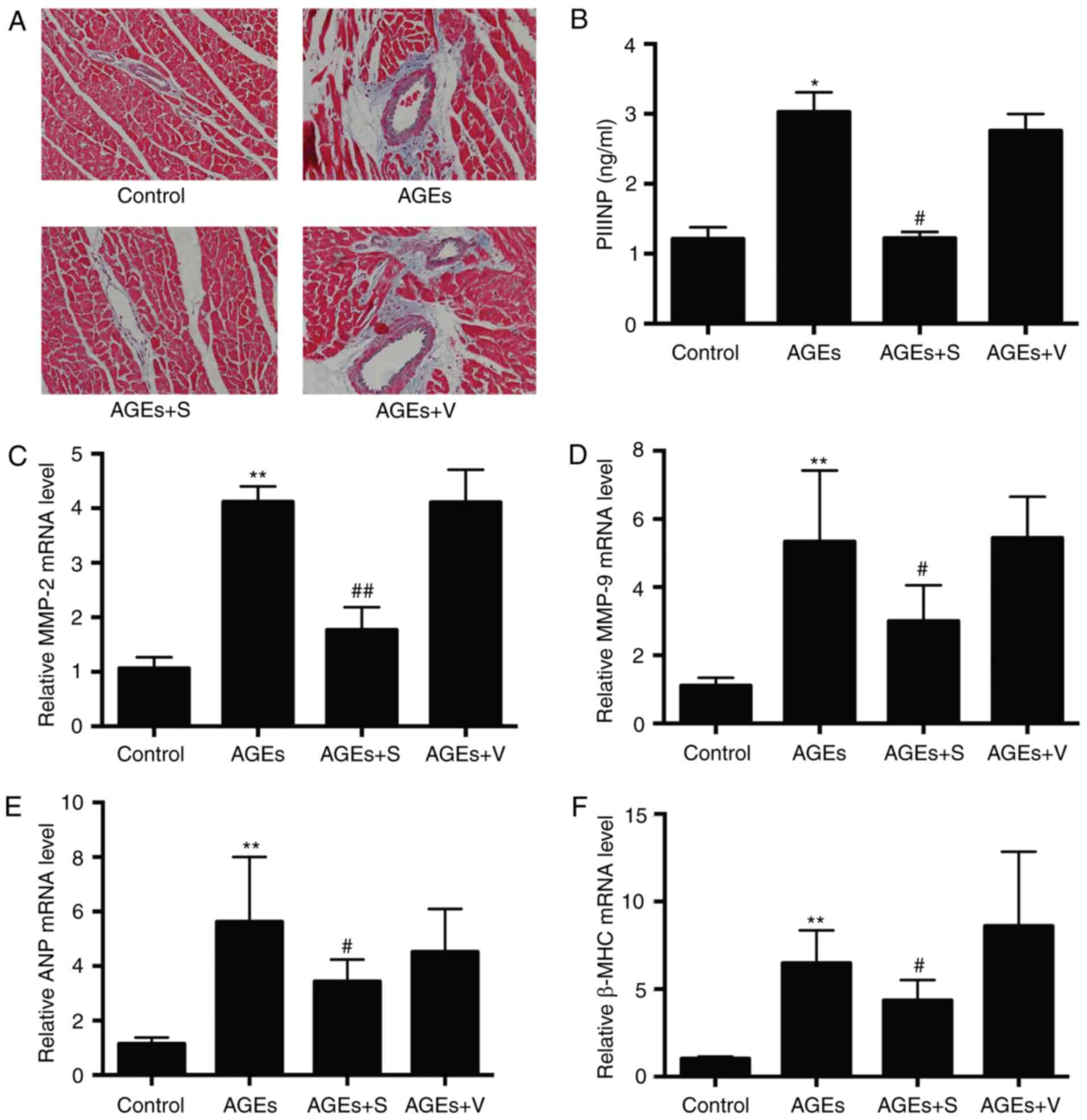 | Figure 2.The effect of profilin-1 shRNA
adenovirus on cardiac remodeling induced by chronic delivery of
AGEs. (A) Representative Masson's trichrome staining images
indicate the fibrotic areas in left ventricles of hearts.
