Introduction
With the increasing threat of acute myocardial
infarction worldwide, the past decades have witnessed an
advancement in interventional technologies and thrombolytic
treatments (1). The goal of these
therapeutic interventions is a timely restoration of the blood
supply to the ischemic tissue, the salvaging of viable myocardium
and limiting the size of myocardial infarction. Typically, the
infarct size is associated with the incidence of short- and
long-term adverse events in patients with acute myocardial
infarction (1–3). However, the reperfusion itself
paradoxically causes further cardiomyocyte death and contractile
dysfunction, and increases infarct size, a phenomenon known as
ischemia/reperfusion (I/R) injury (3,4).
Although the underlying mechanisms of myocardial I/R injury remain
to be fully elucidated, the inflammatory activities triggered by an
acute ischemic injury are hypothesized to be a significant
contributor (3–5).
Mitochondrial DNA (mtDNA) is a double-stranded and
naked circular DNA and resembles bacterial DNA in containing
non-methylated CpG motifs (6).
Accumulating evidence has indicated that mtDNA, when released from
damaged mitochondria into circulation, is able to act as
damage-associated molecular patterns (DAMPs) with inflammation in
several pathological conditions (6–8).
Previously, phosphoinositide-3-kinases (PI3Ks) and their downstream
target, RAC-α serine/threonine-protein kinase (Akt), have been
regarded as a negative feedback signaling pathway in the
progression of inflammatory responses (9,10).
Activation of the PI3K/Akt-dependent pathway has been demonstrated
to protect the heart against I/R injury by downregulating the
expression levels of a series of pro-inflammatory cytokines
(10–12). However, in the context of
myocardial I/R injury, the role of the mtDNA-induced inflammatory
response and its association with PI3Ks/Akt signaling remains to be
elucidated.
Epigallocatechin-3-gallate (EGCG), the most abundant
catechin in green tea, may prevent visceral organ I/R injury
partially due to its anti-inflammatory properties (13,14).
However, more recent findings have also suggested other beneficial
effects of EGCG attributing to its anti-oxidative action for
preventing mitochondrial damage (15). Therefore, the present study was
designed to investigate whether EGCG administered prior to
reperfusion protects the heart against I/R injury, and to determine
its potential mechanisms involving the role of mtDNA and PI3Ks/Akt
signaling pathway in a rat model.
Materials and methods
Animals
The present study was conducted with the approval of
the Institutional Animal Care and Use Committee at West China
Hospital of Sichuan University. A total of fifty 10-week-old male
Wistar rats weighing 220–250 g were obtained from Hua Fu Kang
Experimental Animal Center (Beijing, China). The rats were housed
and acclimated in a specific pathogen-free facility with a 12 h
light:dark cycle and fed with chow food and water ad
libitum.
Myocardial ischemia and reperfusion
preparation
As previously described (11), the rats were anesthetized with
pentobarbital sodium (100 mg/kg, intraperitoneally). Rats were
subsequently placed in a supine position and secured on an electric
heating pad to maintain constant body heat at 37°C. Catheters were
cannulated into the carotid artery and jugular vein for arterial
blood pressure monitoring and drug administration. The
electrocardiogram was monitored with subcutaneous stainless steel
electrodes in the chest. A left thoracotomy through a left
parasternal incision was performed to expose the anterior wall of
the left ventricle. Following a stabilization period of 20 min,
myocardial ischemia was induced by 4-0 silk suture slipknot at the
level of the proximal left anterior descending coronary artery for
30 min. Reperfusion was started by releasing the slipknot and the
heart was harvested following a 2 h reperfusion.
