Introduction
Asthma is a chronic, heterogeneous respiratory
disease characterized by persistent airway inflammation and
variable expiratory airflow limitation. It affects 1–18% of the
global population and inhaled corticosteroids are the primary
treatment (1). However, the
mechanisms underlying the disease occurrence and progression remain
to be fully elucidated. Type 2 inflammation, which is specifically
caused by T helper (Th)2 cells, is thought to have a central role
in asthma pathogenesis, however, recently the importance of the
dysregulation of other types of immune cells has also been
emphasized, including Th17, Th9 and regulatory T (Treg) cells
(2).
As a type of CD4+ T cell, Treg cells are
essential for immune tolerance and the prevention of excessive
inflammation. Tregs are divided into two subsets: Natural Treg
cells (nTregs), which are derived from the thymus and account for
the largest population of Treg cells in vivo; and inducible
Treg cells (iTregs), which are derived from peripheral naïve T
cells under a certain microenvironment (3). Forkhead box protein 3 (FOXP3)
expression in both Treg cell subsets is crucial for their immune
suppressive function (4).
Interleukin (IL)-10 and transforming growth factor (TGF)-β1 are the
major cytokines that are produced by Treg cells. These cells limit
the activation of proinflammatory cytokines, downregulate T
cell-mediated inflammation and inhibit the proliferation of Th2
cells (5). The development of
asthma is controlled by Treg cells as they inhibit the activation
of Th2 cells, prevent the entry of effector immune cells into
inflamed tissue, suppress IgE production and limit Th17-mediated
inflammation (3), functions that
are partially dependent on the cytokines secreted by Treg
cells.
The proviral integration site for Moloney murine
leukemia virus 2 (PIM2) is a type of serine/threonine kinase
belonging to the PIM family, and has an important function in
cellular proliferation, survival and differentiation (6). The majority of studies concerning
PIM2 focus on its participation in hematologic malignancies and
solid tumors, however, a number of studies have also indicated its
role in regulating the immune system (7,8).
These studies demonstrated that PIM2 induced by FOXP3 is essential
for Treg cell expansion and, conversely, PIM2 also inhibits the
suppressive function of Treg cells by phosphorylating FOXP3, which
indicates the complex roles of PIM2 in the regulation of Treg
cells. As Treg cells have an important role in asthma pathogenesis,
PIM2 may also influence the development of asthma inflammation,
however, to the best of our knowledge, no previous studies have
investigated the effect of PIM2 on asthma pathogenesis.
Therefore, the present study investigated the role
of PIM2 in asthma pathogenesis, and the results demonstrated that
PIM2 was overexpressed in peripheral blood mononuclear cells
(PBMCs), and specifically in nTreg cells, from patients with
asthma. In addition, inhibition of PIM2 in asthmatic mice
alleviated airway inflammation, airway hyper-responsiveness (AHR)
and associated symptoms, and these effects of PIM2 may be caused by
decreased expression levels of FOXP3 and IL-10 in Treg cells.
Patients and methods
Subjects
The population consisted of patients with asthma
(n=12; 8 males and 4 females) aged between 16 and 65 years, who
were recruited from Ruijin Hospital (Shanghai, China) between
January 2015 and September 2015. Healthy subjects (n=8; 3 male and
5 female) were included in the present study and were recruited as
volunteers from Ruijin Hospital and School of Medicine, Shanghai
Jiao Tong University (Shanghai, China). The diagnosis of asthma was
made by the pulmonary physicians, according to the Global
Initiative for Asthma guidelines (1). Subjects were excluded if they had
experienced an asthma exacerbation in the previous 4 weeks or a
respiratory infection 1 week prior to enrollment. All subjects gave
written informed consent, and the study was approved by the Ethics
Committee of Ruijin Hospital.
Sample preparation and T cell
isolation
Peripheral blood (20 ml) was obtained in a sodium
heparin vacuum tube. Subsequently, the blood sample was centrifuged
(700 × g for 20 min at 4°C), the supernatant was discarded and the
same volume of PBS was then added. The sample was added onto the
surface of lymphocyte separation medium, Ficoll-Paque PLUS (GE
Healthcare Life Sciences, Little Chalfont, UK), and centrifuged at
700 × g for 20 min at 4°C. PBMCs were collected from the middle
layer and suspended at a density of 2×106 cells/ml in
RPMI-1640 medium with GlutaMAX (Gibco; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) supplemented with 100 U/ml penicillin, 100
µg/ml streptomycin and 10% heat inactivated fetal calf serum
(Gibco; Thermo Fisher Scientific, Inc.). The cells were
subsequently stained at 4°C for 30 min with the following
fluorescently-labeled antibodies: CD4-fluorescein isothiocyanate
(10 µg/ml; 11-0049-41; eBioscience; Thermo Fisher Scientific,
Inc.), CD25-PerCP-Cyanine5.5 (1.25 µg/ml; 45-0259-42; eBioscience;
Thermo Fisher Scientific, Inc.) and CD45RA-allophycocyanin (20
µl/test; 550855; BD Biosciences, Franklin Lakes, NJ, USA) to sort
for nTreg cells (CD4+ CD25+
CD45RA− T cells) and naïve T cells (CD4+
CD25− CD45RA+) on a FACS ARIA II cell sorter.
Prior to cell staining, Fc receptors were blocked by incubating
cells with 10% normal human serum (ImmunoReagents, Inc., Raleigh,
NC, USA) for 20 min at 4°C.
Induction of Th1, Th2 and iTreg
cells
Naïve T cells (1×106 cells/ml) were
stimulated with CD3 and CD28 antibody-coated beads (Dynabeads Mouse
T-activator; Invitrogen; Thermo Fisher Scientific, Inc.) at a 1:1
cell-to-bead ratio for 3 days at 37°C to induce T cell
differentiation. Cultures were supplemented with IL-12 (10 ng/ml),
IL-2 (50 U/ml) and anti-IL-4 (10 µg/ml; 556917) for Th1 induction,
IL-4 (5,000 U/ml), IL-2 (100 U/ml), anti-interferon γ (10 µg/ml;
554699) and anti-IL-12 (10 µg/ml; 555065) for Th2 induction, and
IL-2 (100 U/ml), TGF-β1 (5 ng/ml), anti-IL-4 (10 µg/ml), anti-IFNγ
(10 µg/ml) and anti-IL-12 (10 µg/ml) for iTreg induction, at 37°C
for 3 days. All antibodies and reagents were purchased from BD
Biosciences.
