Introduction
Endometrial carcinoma is the most common malignancy
of the female genital tract and is the fourth most common
malignancy among women worldwide (1). Endometrial adenocarcinoma (EAC) is a
type I endometrial carcinoma, which accounts for ~80% of cases, and
is often associated with obesity, estrogen stimulation and
unfavorable prognosis (2).
Numerous critical genetic events, including microsatellite
instability and alterations in the phosphatase and tensin homolog
signaling pathway, have been revealed to serve essential roles in
EAC progression; however, the precise molecular mechanisms
underlying EAC progression remain unclear (2,3).
The Wnt signaling pathway is a major regulator of
cell proliferation, migration and differentiation, embryogenesis,
adult tissue homeostasis and tumor progression (4). Aberrant activation of the
Wnt/β-catenin signaling pathway contributes to the progression of
numerous major human cancers (5).
Wnt inhibitory factor 1 (WIF1) binds directly to extracellular Wnt
ligands, preventing receptor interactions and inducing β-catenin
degradation. Downregulation of WIF1 has been detected in human
prostate, breast, lung and bladder cancers. Recently, WIF1
silencing caused by promoter hypermethylation was reported in
gastrointestinal, lung and bladder cancers (6–8). In
addition, Wnt pathway activation has been revealed to be involved
in EAC (9); however, to the best
our knowledge, the expression and precise role of WIF1 in EAC
progression have yet to be determined.
The present study aimed to investigate WIF1 gene
promoter methylation, WIF1 mRNA and protein expression, and the
association between WIF1 and the prognosis of patients with EAC. An
EAC cell line was also treated with a demethylating agent to
determine whether demethylation was able to restore WIF1
expression. The role of the WIF1 gene in cell proliferation and
tumor growth of the EAC cell line, KLE, and subsequently, its
effects on the expression of proteins in the non-canonical Wnt
pathway, including c-Myc and extracellular signal-regulated kinase
(ERK), were also evaluated.
Materials and methods
Tissue samples
A total of 106 EAC samples (age, 59.2±10.9 years),
acquired previously by the Department of Obstetrics and Gynecology,
The Second Hospital of Shandong University (Jinan, China) between
January 2006 and June 2008, were provided for use in the present
study. In addition, 50 normal endometrial tissue specimens (age,
40.2±7.3 years) were obtained from women who underwent hysterectomy
or endometrial curettage for non-EAC-associated diseases between
January 2006 and June 2008, such as uterine leiomyoma or prolapse.
None of the patients received chemotherapy or radiation therapy
prior to surgery. The follow-up information for the EAC cases was
collected previously and was obtained for the present study from
the department's follow-up system. The follow-up time was between
14 and 90 months (60.2±19.3 months). The EAC tissues were of
typical histology and were comprised of >70% tumor cells. The
procedures, and the use of all specimens and clinical information,
were approved by the Clinical Research Ethics Committee of the
Second Hospital of Shandong University.
Cell culture and in vitro
demethylation treatment
The human endometrial cancer cell line KLE was
obtained from the American Type Culture Collection (Manassas, VA,
USA). KLE cells were cultured in Dulbecco's modified Eagle's medium
supplemented with 10% heat inactivated fetal bovine serum (both
from Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) at
37°C in a humidified atmosphere containing 5% CO2; the
culture medium was changed every 2–3 days. For demethylation
treatment, 1×106 KLE cells were seeded into 6-well
plates and cultured for 24 h at 37°C. Subsequently, cells were
exposed to 30 µmol/l 5-aza-20-deoxycytidine (5-Aza-dC;
Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) and fresh
demethylating agent was added every 24 h for 7 days at 37°C. Cells
that were not treated with 5-Aza-dC were considered control
cells.
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was isolated using TRIzol reagent
(Invitrogen; Thermo Fisher Scientific, Inc.) from EAC and control
tissues according to the manufacturer's protocol. cDNA was
synthesized using the GoScript reverse transcriptase system and
random hexamer primers (Promega Corporation, Madison, WI, USA)
according to the manufacturer's protocol. RT-qPCR was performed
using a relative quantification protocol by SYBR Fast qPCR Mix
(Takara Bio, Inc., Otsu, Japan). GAPDH was used as an internal
control. The primer sequences were as follows: WIF1 forward,
5′-CCGAAATGGAGGCTTTTGTA-3′ and reverse, 5′-GTGTCTTCCATGCCAACCTT-3′;
GAPDH forward, 5′-ACAACTTTGGTATCGTGGAAGG-3′ and reverse,
5′-GCCATCACGCCACAGTTTC-3′. The thermocycling conditions were as
follows: 95°C for 5 min followed by 40 cycles at 95°C for 30 sec,
52°C for 30 sec and 72°C for 30 sec, and a final extension step at
72°C for 10 min. The relative fold change in mRNA expression
compared with the control was calculated using the comparative Cq
method, 2−ΔΔCq (10).
Genomic DNA extraction, pyrosequencing
and methylation-specific PCR (MSP)
Genomic DNA was isolated from KLE cells and patient
tissues using the Blood and Cell Culture DNA Mini kit (Qiagen,
Inc., Valencia, CA, USA) according to the manufacturer's protocol.
Bisulfite genomic DNA modification and purification was performed
using EpiTect Bisulfite kit (Qiagen, Inc.) according to the
manufacturer's instruction.