Magnification, ×200. (B) Expression of PIIINP in the serum. mRNA
expression of (C) MMP-2, (D) MMP-9, (E) ANP and (F) β-MHC in left
ventricle heart tissues. Data are presented as the mean ± standard
deviation, n=6/group. *P<0.05 and **P<0.01 vs. control;
#P<0.05 and ##P<0.01 vs. AGEs-only
group. shRNA, short hairpin RNA; AGEs, advanced glycation end
products; PIIINP, procollagen type III N-terminal peptide; MMP,
matrix metalloproteinase; ANP, atrial natriuretic peptide; β-MHC,
β-myosin heavy chain; control, rats treated with vehicle saline;
AGEs, rats treated with AGEs; AGEs + S, rats treated with AGEs +
profilin-1 shRNA adenovirus; AGEs + V, rats treated with AGEs +
blank control adenovirus vectors. |
Downregulation of profilin-1
expression attenuates AGE-induced cardiac dysfunction, hypertrophy
and fibrosis
To investigate the role of profilin-1 in AGE-induced
cardiac dysfunction, hypertrophy and fibrosis, the present study
knocked down profilin-1 expression by intravenous delivery of
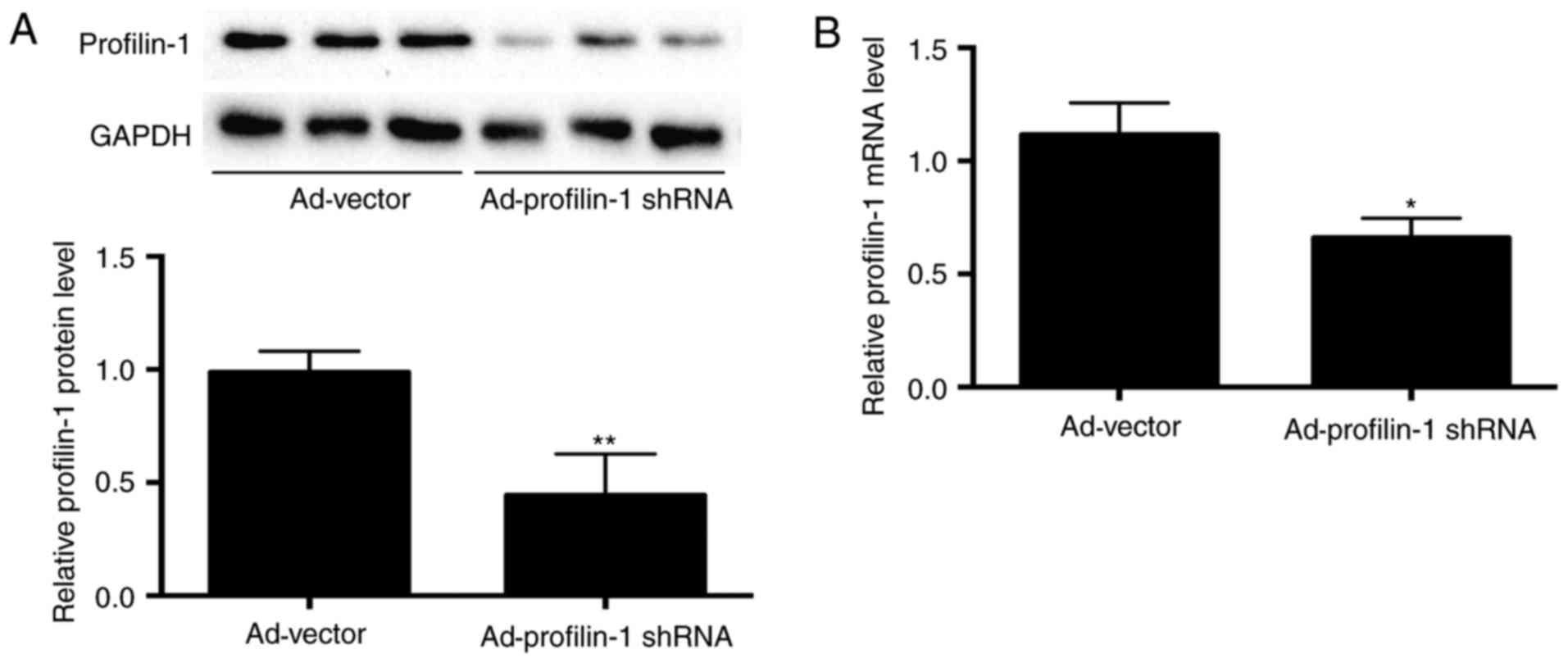
adenovirus expressing profilin-1 shRNA. On day 6 after the second
injection of adenovirus, 50 and 40% decreases in profilin-1
expression at protein and mRNA levels were observed, respectively
(Fig. 4A and B). Knockdown of
profilin-1 expression improved cardiac systolic function (Fig. 1) and reduced cardiac fibrosis, as
indicated by reduced collagen deposition (Fig. 2A) and reduced PIIINP, MMP-2 and
MMP-9 expression (Fig. 2B-D),
compared with the AGEs-only group. In addition, silencing
profilin-1 expression significantly inhibited AGE-induced cardiac
hypertrophy, as indicated by reduced ANP and β-MHC expression
(Fig. 2E and F) compared with the
AGEs-only group.