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA in total PBMCs and individual T cell
subsets was isolated with TRIzol reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) and was reverse transcribed into cDNA with
Reverse Transcription system (Promega Corporation, Madison, WI,
USA), according to manufacturer's protocol. Primers were designed
by Invitrogen and synthesized by BioTNT (Shanghai, China),
according to the manufacturer's protocol. For amplification, a
SYBR-Green I qPCR kit was used (BioTNT). The thermocycling
conditions were as follows: 95°C for 5 min followed by 40 cycles of
95°C for 5 sec, 55°C for 20 sec and 72°C for 20 sec. Each reaction
was performed in triplicate on the ABI ViiA 7 Real-Time PCR system
(Invitrogen; Thermo Fisher Scientific, Inc.) and expression was
normalized using the 2−∆∆Cq method (9) to the expression of the housekeeping
gene β-actin. The specific primers used were as follows: PIM2,
5′-ACTCCAGGTGGCCATCAAAG-3′ (forward) and 5′-TCCATAGCAGTGCGACTTCG-3′
(reverse); and β-actin, 5′-AAGGTGACAGCAGTCGGTT-3′ (forward) and
5′-TGTGTGGACTTGGGAGAG G-3′ (reverse).
Immunocytochemistry
Human nTreg cells were obtained
(1×104-1×105 cells/sample) as aforementioned,
fixed in 4% paraformaldehyde (PFA) for 15 min at room temperature,
permeabilized with 1% Triton-X-100 for 5 min at room temperature
and incubated with 3% H2O2 for 20 min at room
temperature. Cells were subsequently incubated with 5% BSA (Sigma
Aldrich; Merck KGaA, Darmstadt, Germany) for blocking at room
temperature for 20 min and then anti-PIM2 mouse antibody (0.5
µg/ml; MAB4355; R&D Systems, Inc., Minneapolis, MN, USA) at 4°C
overnight. Following three washes with PBS, cells were incubated
with rabbit anti-mouse peroxidase secondary antibody (1:200
dilution; A9044; Sigma Aldrich; Merck KGaA) for 2 h at room
temperature. Following an additional three washes with PBS, cells
were stained with a 3,3′-diaminobenzidine color developing reagent
kit, and nuclei were stained with 1% hematoxylin for 2 min at room
temperature. Samples were examined on a Nikon Eclipse 50i
microscope (Nikon Corporation, Tokyo, Japan) and the images were
analyzed with Image-Pro Plus 6.0 software (Media Cybernetics, Inc.,
Rockville, MD, USA).
Animals and asthma model
A total of 90 Female BALB/c mice were purchased from
Shanghai SLAC laboratory Co., Ltd. (Shanghai, China). Mice were
aged between 6 and 8 weeks-old, weighing between 20 and 22 g, and
were housed at 18–25°C, humidity 50–60%, 0.03% CO2,
12/12 h light/dark cycle and food/water were available ad libitum
and refreshed every 3 days. The mice received an intraperitoneal
injection of 100 µg ovalbumin (OVA; Sigma Aldrich; Merck KGaA) and
2 mg alum (Sigma Aldrich; Merck KGaA) in PBS on days 0, 7 and 14.
On days 25, 26 and 27, the mice were challenge with aerosolized 1%
OVA in PBS for 30 min. Control animals received PBS
intraperitoneally with alum on days 0, 7 and 14, and were
challenged with aerosolized PBS on days 25, 26 and 27. The PIM2
inhibitor (5Z)-5-[[3-(Trifluoromethyl) phenyl]
methylene]-2,4-thiazolidinedione
(Z)-5-(3-trifluoromethylbenzylidene) thiazolidine-2,4-dione (5
µg/µl; Sigma Aldrich; Merck KGaA) was given 30 min prior to
challenge with OVA by intraperitoneal injection to OVA-sensitized
mice on days 25, 26 and 27.
Evaluation of AHR and asthma
symptoms
AHR was assessed by measuring changes of dynamic
lung compliance (Cdyn) in response to increasing doses (0–4 mg/ml)
of acetylcholine (Ach; Shanghai Mengry Biotechnology Co., Ltd.,
Shanghai, China) injected into the tail intravenously in
anesthetized and ventilated mice. AHR was assessed using the
AniRes2005 animal lung function analysis system (Beijing Bestlab
High-Tech Co., Ltd., Beijing, China) 24 h after the last OVA
challenge. Asthma symptoms, including cyanosis, frequently
scratching the mouth/nose/limbs (>10 scratches in 5 sec) and
standing upright were observed during challenge on days 25, 26 and
27. Time to each symptom occurrence (indicated as T1, T2 and T3)
were measured and symptom scores of each mouse on different days
was calculated as [(30 - T1) + (30 - T2) + (30 - T3)]/3.
Bronchoalveolar lavage fluid (BALF),
lung histology and immunohistochemistry (IHC)
Experiments were performed 24 h after the final OVA
challenge. Lungs were lavaged with 1 ml PBS through the trachea
immediately following the assessment of AHR, and the number of
total leukocytes was counted with a hemocytometer, and cell
differentiation was performed on cytospin slides prepared with
Wright-Giemsa staining to differentiate between leukocytes. Cells
were seeded in PBS medium (Beyotime Institute of Biotechnology,
Haimen, China) at 1×105 cells/ml and stained with Fast
Wright and Giemsa Stain kit (Nanjing Jiancheng Technology Co.,
Ltd., Nanjing, China), according to manufacturer's protocol.
Percentages of each cell were counted per sample with a light
microscope: Number of each cell type/every 200 cells ×100%.