Three EAC and two normal tissue samples were
selected to undergo pyrosequencing uing PyroMark Q24 reagents
(Qiagen GmbH, Hilden, Germany) according to the manufacturer's
instructions. The PyroMark MD system and PyroMark CpG software
version 1.0 (Qiagen GmbH) were used to measure the methylation
frequency of the WIF1 promoter CpG sites
(5′-TTTATTTCYCYGGCYTTTTATTGGGCYTATCYTATTG-3′). Methylation was
analyzed using MSP and the following MSP primers were used:
Methylated WIF1 forward, 5′-CGTTTTATTGGGCGTATCGT-3′ and reverse,
5′-ACTAACGCGAACG-AAATACGA-3′; and unmethylated WIF1 forward,
5′-GGGTGTTTTATTGGG-TGTATTGT-3′ and reverse,
5′-AAACCAACAATCAACAAAAC-3′. PCR was performed in a 30 µl reaction
volume using HotStarTaq DNA polymerase (Qiagen GmbH). The
thermocycling conditions were as follows: 95°C for 5 min followed
by 35 cycles at 95°C for 30 sec, 60°C for 30 sec and 72°C for 30
sec, and a final extension at 72°C for 5 min. Products were then
separated by 2% agarose gel electrophoresis and visualized by
ethidium bromide under ultraviolet illumination. The methylation
rate was calculated using the following equation:
Methylated/(methylated + unmethylated). Methylation >20% was set
as the cut off value.
Immunohistochemistry (IHC)
Formalin-fixed, paraffin-embedded tissue blocks were
cut into 4-µm sections and mounted onto charged glass slides,
deparaffinized and rehydrated in a graded series of ethanol.
Antigen retrieval was performed in 1 mmol/l EDTA solution (pH 8.0)
at 98°C for 15 min. Endogenous peroxidase activity was blocked with
3.0% hydrogen peroxide for 5 min at room temperature. Sections were
then blocked in 10% normal goat serum with 1% bovine serum albumin
(both from Sigma-Aldrich; Merck KGaA) in Tris-buffered saline (TBS)
with 0.025% Tween-20 (TBST) for 1 h at 37°C. Subsequently, the
sections were incubated with anti-WIF1 rabbit monoclonal antibody
(1:250; ab89935; Abcam, Cambridge, MA, USA) in blocking buffer at
4°C overnight. Following three washes, the sections were incubated
with horseradish peroxidase-conjugated goat anti-rabbit secondary
antibody for 1 h (ab6721, 1:500 dilution; Abcam). Visualization was
performed using the DAB peroxidase substrate kit (cat no. SK-4705;
Vector Laboratories, Inc., Burlingame, CA, USA) in Milli-Q purified
water (EMD Millipore, Billerica, MA, USA). The slides were
counterstained with Mayer's hematoxylin at room temperature, and
the sections were dehydrated, cleared, mounted and observed by
light microscopy. Negative controls were run on all sections by
incubating with 3% normal goat serum in TBST, generated against
unassociated antigens. Finally, the results of IHC were
semi-quantified; the percentage of positive staining was scored as
follows: i) 0, ii) 1 (1–25%), iii) 2 (26–50%), iv) 3 (51–75%) and
v) 4 (76–100%). The intensity of positive staining was scored as
follows: i) 0 (negative), ii) 1 (weak), iii) 2 (moderate) and iv) 3
(strong). A percentage >50% and a moderate intensity score were
used as the positive cut-off criteria.
Western blotting
Briefly, endometrial cancer KLE cells were lysed for
30 min at 4°C in lysis buffer (Sigma-Aldrich; Merck KGaA). The
bicinchoninic acid method was used to determine the protein
concentration. Equal amounts of protein extracts (10 µg) were
separated by 10% SDS-PAGE and were transferred to polyvinylidene
difluoride membranes (Bio-Rad Laboratories, Inc., Hercules, CA,
USA), which were blocked with 5% BSA at room temperature for 1 h.
The membranes were then incubated overnight at 4°C with: Anti-WIF1
(ab89935, 1:500 dilution), anti-Wnt family member 1 (ab15251, 1:500
dilution) (both from Abcam), anti-ERK/p-ERK (#4695/#4370, 1:500
dilution), anti-c-Myc (#13987, 1:500 dilution) (all from Cell
Signaling Technology, Inc., Danvers, MA, USA) or anti-β-actin
(A2066, 1:1000 dilution; Sigma-Aldrich; Merck KGaA) antibodies.
Following washing, the membranes were incubated with anti-rabbit or
anti-mouse HRP-labeled secondary antibodies (#7074 or #7076,
1:2,000 dilution; Cell Signaling Technology, Inc.) at room
temperature for 2 h. Finally, the blots were developed using an
enhanced chemiluminescence detection system (Amersham Life Science;
GE Healthcare, Chicago, IL, USA). The expression of β-actin served
as the loading control. Densitometric analyses were performed using
Bio-Rad Quantity One software version 4.3.0 (Bio-Rad Laboratories,
Inc.). The intensity of the bands of each treatment was compared
with the intensity of the control.
Plasmid construction and cell
transfection
The full-length WIF1 cDNA was PCR cloned,
sequence-verified and further subcloned into the retroviral
expression vector pBABE-puro (Addgene, Inc., Cambridge, MA, USA) to
generate pBABE-puro-WIF1. For stable transfection, KLE cells were
seeded into 24-well culture plates at a density of 3×104
cells/well and transfected with pBABE-puro-WIF1 or pBABE-puro empty
vector using Lipofectamine 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol.