Nuclear factor (NF)-κB and Rho
signaling pathways are involved in AGE-induced cardiac injury
Increasing evidence has indicated that AGEs activate
various signaling pathways, including NF-κB and Rho/Rho-associated
protein kinase (ROCK), and induces the expression of genes
associated with diabetic complications by binding to RAGE (2,25,26).
Due to the important role of the NF-κB signaling pathway in
regulating oxidative stress, fibrosis, hypertrophy and apoptosis,
and as the Rho/ROCK pathway in the actin cytoskeleton are reported
to be involved DCM, p65 and Rho were selected for further
investigation in the present study. Following chronic treatment
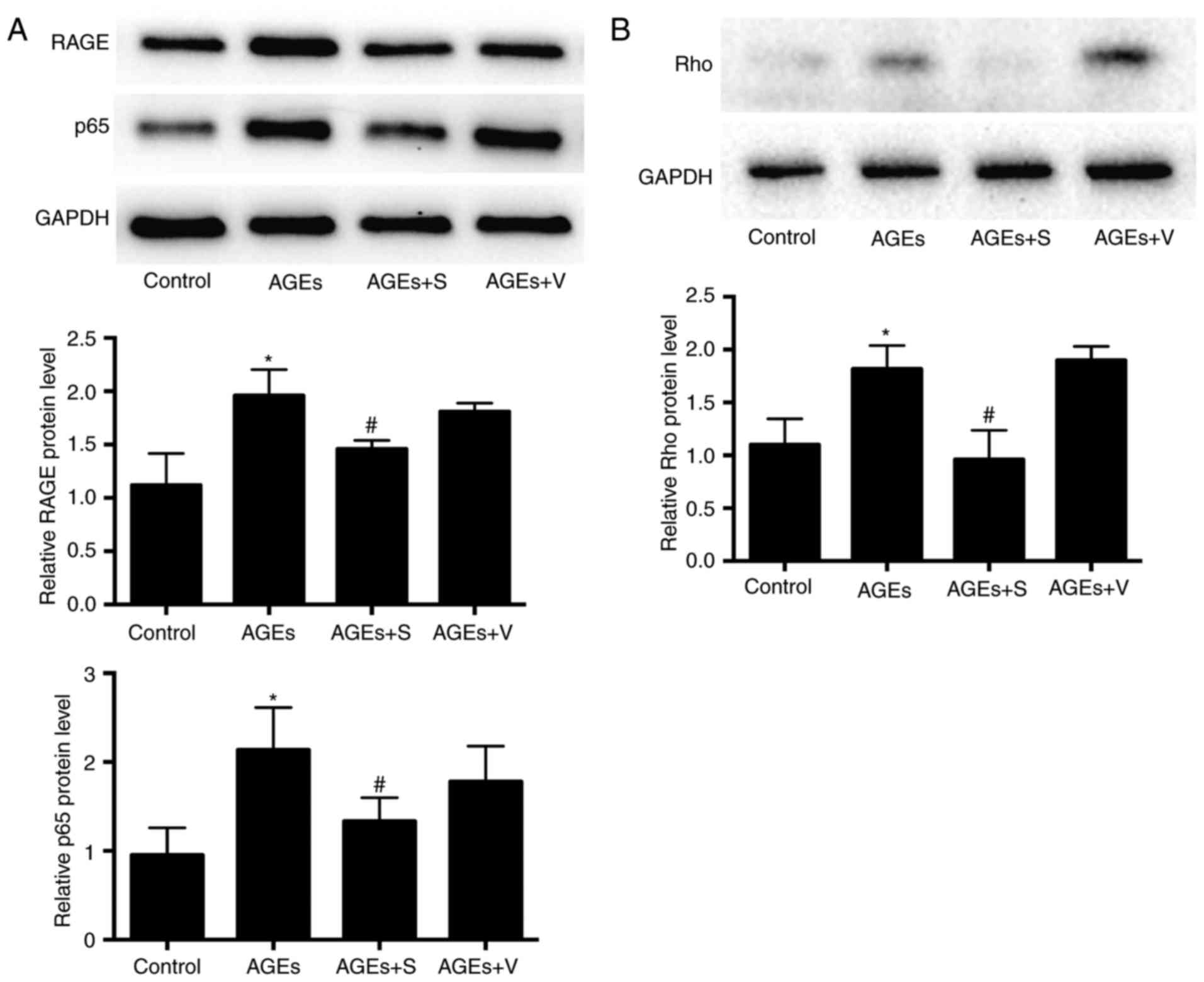
with AGEs for 8 weeks, a significant increase in the expression of
RAGE, p65 and Rho at the protein level was observed in left
ventricle heart tissues, compared with the control group (Fig. 5). Notably, blockade of profilin-1
significantly reduced the expression of RAGE, p65 and Rho compared
with the AGEs-only group (Fig.
5).
Discussion
The primary findings of the present study were as
follows: Chronic injection of AGEs markedly upregulated the
expression of profilin-1 in vivo, which was associated with
abnormal cardiac structure and function, potentially via the
activation of NF-κB and Rho signaling pathways; and knocking down
the expression of profilin-1 attenuated AGE-induced myocardial
injury, including cardiac dysfunction, hypertrophy and fibrosis.
These results indicate that profilin-1 may represent a crucial