The left upper lobes of the lungs were fixed in 2 ml
4% PFA at 4°C for 12–24 h and embedded in paraffin. Prepared
sections (4 µm) were stained with 0.5% hematoxylin (10 min) and
0.5% eosin (30 sec) at room temperature using standardized
protocols and analyzed with a Nikon Eclipse 50i microscope. IHC was
performed on 4 µm-PFA fixed sections. Antigen retrieval was
performed with 0.01 M citric acid buffer (pH 6.0) for 15 min at
95°C prior to rehydration in descending alcohol series. Sections
were subsequently blocked with 5% goat serum (Beyotime) for 20 min
at room temperature. Endogenous peroxidase/phosphatase activity was
blocked with 3% hydrogen peroxide prior to incubation overnight at
4°C with anti-PIM2 (2 µg/ml; sc13514; Santa Cruz Biotechnology,
Inc., Dallas, TX, USA) and anti-FOXP3 (1:500; SAB5300461;
Sigma-Aldrich; Merck KGaA). Subsequently, sections were incubated
for 30 min at 37°C with anti-mouse IgG-peroxidase secondary
antibody (1:100; A0168; Sigma Aldrich; Merck KGaA).
3,3′-Diaminobenzidine was applied for chromogen detection. Sections
were counterstained with 0.5% hematoxylin and eosin for 30 sec at
room temperature and visualized with the Nikon Eclipse 50i upright
microscope (magnification, ×200). Lung inflammation was scored as
described previously (10).
Expression levels of PIM2 and FOXP3 in lung tissues were evaluated
by Image-Pro Plus 6.0, and recorded as the mean optical
density.
ELISA
Experiments were performed 24 h after the last OVA
challenge. A total of 0.5–0.8 ml blood was drawn from each mouse.
Serum was collected following centrifugation at 1,000 × g for 5 min
at 4°C. Concentrations of cytokines and OVA specific IgE in the
BALF and serum of mice were measured by ELISA, according to the
protocols of each ELISA kit (FOXP3, MR45686; IL-4, MR63901; IL-5,
MR63900; TRG-β, MR63871; IL-10, MR63912; OVA specific IgE, MR64027;
Shanghai Mengry Biotechnology Co., Ltd.).
Statistical analysis
Data are presented as the mean ± standard deviation
for continuous variants. All analyses were performed using SPSS
(version 20; IBM Corp., Armonk, NY, USA). Student's t-test was used
to determine differences between two groups. For comparisons
between multiple groups, one-way analysis of variance (post hoc
Tukey test) was used. Nonparametric analyses, using the
Mann-Whitney U test or Kruskal-Wallis test (post hoc Dunn's test),
were applied if the distributions of numerical data were not
normal. Comparisons were made using Pearson's Chi-square test if
the data were categorical variants. P<0.05 was considered to
indicate a statistically significant difference.
Results
Baseline characteristics of
subjects
Asthma patients (n=12) and healthy subjects (n=8)
were included in the present study. The age of the asthma patients
was 47±10 years, with no significant difference compared with
healthy controls (38±9 years; P>0.05). A total of 8 patients
with asthma were male, which was comparable to healthy subjects (3
males; P>0.05).
PIM2 is highly expressed in asthma
patients and primarily located in Treg cells