Transfection was performed at a DNA concentration of 800 ng/well.
Following transfection, stable clones were selected using puromycin
(2 µg/ml) starting at 48 h post-transfection, and the expression of
the transgene was detected by western blotting and RT-qPCR.
Proliferation assay
To determine the effects of WIF1 on the
proliferation of KLE cells, cells (4×103/well)
transfected as aforementioned with pBABE-WIF1 and empty vector were
seeded in 96-well culture plates in complete medium in triplicate
and incubated at 37°C with 5% CO2 in a humidified
incubator for 1–6 days. Cell proliferation assay was performed
using Cell Counting Kit-8 (CCK-8; Dojindo Molecular Technologies,
Inc., Rockville, MD, USA) every day according to the manufacturer's
protocol. Absorbance was measured at 450 nm. Data are presented as
the mean ± standard deviation of three separate experiments. For
demethylation detection, KLE cells (3×103/well) were
plated into 96-well plates and incubated with complete medium or
medium containing 5 µmol 5-Aza-dC for 1–7 days, then cells
underwent CCK-8 assay according to the aforementioned protocol.
Tumorigenesis in vivo
For in vivo experiments, 1×107
pBABE-WIF1-KLE and control KLE cells were injected subcutaneously
into 12 athymic nude female mice (n=6/group; age, 4 weeks; weight,
18–20 g). The mice were supplied by Beijing Vital River Laboratory
Animal Technology Co., Ltd. (Beijing, China). Animals were housed
in the animal facility of the Medical College of Shandong
University (Shandong, China), at 26–28°C, 40–60% humidity and a
12/12 h light/dark cycle. Food and water, supplied by Beijing Keao
Xieli Feed Co., Ltd. (Beijing, China), were autoclaved prior to
feeding and were freely available. Tumor growth was monitored by
measurement of the tumor diameter every 4 days from days 14 to 34
following injection. For each time point, the mean tumor volume was
calculated for each group of 6 mice. After 34 days, all 12 mice
were sacrificed by neck dislocation, tumors were collected and