Experimental protocol
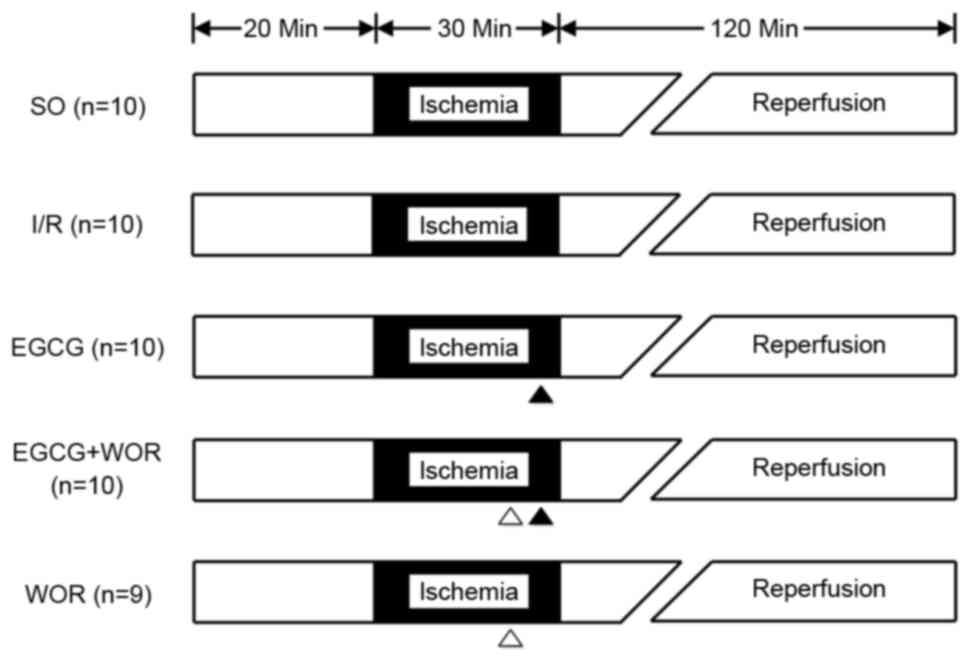
The Wistar rats were randomly allocated into five
groups (n=10 in each group; Fig.
1): i) Sham-operated control (SO) group (rats were subjected to
surgical manipulation without any induction of I/R injury); ii) I/R
group (rats were subjected to the coronary artery occlusion for 30
min followed by 2 h reperfusion); iii) EGCG group (EGCG 10 mg/kg,
administered intravenously at 5 min prior to the onset of
reperfusion); iv) EGCG + wortmannin (WOR) group (WOR, a PI3K
inhibitor, 0.6 mg/kg + EGCG 10 mg/kg administered intravenously at
10 min prior to the reperfusion); and v) WOR group (WOR alone
intravenously administered at 10 min prior to the reperfusion). The
dosage of EGCG and WOR was determined according to previous studies
(11–13).
Measurement of myocardial injury and
infarct size
At the end of reperfusion, blood samples were
collected and centrifuged at 500 × g for 15 min at 4°C to assess
the serum levels of lactate dehydrogenase (LDH) and creatine kinase
(CK) by using commercial assay kits (Nanjing Jiancheng
Bioengineering Institute, Nanjing, China). The hearts were
subsequently harvested and the left anterior descending artery was
occluded again. Diluted fluorescent polymer microspheres (Duke
Scientific Corporation, Palo Alto, CA, USA) were infused into the
aorta to demarcate the infarct area. Subsequent to being frozen at
−20°C, the hearts were cut into 2 mm transverse sections and
incubated in 1% 2,3,5-triphenyltetrazolium chloride (Sigma-Aldrich;
Merck KGaA, Darmstadt, Germany) at 37°C for 20 min. The infarct
regions and the area at risk were quantified using Image-Pro Plus
(version 3.0; Media Cybernetics, Silver Spring, MD, USA). The
infarct regions and area at risk were converted into volumes by
multiplying them by section thickness.
Western blot analysis
Western blot analysis of the protein levels for
phosphorylated (p)-p85, total (t)-Akt and p-Akt in myocardial
tissues was carried out as previously reported (14). Antibodies against p-p85 (1:1,000;
catalog no. ab182651; Abcam, Cambridge, MA, USA), p-Akt (1:500;
catalog no. 4051; Cell Signaling Technology, Inc., Danvers, MA,
USA) and t-Akt (1:1,000; catalog no. 2920; Cell Signaling
Technology, Inc.) were used to probe the membranes, followed by
incubation with a horseradish peroxidase-conjugated secondary
antibody (1:500; catalog no. 111-035-003; Jackson ImmunoResearch
Laboratories, West Grove, PA, USA). β-actin (1:300; catalog no.