PIM2 has been reported to be expressed ubiquitously,
with the highest levels in the brain and lymphoid tissues (11). In addition, PIM2 is established to
be upregulated in various hematologic malignancies, including acute
or chronic myeloid leukemia, acute lymphoblastic leukemia, multiple
myeloma and lymphoma (12),
however, its expression pattern in human T lymphocytes is not
clear. The present study isolated naïve T cells and nTreg cells
from healthy human PBMCs and induced naïve T cells into Th1, Th2
and iTreg cells by different culture conditions, the results
demonstrated that PIM2 was primarily expressed in Treg cells,
particularly nTreg cells (Fig.
1A).
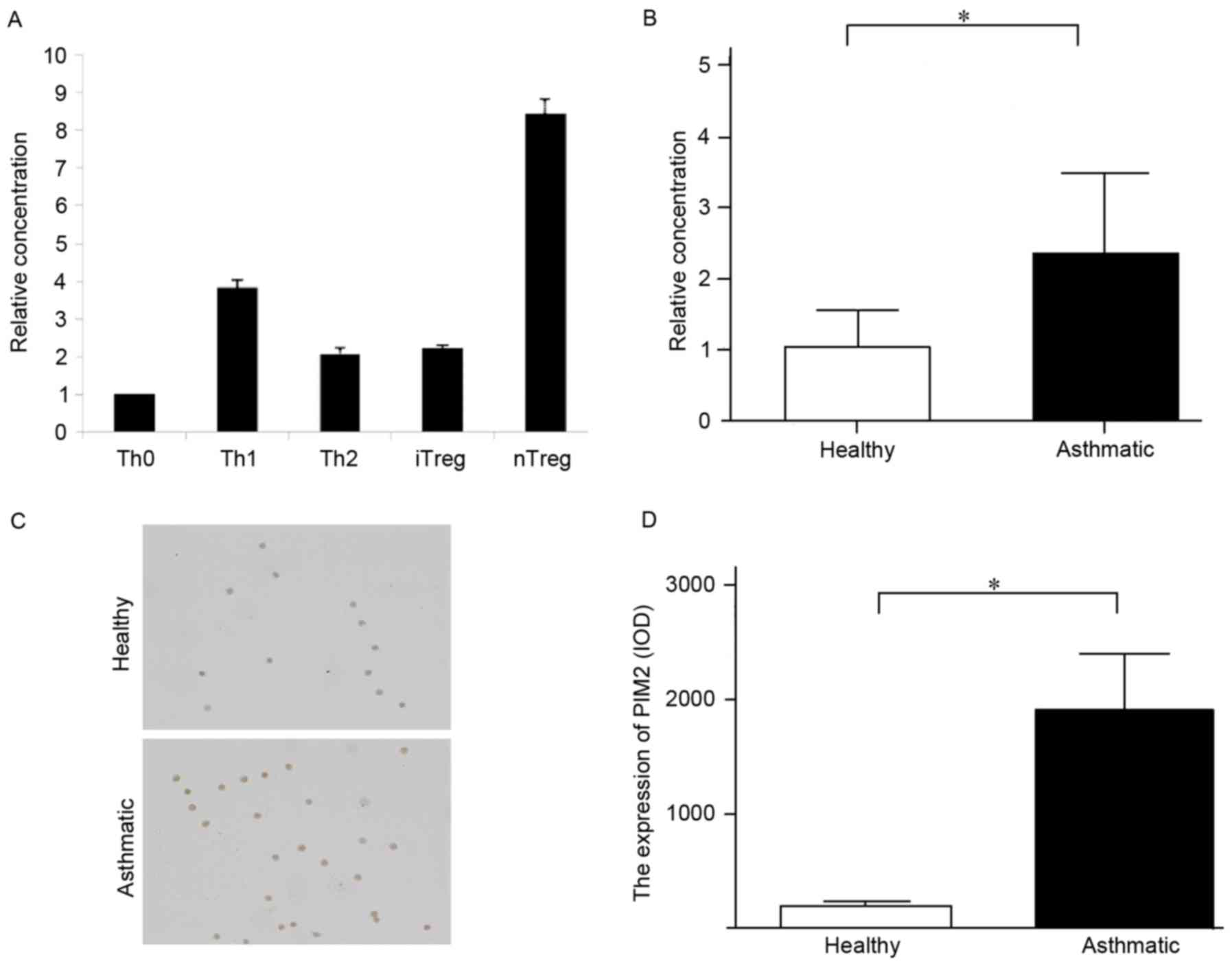 | Figure 1.The expression of PIM2 in PBMCs and
Treg cells from patients with asthma was higher compared with
healthy subjects. (A) Expression levels of PIM2 mRNA in different
types of T cells in healthy subjects. (B) Expression level of PIM2
mRNA in PBMCs. (C) Representative images (magnification, ×200) of
PIM2 expression in nTreg cells from patients with asthma and
healthy subjects, as measured by immunocytochemistry. (D)
Quantified PIM2 expression levels in Treg cells, as measured by
immunocytochemistry. *P<0.05, as indicated. PIM2, proviral
integration site for Moloney murine leukemia virus 2; PBMCs,
peripheral blood mononuclear cells; Tregs, T regulatory cells; Th,
T helper cells; iTregs, inducible Tregs; nTregs, natural Tregs;
IOD, integrated optical density. |
To the best of our knowledge, no previous study has
investigated the PIM2 expression level in patients with asthma, and
the present study demonstrated that the mRNA and protein expression
levels of PIM2 in PBMCs and nTregs, respectively, were higher in
patients with asthma compared with healthy subjects (Fig. 1B-D), indicating that PIM2 may have
an important role in asthma pathogenesis.
PIM2 is essential for airway
inflammation and AHR
As PIM2 was highly expressed in patients with
asthma, an asthma mouse model and PIM2 inhibitor were used to
investigate the role of PIM2 in asthma pathogenesis. The mice were
sensitized and challenged with OVA, and the PIM2 inhibitor was
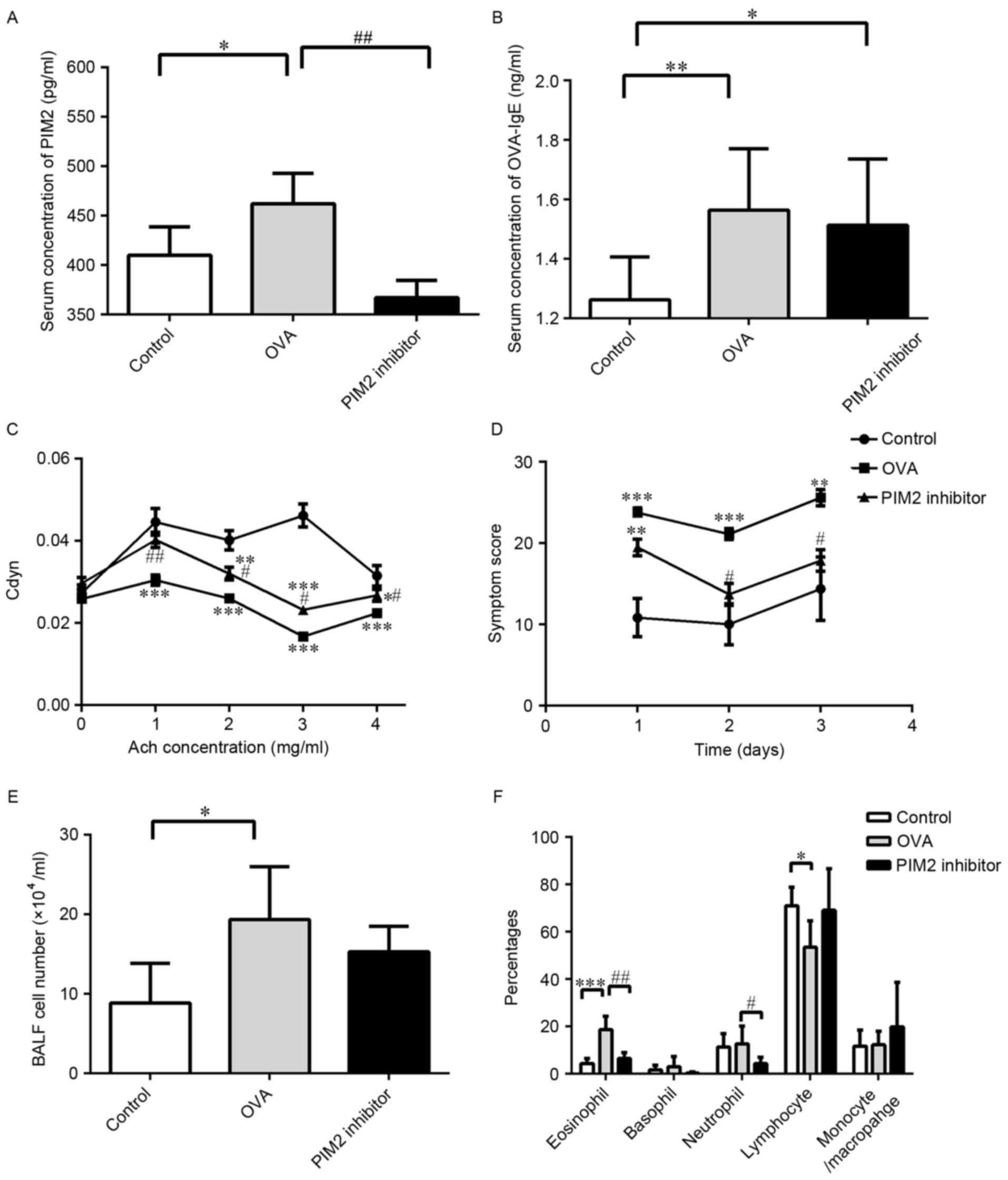
added during the challenge period. Serum levels of PIM2 were
determined in control, OVA and PIM2 inhibitor mice, and levels were
significantly increased in the OVA treatment group compared with
the control group, while levels were significantly decreased in the
OVA + PIM2 inhibitor group compared with the OVA-only group
(Fig. 2A). In addition, the serum
levels of OVA-specific IgE were measured, with levels significantly
increased in the OVA group compared with controls. However,
although PIM2 inhibitor reduced the levels compared with the
OVA-only group, this reduction was not statistically significant
(Fig. 2B). Furthermore, the
results indicated that PIM2 inhibition improved AHR (indicated as
Cdyn) and asthma-associated symptoms significantly, compared with
OVA-treated mice only (Fig. 2C and
D).