pictures were acquired. All animal experiments were performed with
the approval of the Animal Care and Use Committee of Shandong
University (Shandong, China).
Statistical analysis
Statistical analysis was performed using SPSS
software (version 20.0; IBM Corp., Armonk, NY, USA). Differences in
the frequencies of WIF1 methylation or expression between groups
were examined using the Pearson's χ2 and Fisher exact
probability tests. The survival curves were estimated using the
Kaplan-Meier estimate, and the difference in their distribution was
evaluated by a log-rank test. Disease-specific survival time was
determined by comparing the diagnosis date with the date of
mortality caused by EAC. Comparisons in cell density, and the
relative levels of mRNA expression, protein expression and tumor
size between the two different transfection groups were presented
as the mean ± standard deviation from at least three independent
experiments, and were analyzed using a unpaired Student's t-test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Pyrosequencing, methylation and mRNA
expression of WIF1 in primary EAC
The methylation and mRNA expression status of WIF1
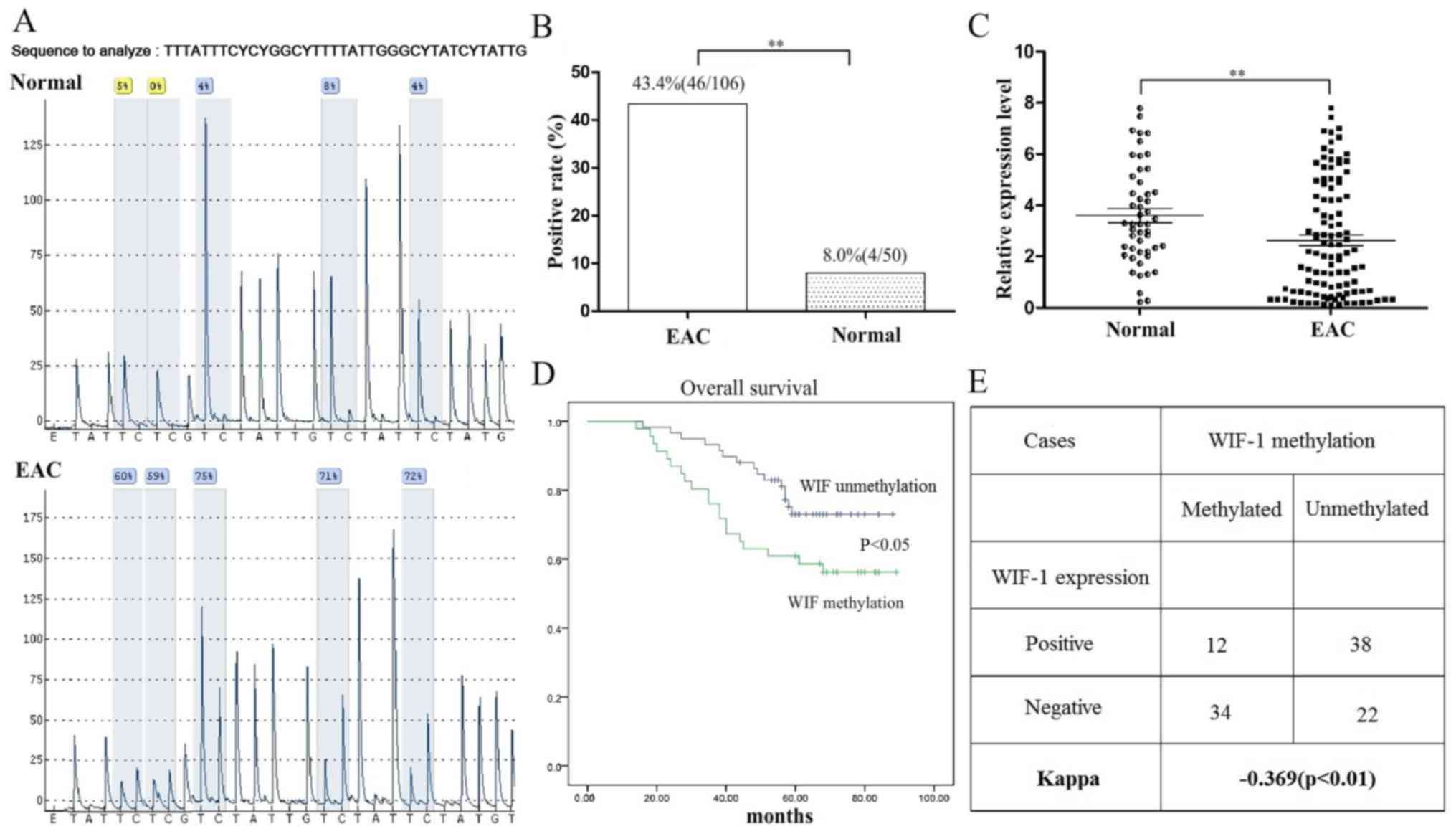
in EAC and control endometrial tissues was determined (Fig. 1). Two out of three EAC cases
exhibited significantly high methylation in five CpG sites
(Fig. 1A). WIF1 methylation was
observed in 46 of the 106 cases (43.4%) of EAC tumor tissue by MSP
(Table I); however, methylation
was only observed in 4 out of 50 (8%) normal endometrial tissue
samples; this difference in methylation was statistically
significant (P<0.05; Fig. 1B).
WIF1 mRNA was detectable in 47.2% (50/106) of neoplastic EAC
tissues, which was significantly lower than that observed in
non-neoplastic tissues [41/50 (82%); Fig. 1C]. The WIF1 methylation positivity
was inversely related to WIF1 expression positivity (P<0.01;
Fig. 1E).
 | Table I.Clinical, pathological and molecular
characteristics of endometrial adenocarcinoma cases. |
Table I.