mediator in AGE-induced cardiac injury, which may be developed as a
potential therapeutic target for patients with diabetes to protect
against heart failure.
Cardiac hypertrophy and fibrosis are the major
features of DCM, and result in ventricular dysfunction and heart
failure (27,28). Evidence has indicated that
increased and accelerated formation of AGEs has emerged as an
important contributor to the onset and development of cardiac
hypertrophy and fibrosis (2).
Thus, identifying the mechanisms mediated by AGEs is essential for
the development of novel therapeutic targets for the treatment of
DCM. It was previously reported that intraperitoneal or tail vein
injection of AGEs caused vascular impairment and remodeling
(29,30), and ~70% of the injected
125I-AGE irreversibly bound to heart muscle tissues
(30). Therefore, we hypothesized
that increasing the circulating concentration of AGEs by tail vein
injection may mimic myocardial injury in vivo. The results
of the present study demonstrated that chronic injection of
exogenous AGEs for 8 weeks markedly decreased cardiac contractile
function, and induced cardiac hypotrophy and fibrosis (Figs. 1 and 2). However, no alterations in the
circulating levels of glucose and lipids were observed (Table II), which indicates that AGEs may
have an independent effect on myocardium injury, in the absence of
hyperglycemia and hyperlipidemia.
It is established that profilin-1 is a highly
conserved actin binding protein that has a prominent role in
various cellular processes (10).