Additionally, increases in BALF cell numbers induced
by OVA were marginally decreased following PIM2 inhibition
(Fig. 2E), and the percentages of
BALF eosinophils and neutrophils were significantly reduced
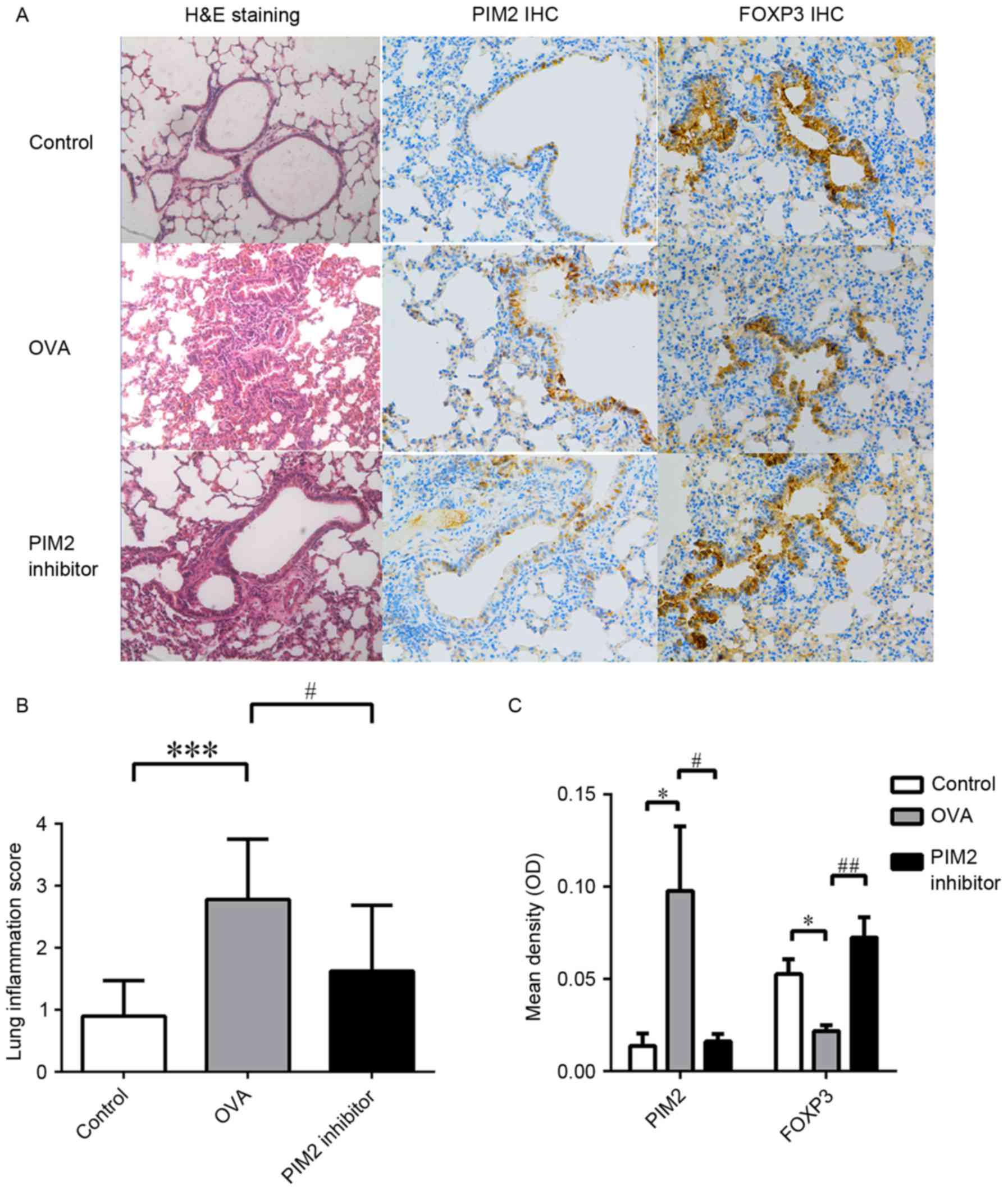
following PIM2 inhibition in OVA-treated mice (Fig. 2F). In addition, H&E staining of
lung tissues from mouse models demonstrated that PIM2 inhibition
alleviated airway inflammation compared with the OVA-only group
(Fig. 3A and B). These results
indicate an important role of PIM2 in inducing airway inflammation
and AHR, however, the underlying mechanism was yet to be
determined.