Clinical, pathological and molecular
characteristics of endometrial adenocarcinoma cases.
| Characteristic | Total EAC cases
n=106 | WIF1 methylated cases
n=46 | WIF1 unmethylated
cases n=60 |
|---|
| Age, years (mean ±
standard deviation) | 59.2±10.9 | 59.7±10.3 | 58.1±11.5 |
| Grade of cancer
(n) |
|
|
|
| 1 | 53 (50.0%) | 21 | 32 |
| 2 | 35 (33.0%) | 16 | 19 |
| 3 | 18 (17.0%) | 9 | 9 |
| FIGO stage (n) |
|
|
|
| I | 40 (37.7%) | 13 | 27 |
| II | 13 (12.3%) | 6 | 7 |
| III | 37 (34.9%) | 19 | 18 |
| IV | 16 (15.1%) | 8 | 8 |
| Adjuvant therapy
(n) |
|
|
|
| No | 46 (43.4%) | 17 | 29 |
| Yes | 60 (56.7%) | 29 | 31 |
Prognostic value of WIF1 expression
and methylation
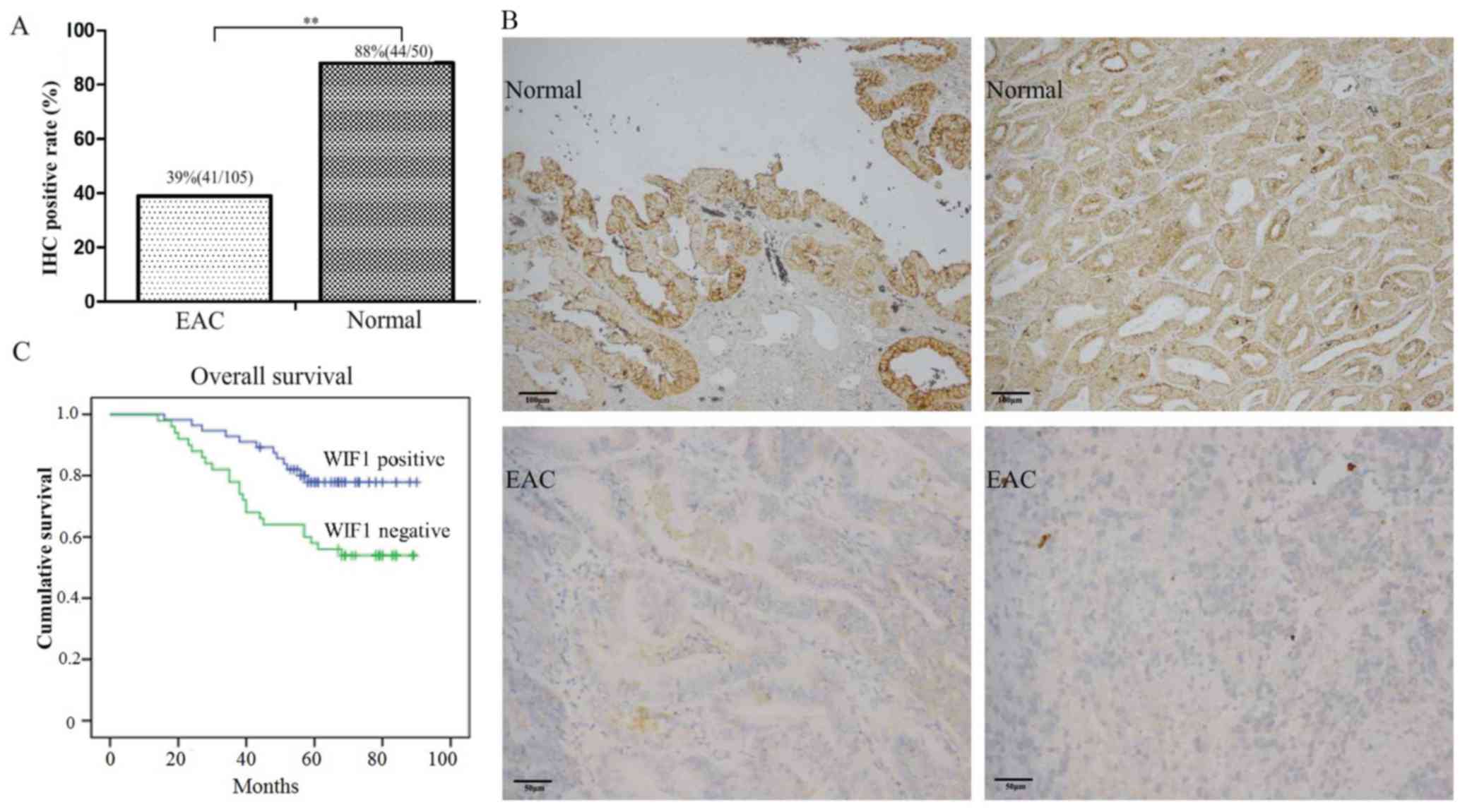
The protein expression levels of WIF1 in primary EAC
and non-tumorous endometrial tissues are presented in Fig. 2A and B. WIF1 expression status was
defined as positive or negative based on WIF1 protein level, in
order to provide a prognostic value for the disease. Patients
bearing tumors with WIF1 methylation or negative WIF1 expression
had a shorter overall survival following surgical resection of
primary tumors (Figs. 1D and
2C). The 5-year overall survival
rates were 53.9% for patients with tumors exhibiting negative
expression of WIF1, as compared with 77.9% for those with positive
WIF1 expression (Fig. 2C).
Demethylation of the WIF1 promoter
restores its expression in the EAC cell line KLE and inhibits cell
proliferation
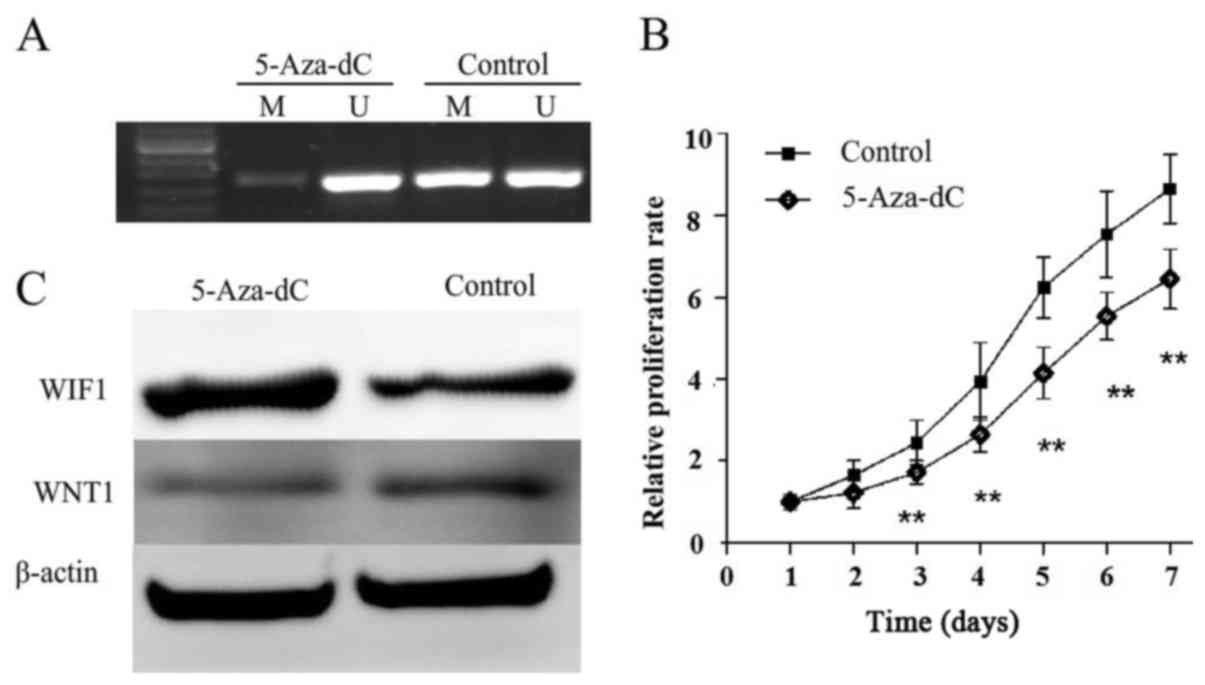
The methylation and expression of WIF1 were detected
in KLE cells with or without 5-Aza-dC treatment, in order to
identify whether WIF1 silencing is associated with promoter
hypermethylation (Fig. 3). KLE
cells were treated with 5-Aza-dC, which resulted in a decrease in
methylation and the restorationof WIF1 expression (Fig. 3A and C). WNT1, which is the
downstream factor of WIF1, was inhibited by restoration of WNT1
(Fig. 3C). In addition, cell
proliferation was significantly reduced following 5-Aza-dC
treatment in KLE cells (Fig.