Increasing evidence indicates that, in addition to regulating actin
polymerization, profilin-1 is an important molecule associated with
pathological hypertrophy and fibrosis. It was previously reported
that profilin-1 was upregulated 2–3-fold in hypertrophic vascular
tissues and ventricular cardiomyocytes (17,18),
and overexpression of profilin-1 directly caused vascular and
cardiac hypertrophy and fibrosis by activating mitogen-activated
protein kinase or extracellular signal-regulated kinase 1/2
signaling pathways (17). By
contrast, suppression of profilin-1 expression conferred protection
against hypertrophic insult. Notably, profilin-1
overexpression-associated ventricular hypertrophy is not only
observed in the ultra-early stage (21), but also at later stages (19), which indicates that profilin-1 may
function as an important regulator throughout the progression of
the pathological cardiac hypertrophy. In the present study, the
results demonstrated that chronic injection of AGEs significantly
upregulated the mRNA and protein expression of profilin-1 in the
heart, which was accompanied by cardiac hypertrophy, fibrosis and
impaired heart function. In addition, AGE-induced cardiac fibrosis
was particularly prominent in the area surrounding vessels, which
is a feature of both human patients and animal models of DCM
(27). However, silencing
profilin-1 expression markedly attenuated the observed AGE-induced
cardiac injury.
Previous studies have provided evidence that
demonstrates that the activation of NF-κB and Rho/ROCK signaling
pathways in the heart contributes to the development of DCM, and
targeting NF-κB and Rho/ROCK signaling may improve diabetic cardiac
function in patients with diabetes (31–35).
The results of the present study demonstrated that chronic
intravenous delivery of exogenous AGEs increased the protein
expression of p65 and Rho in heart tissue, and increased protein
expression of RAGE was also observed, compared with the control
group. Our previous study indicated that profilin-1 may function as
a common cellular molecule, and may be downstream of protein kinase
C or NF-κB signaling pathways mediated by AGEs in endothelial cells
(12). Therefore, we hypothesized
that AGEs may activate NF-κB and Rho signaling pathways via RAGE,
which results in increased profilin-1 expression. Notably, in the
present study, inhibition of profilin-1 expression decreased the
protein expression of p65 and Rho, and ameliorated cardiac injury,
which indicates a potential positive feedback loop between
profilin-1 and p65, and profilin-1 and Rho. Previous studies have
demonstrated that in diabetic cardiomyocytes, actin polymerization
is significantly increased (32),
and the actin cytoskeleton has an important role in sustaining ROS
production (31). Therefore, it
may be inferred that increased profilin-1 expression induced by
AGEs may cause accumulated actin polymerization and, in turn,
excess ROS production, which subsequently activates NF-κB and Rho
to form a positive loop.
In conclusion, the present study demonstrates that
chronic delivery of AGEs promoted cardiac fibrosis and hypertrophy,
impaired systolic function and upregulated profilin-1 expression,
which may occur via NF-κB or Rho signaling pathways. Reducing
profilin-1 expression may aid the prevention of heart injuries in
patients with diabetes. Therefore, targeting AGE-induced profilin-1
expression may represent a novel therapeutic target to treat
diabetic heart failure.
Acknowledgements
This study was supported by the Central South
University Innovation Foundation for postgraduates (grant no.
2015zzts111, awarded to Dr Dafeng Yang), the National Natural
Science Foundation of China (grant no. 81000140, awarded to Dr
Meifang Chen; grant nos. 81570320 and 81570334, awarded to Dr
Tianlun Yang) and the Xiangya Eminent Doctor Project (grant no.
013, awarded to Dr Tianlun Yang). The authors thank Dr Xinghui Sun
(Department of Biochemistry, University of Nebraska, Lincoln, NE,
USA) and Dr Shao Liu (Department of Pharmacy, Xiangya Hospital,
Central South University, Changsha, China) for their assistance
with revision of the manuscript.
References
|
1
|
Miki T, Yuda S, Kouzu H and Miura T:
Diabetic cardiomyopathy: Pathophysiology and clinical features.