PIM2 inhibits IL-10 production by
regulating FOXP3
A previous study demonstrated the role of PIM2 in
regulating the function of Treg cells by mediating FOXP3
phosphorylation (8), however, its
influence on FOXP3 expression and downstream cytokines, including
TGF-β1 and IL-10, in asthma mouse models has not previously been
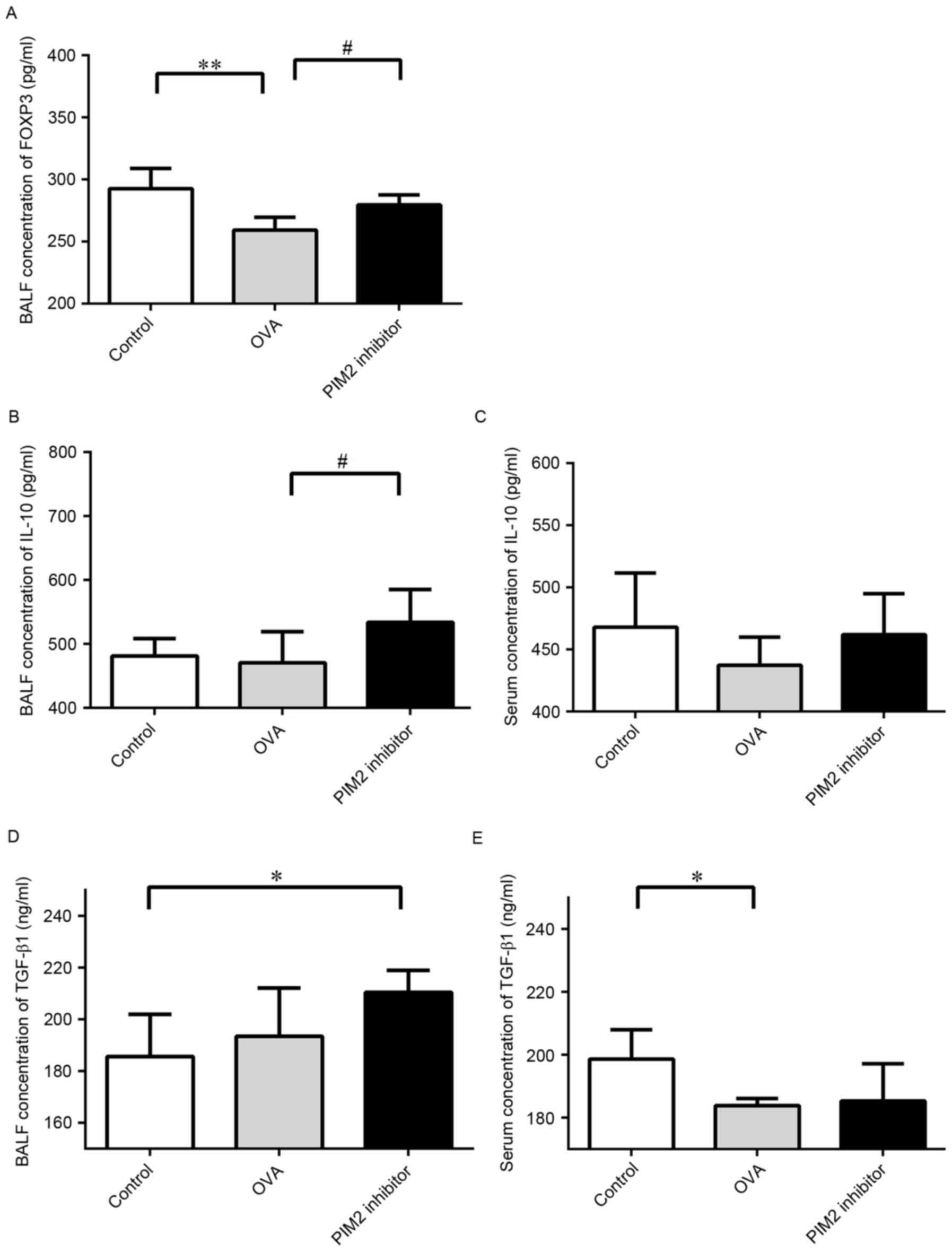
investigated. The present study demonstrated that PIM2 inhibition
led to increased expression levels of FOXP3 in lung tissues
(Fig. 3A and C) and BALF compared
with OVA-only treated mice (Fig.
4A), indicating that PIM2 may downregulate FOXP3 expression in
allergic asthma.
Additionally, BALF and serum IL-10 levels were
increased following PIM2 inhibition in OVA-treated mice (Fig. 4B and C), however, the expression
levels of TGF-β1 in BALF and serum were not altered by PIM2
inhibition in asthmatic mice (Fig. 4D
and E). These results indicate that PIM2 may inhibit IL-10
production, and not TGF-β1 production, to suppress FOXP3
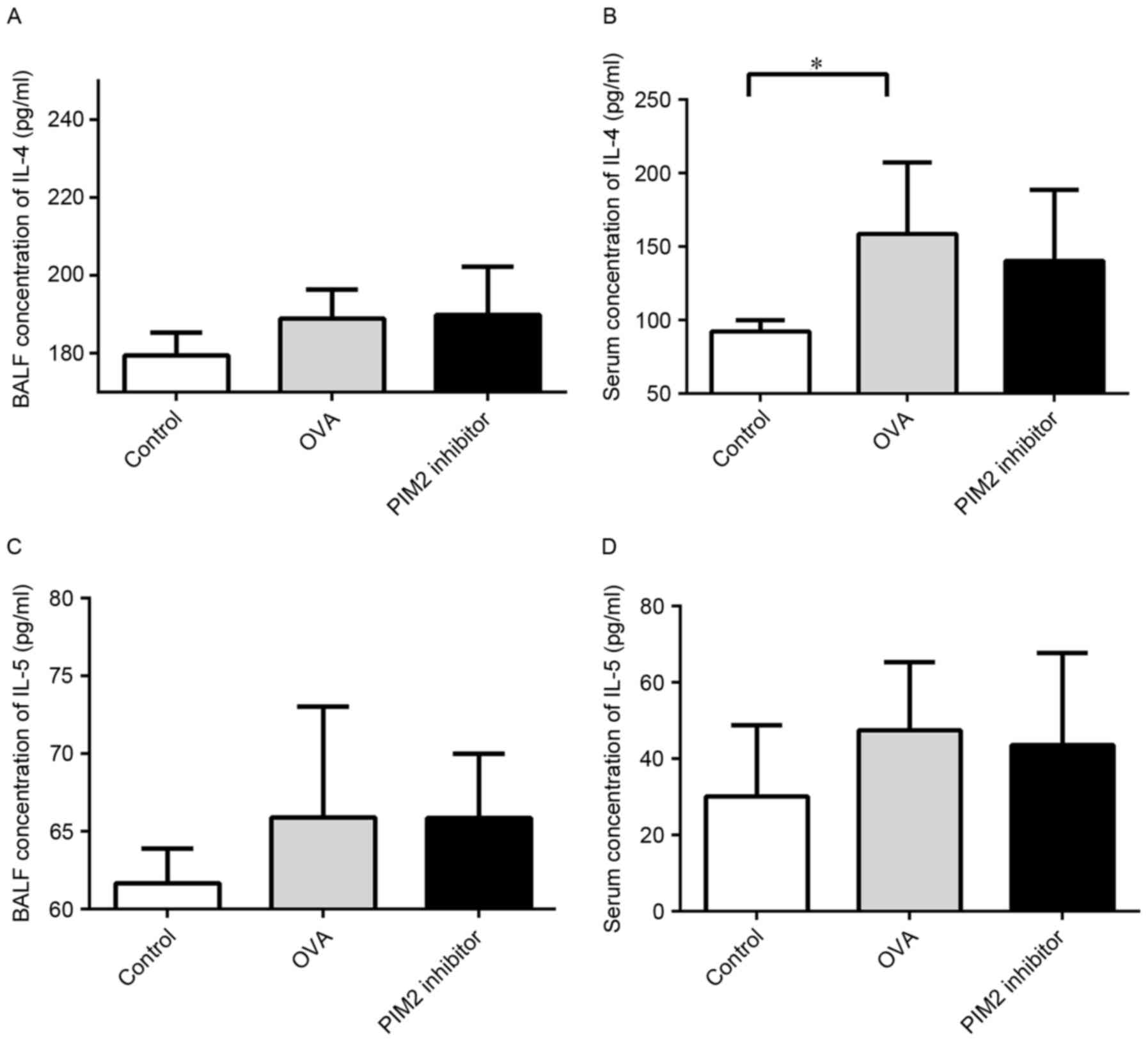
expression. Additionally, the expression levels of Th2 cytokines in
the BALF and serum, including IL-4 and IL-5, were not altered
following PIM2 inhibition in OVA-treated mice (Fig. 5).