3B).
WIF1 reduces cell proliferation by
reducing c-Myc and phosphorylated (p)-ERK in vitro
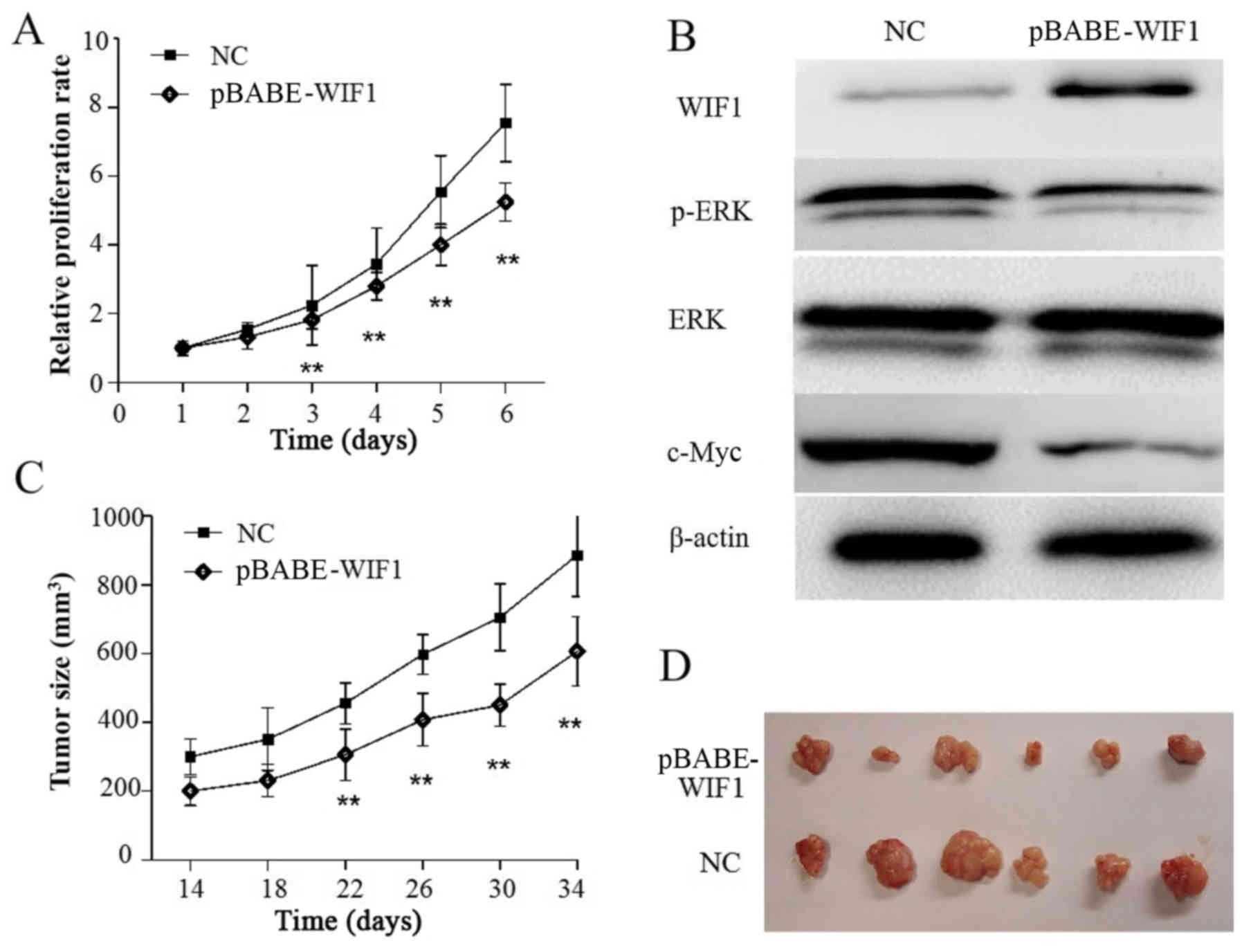
To examine the effects of restored WIF1 on KLE cell
proliferation, cells were stably transfected with pBABE-WIF1 or
empty vector. CCK-8 assay revealed that the proliferation of KLE
cells transfected with pBABE-WIF1 was significantly inhibited
compared with the cells transfected with the control (P<0.05;
Fig. 4A). When compared with the
control group, western blotting demonstrated that WIF1
overexpression in KLE cells led to downregulation of c-Myc and
p-ERK (Fig. 4B). Collectively,
these results indicated that WIF1-induced suppression of
proliferation in KLE cells may be associated with the inhibition of
the transcriptional factor c-Myc and the phosphorylation of
ERK.