Heart Fail Rev. 18:149–166. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bodiga VL, Eda SR and Bodiga S: Advanced
glycation end products: Role in pathology of diabetic
cardiomyopathy. Heart Fail Rev. 19:49–63. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sveen KA, Nerdrum T, Hanssen KF, Brekke M,
Torjesen PA, Strauch CM, Sell DR, Monnier VM, Dahl-Jørgensen K and
Steine K: Impaired left ventricular function and myocardial blood
flow reserve in patients with long-term type 1 diabetes and no
significant coronary artery disease: Associations with protein
glycation. Diab Vasc Dis Res. 11:84–91. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cooper ME: Importance of advanced
glycation end products in diabetes-associated cardiovascular and
renal disease. Am J Hypertens. 17:31S–38S. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ko SY, Lin IH, Shieh TM, Ko HA, Chen HI,
Chi TC, Chang SS and Hsu YC: Cell hypertrophy and MEK/ERK
phosphorylation are regulated by glyceraldehyde-derived AGEs in
cardiomyocyte H9c2 cells. Cell Biochem Biophys. 66:537–544. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li SY, Sigmon VK, Babcock SA and Ren J:
Advanced glycation endproduct induces ROS accumulation, apoptosis,
MAP kinase activation and nuclear O-GlcNAcylation in human cardiac
myocytes. Life Sci. 80:1051–1056. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Guo R, Liu W, Liu B, Zhang B, Li W and Xu
Y: SIRT1 suppresses cardiomyocyte apoptosis in diabetic
cardiomyopathy: An insight into endoplasmic reticulum stress
response mechanism. Int J Cardiol. 191:36–45. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Brouwers O, de Vos-Houben JM, Niessen PM,
Miyata T, van Nieuwenhoven F, Janssen BJ, Hageman G, Stehouwer CD
and Schalkwijk CG: Mild oxidative damage in the diabetic rat heart
is attenuated by glyoxalase-1 overexpression. Int J Mol Sci.
14:15724–15739. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pernier J, Shekhar S, Jegou A, Guichard B
and Carlier MF: Profilin interaction with actin filament barbed end
controls dynamic instability, capping, branching, and motility. Dev
Cell. 36:201–214. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Witke W: The role of profilin complexes in
cell motility and other cellular processes. Trends Cell Biol.
14:461–469. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jockusch BM, Murk K and Rothkegel M: The
profile of profilins. Rev Physiol Biochem Pharmacol. 159:131–149.
2007.PubMed/NCBI
|
|
12
|
Li Z, Zhong Q, Yang T, Xie X and Chen M:
The role of profilin-1 in endothelial cell injury induced by
advanced glycation end products (AGEs). Cardiovasc Diabetol.
12:1412013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Romeo G, Frangioni JV and Kazlauskas A:
Profilin acts downstream of LDL to mediate diabetic endothelial
cell dysfunction. FASEB J. 18:725–727. 2004.PubMed/NCBI
|
|
14
|
Cheng JF, Ni GH, Chen MF, Li YJ, Wang YJ,
Wang CL, Yuan Q, Shi RZ, Hu CP and Yang TL: Involvement of
profilin-1 in angiotensin II-induced vascular smooth muscle cell
proliferation. Vascul Pharmacol. 55:34–41. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jin HY, Song B, Oudit GY, Davidge ST, Yu
HM, Jiang YY, Gao PJ, Zhu DL, Ning G, Kassiri Z, et al: ACE2
deficiency enhances angiotensin II-mediated aortic profilin-1
expression, inflammation and peroxynitrite production. PLoS One.
7:e385022012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Caglayan E, Romeo GR, Kappert K, Odenthal
M, Südkamp M, Body SC, Shernan SK, Hackbusch D, Vantler M,
Kazlauskas A and Rosenkranz S: Profilin-1 is expressed in human
atherosclerotic plaques and induces atherogenic effects on vascular
smooth muscle cells. PLoS One. 5:e136082010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kooij V, Viswanathan MC, Lee DI, Rainer
PP, Schmidt W, Kronert WA, Harding SE, Kass DA, Bernstein SI, Van
Eyk JE and Cammarato A: Profilin modulates sarcomeric organization
and mediates cardiomyocyte hypertrophy. Cardiovasc Res.
110:238–248. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhao SH, Qiu J, Wang Y, Ji X, Liu XJ, You
BA, Sheng YP, Li X and Gao HQ: Profilin-1 promotes the development
of hypertension-induced cardiac hypertrophy. J Hypertens.
31:576–586. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Elnakish MT, Hassanain HH and Janssen PM:
Vascular remodeling-associated hypertension leads to left
ventricular hypertrophy and contractile dysfunction in profilin-1
transgenic mice. J Cardiovasc Pharmacol. 60:544–552. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hein S, Kostin S, Heling A, Maeno Y and
Schaper J: The role of the cytoskeleton in heart failure.