Discussion
PIM2 has been primarily recognized as an oncogene
since its discovery, however, its role in regulating the immune
system has not been extensively investigated. The present study
investigated its role in asthma development and demonstrated that
PIM2 may have an important role in asthma pathogenesis. In patients
with asthma, PIM2 expression levels were elevated, particularly in
nTreg cells. Furthermore, results from an animal model of asthma
further confirmed the critical role of PIM2 in promoting airway
inflammation (both eosinophilic and neutrophilic) and AHR, which
may occur via the regulation of FOXP3 and IL-10 expression in Treg
cells.
Although no previous study has elucidated the role
of PIM2 in asthma pathogenesis, the following observations have
indicated the potential of a role for PIM2 in asthma: PIM2 is
induced upon IL-4 release to promote T cell growth and survival,
indicating its role in regulating adaptive immune system (8); it has been reported that PIM1, which
is 61% homologous to PIM2 (11),
has the ability to affect airway inflammation, AHR and the
production of type 2 cytokines in asthmatic mice, indicating that
PIM2 may exhibit a similar effect on asthma development; PIM2 has
been recognized as a molecule that may lead to mammalian target of
rapamycin complex 1 (mTORC1) activation, which inhibits Treg
differentiation (7,13,14);
and, conversely, the inhibition of PIM2 was demonstrated to enhance
the function of Treg cells in vitro (8). It is established that Treg cells are
important regulators in excessive immune responses, such as
persistent airway inflammation in patients with asthma, and these
reports have indicated that PIM2 may have an important function in
asthma pathogenesis by regulating the proliferation,
differentiation and function of Treg cells.
Overexpression of PIM2 is commonly reported in
hematologic and solid malignancies, including acute or chronic
leukemia, lymphomas, and prostate and liver cancers, however, no
previous study has measured its expression level in patients with
asthma (11,15). In healthy subjects, PIM2 is
constitutively expressed in lymphoid cells (16,17),
and a previous report demonstrated high expression of PIM2 in nTreg
cells (18). Therefore, the
present study collected PBMCs from asthmatic and healthy subjects
in order to measure the difference in PIM2 expression level between
asthmatic and healthy subjects. The results demonstrated that PIM2
was overexpressed in patients with asthma compared with healthy
subjects in nTreg cells, which was consistent with our hypothesis
that PIM2 may influence asthma development by regulating Treg
cells.
To further confirm our hypothesis, animal
experiments were performed. The results demonstrated that PIM2
inhibition alleviated airway inflammation and AHR, and reversed the
downregulated FOXP3 and IL-10 expression levels in asthmatic mice.
Unlike PIM1, which is associated with the survival of eosinophils,
PIM2 has not been previously reported to influence eosinophil
survival in vitro or in vivo (19). Therefore, the reduced percentage of
eosinophils in BALF following PIM2 inhibition in asthmatic mice may
be the result of enhanced Treg cell function or cytokine secretion,
which may inhibit the function of type 2 cytokines, including IL-4
and IL-5, but not the expression levels of these cytokines, as PIM2
inhibition appeared to exert no direct influence on the expression
levels of IL-4 and IL-5 in the present study.
Although the association between PIM2 and Treg cells
is established, the molecular mechanisms underlying this
association remain unclear. A previous study reported that PIM2
suppressed the function of Treg cells by phosphorylating the FOXP3
N-terminal domain (8), however,
this may be only one of several mechanisms. For example, PIM2 is
also reported to upregulate mTORC expression (20), which has a negative effect on Treg
cell function and differentiation (4). In addition, increased expression of
PIM2 was also demonstrated to be associated decreased phosphatase
and tensin homolog deleted on chromosome 10, which is highly
expressed in Treg cells and regulates their differentiation
(4,21). The present study has demonstrated
that PIM2 may influence the expression of FOXP3 and IL-10, both of
which represent the function of Treg cells, however, the precise
underlying molecular mechanisms behind this phenomenon remain
unclear, and further studies are required. In addition, previous
studies have indicated the important role of TGF-β1 in asthma
pathogenesis and airway remodeling (22,23),
however, in the current study, its expression level was increased
marginally in BALF and decreased significantly in the serum of
asthmatic mice compared with the control group. This result may be
due to relative acute and early phase of asthma in this mouse
model, in which airway remodeling was not predominant. Increased
eosinophils in the airway may secret small amounts of this cytokine
leading to the increased expression of this cytokine observed in
the BALF. Conversely, systemic functional deficiency of Treg cells
may be responsible for the reduced secretion of TGF-β1 observed in
the serum.