WIF1 inhibits tumor growth of
xenografts
To examine whether WIF1 overexpression could
suppress KLE cell growth in vivo, KLE cells transfected with
pbabe-puro-WIF1 or pbabe-puro empty vector were subcutaneously
inoculated into nude mice (n=6/group). The results demonstrated
that the size of tumors derived from pBABE-WIF1-overexpressing
cells was significantly smaller than those in the control group
(P<0.05; Fig. 4C and D).
Discussion
The Wnt signaling pathway, which is evolutionarily
well conserved, has a crucial role in embryonic development and in
numerous cancer-associated processes (11,12).
Alterations in the Wnt/β-catenin signaling pathway have been
identified in ≤50% of EAC cases (13). Nuclear accumulation of β-catenin,
which is an activated factor of Wnt/β-catenin signaling, has been
detected in ≤47% of EAC cases (13–15).
The gain-of-function mutation present in the β-catenin gene appears
to be an important mechanism by which Wnt/β-catenin signaling is
activated, and has been observed in ~25% of EAC cases (9,15,16).
WIF1 is a secreted Wnt antagonist, which can
directly bind to Wnts in order to prevent signaling (17,18).
WIF1 is often downregulated by promoter hypermethylation in
numerous types of human cancer, including prostate (19), cervix (20), esophagus (21) and others (22,23).
The present study demonstrated that WIF1 downregulation is a
widespread event in human EAC oncogenesis, which occurs frequently
due to promoter hypermethylation. This is consistent with previous
studies in other cancer types (19–23),
which reported that promoter methylation is the major mechanism for
the inactivation of this tumor suppressor gene (24). The frequency of WIF1 methylation in
primary EAC identified in the present study was 43.4%. To further
explore the role of DNA methylation in the transcriptional
repression of the WIF1 gene, KLE cells were treated with 5-Aza-dC.
The results demonstrated that 5-Aza-dC was able to restore the
expression of WIF1 in the KLE cell line and inhibit cell
proliferation.
In the present study, WIF1 protein expression was
associated with good survival rates in EAC. This is similar to a
previous study in hepatocellular carcinoma (HCC), which reported
that WIF1 protein expression, but not methylation of WIF1, is a
predictor of good patient outcomes in those undergoing resection of
HCC (25). In addition, WIF1
methylation was reported to act as a strong prognostic factor in
overall survival, and the increased methylation index was
significantly associated with decreased relapse-free survival and
overall survival of non-small cell lung cancer (26). However, increased WIF1 promoter
methylation and decreased WIF1 protein expression were not
associated with human astrocytoma patient survival (27).
By transfecting cell lines with a WIF1-expressing
plasmid, the present study has demonstrated that ectopic expression
of WIF1 in EAC cell lines may inhibit endometrial cancer cell
proliferation. This inhibition of cell proliferation is consistent
with the inhibition of cell growth by WIF1 as previously shown in
other cancer cell lines, indicating that WIF1 acts as a functional
tumor suppressor (6,28). The present study also provided
evidence to suggest that WIF1 exerts its tumor suppressor functions
by downregulating the intracellular protein levels of c-Myc and
p-ERK, which are important Wnt/β-catenin transcriptional target
genes.
In conclusion, to the best of our knowledge, the
present study is the first to report that the WIF1 gene is
frequently hypermethylated in EAC, which is an important mechanism
to silence WIF1 gene expression. Conversely, restoration of WIF1
expression in EAC cells was able to inhibit cell growth in
vitro and in vivo. These results suggested that WIF1 may
be frequently inactivated by promoter methylation and therefore,
may be considered a candidate tumor suppressor in EAC.
Acknowledgements
The present study was supported by the Youth Fund of
the Second Hospital of Shandong University (grant no.
26010275618056).
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2015. CA Cancer J Clin. 65:5–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Murali R, Soslow RA and Weigelt B:
Classification of endometrial carcinoma: More than two types.
Lancet Oncol. 15:e268–e278. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bokhman JV: Two pathogenetic types of
endometrial carcinoma. Gynecol Oncol. 15:10–17. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
MacDonald BT, Tamai K and He X:
Wnt/beta-catenin signaling: Components, mechanisms, and diseases.
Dev Cell. 17:9–26. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Polakis P: Wnt signaling in cancer. Cold
Spring Harbor perspectives in biology. 4:pii:a0080522012.
View Article : Google Scholar
|
|
6
|
Taniguchi H, Yamamoto H, Hirata T,
Miyamoto N, Oki M, Nosho K, Adachi Y, Endo T, Imai KA and Shinomura
Y: Frequent epigenetic inactivation of Wnt inhibitory factor-1 in
human gastrointestinal cancers. Oncogene. 24:7946–7952. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yoshino M, Suzuki M, Tian L, Moriya Y,
Hoshino H, Okamoto T, Yoshida S, Shibuya K and Yoshino I: Promoter
hypermethylation of the p16 and Wif-1 genes as an independent
prognostic marker in stage IA non-small cell lung cancers. Int J
Oncol. 35:1201–1209. 2009.PubMed/NCBI
|
|
8
|
Urakami S, Shiina H, Enokida H, Kawakami
T, Tokizane T, Ogishima T, Tanaka Y, Li LC, Ribeiro-Filho LA,
Terashima M, et al: Epigenetic inactivation of Wnt inhibitory
factor-1 plays an important role in bladder cancer through aberrant
canonical Wnt/beta-catenin signaling pathway. Clin Cancer Res.