Cardiovasc Res. 45:273–278. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhao SH, Gao HQ, Ji X, Wang Y, Liu XJ, You
BA, Cui XP and Qiu J: Effect of ouabain on myocardial
ultrastructure and cytoskeleton during the development of
ventricular hypertrophy. Heart Vessels. 28:101–113. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wu S, Song T, Zhou S, Liu Y, Chen G, Huang
N and Liu L: Involvement of Na+/H+ exchanger
1 in advanced glycation end products-induced proliferation of
vascular smooth muscle cell. Biochem Biophys Res Commun.
375:384–389. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
National Institutes of Health Guide for
the Care and Use of Laboratory Animals. National Academies Press.
85-23 revised. Washington, DC: 1996, https://grants.nih.gov/grants/olaw/guide-for-the-care-and-use-of-laboratory-animals.pdf
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Nielsen JM, Kristiansen SB, Nørregaard R,
Andersen CL, Denner L, Nielsen TT, Flyvbjerg A and Bøtker HE:
Blockage of receptor for advanced glycation end products prevents
development of cardiac dysfunction in db/db type 2 diabetic mice.
Eur J Heart Fail. 11:638–647. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ma H, Li SY, Xu P, Babcock SA, Dolence EK,
Brownlee M, Li J and Ren J: Advanced glycation endproduct (AGE)
accumulation and AGE receptor (RAGE) up-regulation contribute to
the onset of diabetic cardiomyopathy. J Cell Mol Med. 13:1751–1764.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Russo I and Frangogiannis NG:
Diabetes-associated cardiac fibrosis: Cellular effectors, molecular
mechanisms and therapeutic opportunities. J Mol Cell Cardiol.
90:84–93. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bando YK and Murohara T: Diabetes-related
heart failure. Circ J. 78:576–583. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Soro-Paavonen A, Zhang WZ, Venardos K,
Coughlan MT, Harris E, Tong DC, Brasacchio D, Paavonen K,
Chin-Dusting J, Cooper ME, et al: Advanced glycation end-products
induce vascular dysfunction via resistance to nitric oxide and
suppression of endothelial nitric oxide synthase. J Hypertens.
28:780–788. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Vlassara H, Fuh H, Makita Z, Krungkrai S,
Cerami A and Bucala R: Exogenous advanced glycosylation end
products induce complex vascular dysfunction in normal animals: A
model for diabetic and aging complications. Proc Natl Acad Sci USA.
89:12043–12047. 1992; View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Soliman H, Gador A, Lu YH, Lin G, Bankar G
and MacLeod KM: Diabetes-induced increased oxidative stress in
cardiomyocytes is sustained by a positive feedback loop involving
Rho kinase and PKCβ2. Am J Physiol Heart Circ Physiol.
303:H989–H1000. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lin G, Craig GP, Zhang L, Yuen VG, Allard
M, McNeill JH and MacLeod KM: Acute inhibition of Rho-kinase
improves cardiac contractile function in streptozotocin-diabetic
rats. Cardiovasc Res. 75:51–58. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhou H, Li YJ, Wang M, Zhang LH, Guo BY,
Zhao ZS, Meng FL, Deng YG and Wang RY: Involvement of RhoA/ROCK in
myocardial fibrosis in a rat model of type 2 diabetes. Acta
Pharmacol Sin. 32:999–1008. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lorenzo O, Picatoste B, Ares-Carrasco S,
Ramírez E, Egido J and Tuñón J: Potential role of nuclear factor κB
in diabetic cardiomyopathy. Mediators Inflamm. 2011:6520972011.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Thomas CM, Yong QC, Rosa RM, Seqqat R,
Gopal S, Casarini DE, Jones WK, Gupta S, Baker KM and Kumar R:
Cardiac-specific suppression of NF-κB signaling prevents diabetic
cardiomyopathy via inhibition of the renin-angiotensin system. Am J
Physiol Heart Circ Physiol. 307:H1036–H1045. 2014. View Article : Google Scholar : PubMed/NCBI
|