In conclusion, the present study is the first to
demonstrate the critical role of PIM2 in asthma pathogenesis, and
this effect may be dependent on Treg cells and the secretion of
IL-10 by Tregs, which suppresses excessive airway inflammation in
patients with asthma. As the number of studies that have
investigated the association between asthma pathogenesis and PIM2
is limited, further studies should be performed to determine the
mechanisms underlying the association and any therapeutic potential
of this kinase.
Acknowledgements
The present study was funded by Shanghai Municipal
Education Commission (grant no. 14ZZ107) and the National Natural
Science Foundation of China (grant nos. 81270083 and 81470216). The
abstract was presented as the American Thoracic Society 2016
International Conference May 13–18, 2016 in San Francisco, CA and
published as abstract no. A6701 in the American Journal of
Respiratory and Critical Care Medicine 193, 2016.
References
|
1
|
From the global strategy for asthma
management and prevention, global initiative for asthma (GINA).
2015.http://ginasthma.org/wp-content/uploads/2016/01/GINA_Report_2015_Aug11-1.pdfDecember.
2015
|
|
2
|
Hirahara K and Nakayama T: CD4+ T-cell
subsets in inflammatory diseases: Beyond the Th1/Th2 paradigm. Int
Immunol. 28:163–171. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Stelmaszczyk-Emmel A: Regulatory T cells
in children with allergy and asthma: It is time to act. Respir
Physiol Neurobiol. 209:59–63. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kasper IR, Apostolidis SA, Sharabi A and
Tsokos GC: Empowering regulatory T cells in autoimmunity. Trends
Mol Med. 22:784–797. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Qiao YC, Shen J, Hong XZ, Liang L, Bo CS,
Sui Y and Zhao HL: Changes of regulatory T cells, transforming
growth factor-beta and interleukin-10 in patients with type 1
diabetes mellitus: A systematic review and meta-analysis. Clin
Immunol. 170:61–69. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Uddin N, Kim RK, Yoo KC, Kim YH, Cui YH,
Kim IG, Suh Y and Lee SJ: Persistent activation of STAT3 by
PIM2-driven positive feedback loop for epithelial-mesenchymal
transition in breast cancer. Cancer Sci. 106:718–725. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yin G, Li Y, Yang M, Cen XM and Xie QB:
Pim-2/mTORC1 pathway shapes inflammatory capacity in rheumatoid
arthritis synovial cells exposed to lipid peroxidations. Biomed Res
Int. 2015:2402102015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Deng G, Nagai Y, Xiao Y, Li Z, Dai S,
Ohtani T, Banham A, Li B, Wu SL, Hancock W, et al: Pim-2 kinase
influences regulatory T cell function and stability by mediating
Foxp3 protein N-terminal phosphorylation. J Biol Chem.
290:20211–20220. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Massoud AH, Charbonnier LM, Lopez D,
Pellegrini M, Phipatanakul W and Chatila TA: An asthma-associated
IL4R variant exacerbates airway inflammation by promoting
conversion of regulatory T cells to TH17-like cells. Nat Med.
22:1013–1022. 2016. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Narlik-Grassow M, Blanco-Aparicio C and
Carnero A: The PIM family of serine/threonine kinases in cancer.
Med Res Rev. 34:136–159. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Keeton EK, McEachern K, Dillman KS,
Palakurthi S, Cao Y, Grondine MR, Kaur S, Wang S, Chen Y, Wu A, et
al: AZD1208, a potent and selective pan-Pim kinase inhibitor,
demonstrates efficacy in preclinical models of acute myeloid
leukemia. Blood. 123:905–913. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li MO and Rudensky AY: T cell receptor
signalling in the control of regulatory T cell differentiation and
function. Nat Rev Immunol. 16:220–233. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang XH, Yu HL, Wang FJ, Han YL and Yang
WL: Pim-2 modulates aerobic glycolysis and energy production during
the development of colorectal tumors. Int J Med Sci. 12:487–493.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Alvarado Y, Giles FJ and Swords RT: The
PIM kinases in hematological cancers. Expert Rev Hematol. 5:81–96.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yang J, Li X, Hanidu A, Htut TM, Sellati
R, Wang L, Jiang H and Li J: Proviral integration site 2 is
required for interleukin-6 expression induced by interleukin-1,
tumour necrosis factor-α and lipopolysaccharide. Immunology.
131:174–182. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Brault L, Gasser C, Bracher F, Huber K,
Knapp S and Schwaller J: PIM serine/threonine kinases in the
pathogenesis and therapy of hematologic malignancies and solid
cancers. Haematologica. 95:1004–1015. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Basu S, Golovina T, Mikheeva T, June CH
and Riley JL: Cutting edge: Foxp3-mediated induction of pim 2
allows human T regulatory cells to preferentially expand in
rapamycin. J Immunol. 180:5794–5798. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shin YS, Takeda K, Shiraishi Y, Jia Y,
Wang M, Jackson L, Wright AD, Carter L, Robinson J, Hicken E and
Gelfand EW: Inhibition of Pim1 kinase activation attenuates
allergen-induced airway hyperresponsiveness and inflammation. Am J
Respir Cell Mol Biol. 46:488–497. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ezell SA, Wang S, Bihani T, Lai Z,
Grosskurth SE, Tepsuporn S, Davies BR, Huszar D and Byth KF:
Differential regulation of mTOR signaling determines sensitivity to
AKT inhibition in diffuse large B cell lymphoma. Oncotarget.
7:9163–9174. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Asanuma S, Tanaka J, Sugita J, Kosugi M,
Shiratori S, Wakasa K, Shono Y, Shigematsu A, Kondo T, Kobayashi T,
et al: Expansion of CD4(+) CD25(+) regulatory T cells from cord
blood CD4(+) cells using the common γ-chain cytokines (IL-2 and
IL-15) and rapamycin. Ann Hematol. 90:617–624. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ojiaku CA, Yoo EJ and Panettieri RA Jr:
Transforming growth factor β1 function in airway remodeling and
hyperresponsiveness. The missing link? Am J Respir Cell Mol Biol.
56:432–442. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Koćwin M, Jonakowski M, Przemęcka M, Zioło
J, Panek M and Kuna P: The role of the TGF-SMAD signalling pathway
in the etiopathogenesis of severe asthma. Pneumonol Alergol Pol.
84:290–301. 2016. View Article : Google Scholar : PubMed/NCBI
|