12:383–391. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liu Y, Patel L, Mills GB, Lu KH, Sood AK,
Ding L, Kucherlapati R, Mardis ER, Levine DA, Shmulevich I, et al:
Clinical significance of CTNNB1 mutation and Wnt pathway activation
in endometrioid endometrial carcinoma. J Natl Cancer Inst.
106:pii:dju2452014. View Article : Google Scholar
|
|
10
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Huelsken J, Vogel R, Erdmann B, Cotsarelis
G and Birchmeier W: Beta-catenin controls hair follicle
morphogenesis and stem cell differentiation in the skin. Cell.
105:533–545. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Weigelt B and Banerjee S: Molecular
targets and targeted therapeutics in endometrial cancer. Curr Opin
Oncol. 24:554–563. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Klaus A and Birchmeier W: Wnt signalling
and its impact on development and cancer. Nat Rev Cancer.
8:387–398. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dedes KJ, Wetterskog D, Ashworth A, Kaye
SB and Reis-Filho JS: Emerging therapeutic targets in endometrial
cancer. Nat Rev Clin Oncol. 8:261–271. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Matias-Guiu X and Prat J: Molecular
pathology of endometrial carcinoma. Histopathology. 62:111–123.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
McConechy MK, Ding J, Cheang MC, Wiegand
K, Senz J, Tone A, Yang W, Prentice L, Tse K, Zeng T, et al: Use of
mutation profiles to refine the classification of endometrial
carcinomas. J Pathol. 228:20–30. 2012.PubMed/NCBI
|
|
17
|
Kawano Y and Kypta R: Secreted antagonists
of the Wnt signalling pathway. J Cell Sci. 116:2627–2634. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tamai K, Semenov M, Kato Y, Spokony R, Liu
C, Katsuyama Y, Hess F, Saint-Jeannet JP and He X:
LDL-receptor-related proteins in Wnt signal transduction. Nature.
407:530–535. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yee DS, Tang Y, Li X, Liu Z, Guo Y,
Ghaffar S, McQueen P, Atreya D, Xie J, Simoneau AR, et al: The Wnt
inhibitory factor 1 restoration in prostate cancer cells was
associated with reduced tumor growth, decreased capacity of cell
migration and invasion and a reversal of epithelial to mesenchymal
transition. Mol Cancer. 9:1622010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ramachandran I, Thavathiru E, Ramalingam
S, Natarajan G, Mills WK, Benbrook DM, Zuna R, Lightfoot S, Reis A,
Anant S and Queimado L: Wnt inhibitory factor 1 induces apoptosis
and inhibits cervical cancer growth, invasion and angiogenesis in
vivo. Oncogene. 31:2725–2737. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yang SH, Li SL, Dong ZM and Kan QC:
Epigenetic inactivation of Wnt inhibitory factor-1 in human
esophageal squamous cell carcinoma. Oncol Res. 20:123–130. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kawakami K, Hirata H, Yamamura S, Kikuno
N, Saini S, Majid S, Tanaka Y, Kawamoto K, Enokida H, Nakagawa M
and Dahiya R: Functional significance of Wnt inhibitory factor-1
gene in kidney cancer. Cancer Res. 69:8603–8610. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kansara M, Tsang M, Kodjabachian L, Sims
NA, Trivett MK, Ehrich M, Dobrovic A, Slavin J, Choong PF, Simmons
PJ, et al: Wnt inhibitory factor 1 is epigenetically silenced in
human osteosarcoma, and targeted disruption accelerates
osteosarcomagenesis in mice. J Clin Invest. 119:837–851. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Malinauskas T, Aricescu AR, Lu W, Siebold
C and Jones EY: Modular mechanism of Wnt signaling inhibition by
Wnt inhibitory factor 1. Nat Struct Mol Biol. 18:886–893. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Huang L, Li MX, Wang L, Li BK, Chen GH, He
LR, Xu L and Yuan YF: Prognostic value of Wnt inhibitory factor-1
expression in hepatocellular carcinoma that is independent of gene
methylation. Tumour Biol. 32:233–240. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yoshino M, Suzuki M, Tian L, Moriya Y,
Hoshino H, Okamoto T, Yoshida S, Shibuya K and Yoshino I: Promoter
hypermethylation of the p16 and Wif-1 genes as an independent
prognostic marker in stage IA non-small cell lung cancers. Int J
Oncol. 35:1201–1209. 2009.PubMed/NCBI
|
|
27
|
Kim SA, Kwak J, Nam HY, Chun SM, Lee BW,
Lee HJ, Khang SK and Kim SW: Promoter methylation of WNT inhibitory
factor-1 and expression pattern of WNT/β-catenin pathway in human
astrocytoma: Pathologic and prognostic correlations. Mod Pathol.
26:626–639. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang J, Zhou B, Liu Y, Chen K, Bao P,
Wang Y, Wang J, Zhou Z, Sun X and Li Y: Wnt inhibitory factor-1
functions as a tumor suppressor through modulating Wnt/β-catenin
signaling in neuroblastoma. Cancer Lett. 348:12–19. 2014.
View Article : Google Scholar : PubMed/NCBI
|