A0A068F1Y2; Abmart (Shanghai) Co., Ltd., Shanghai, China,) was used
for normalization. The reactive bands were visualized using the
ECL-Plus reagent (Amersham Biosciences Corp., Piscataway, NJ, USA).
The density of each reactive band was quantified using the Labworks
image acquisition platform and its related analytic software
(GDS8000; UVP, Inc., Upland, CA, USA).
DNA isolation and quantitative
polymerase chain reaction (qPCR) of mtDNA
As previously reported (16), the whole plasma DNA was isolated
from plasma using the DNeasy Blood and Tissue kit (69504; Qiagen
Inc., Valencia, CA, USA). A total of 50 µl plasma samples were
added to 50 µl PBS and centrifuged at 700 × g at 4°C for 5 min to
obtain the supernatant. All procedures were performed according to
the manufacturer's protocols. Plasma mtDNA levels were measured by
SYBR-Green dye-based qPCR assay (45 cycles of 95°C for 15 sec and
60°C for 1 min) using a PRISM 7300 sequence detection system. The
primer sequences were rat NADH dehydrogenase 1 gene (mtDNA):
Forward CGC CTG ACC AAT AGC CAT AA and reverse ATT CGA CGT TAA AGC
CTG AGA. All samples were measured with standards at the same time
and standard mtDNA was diluted in 10-fold serial dilutions. All
measurements were conducted three times for quality control.
Concentration of plasma mtDNA was converted to copy number via a
DNA copy number calculator (cels.uri.edu/gsc/cndna.html; University of Rhode
Island Genomics and Sequencing Center). Plasma mtDNA levels were
shown in copies per µl plasma according to the following formula:
c=QxVDNA/VPCRx1/Vext, where c is
the concentration of plasma mtDNA (copies/µl), Q is the quantity of
DNA measured by RT-PCR, VDNA is the total volume of
plasma DNA solution obtained from the extraction (200 µl),
VPCR is the volume of plasma DNA solution for RT-PCR (1
µl) and Vext is the volume of plasma used for the
extraction (50 µl).
Assessment of inflammatory
cytokines
Expression levels of TNF-α and IL-6 and −8 in
myocardial tissue supernatants were quantified by using specific
ELISA kits (BioSource International; Thermo Fisher Scientific,
Inc., Waltham, MA, USA), according to the manufacturer's protocols.
Additionally, total RNA was extracted from myocardial tissues with
Trizol, according to the manufacturer's protocol (Takara Bio, Inc.,
Otsu, Japan). The mRNA from each tissue sample was reverse
transcribed with PrimeScript RT Master Mix (Takara Bio, Inc.). qPCR
amplifications (45 cycles of 95°C for 15 sec and 55°C for 1 min)
were carried out using the ABI 7500 system (Applied Biosystems;
Thermo Fisher Scientific, Inc.). The primers used were as follows:
TNF-α forward 5′-CTGAACTTCGGGGTGATCGG-3′ and reverse
5′-GGCTTGTCACTCGAATTTTGAGA-3′; IL-6 forward
5′-CTTCCAGCCAGTTGCCTTCT-3′ and reverse 5′-GAGAGCATTGGAAGTTGGGG-3′;
IL-8 forward 5′-AGCTTCCTTGTGCAAGTGTCT-3′ and reverse
5′-GACAGCCCAGGTCAAAGGTT-3′; and β-actin forward
5′-AGAGGGAAATCGTGCGTGAC-3′ and reverse 5′-CAATAGTGATGACCTGGCCGT-3′.
The amount of mRNA for each gene was normalized by β-actin and the
relative expression levels were calculated using the
2−ΔΔCq method (17).
Statistical analysis
Results are expressed as the mean ± standard
deviation. Differences in hemodynamic parameters and infarct
measurements between groups were determined by one-way analysis of
variance with Tukey's honest significant difference post hoc test.
χ2 analysis was used to test the differences in the
incidence of ventricular arrhythmias. The Kruskal-Wallis test was
used to compare the means for all nonparametric scores between
groups. The Pearson correlation coefficient test was performed. All
analyses were performed using SPSS (version 19.0; IBM SPSS, Armonk,
NY, USA) and P<0.05 was considered to indicate a statistically
significant difference.
Results
Heart extraction
A total of 49 rat hearts were successfully harvested
for the experiment. One rat in the WOR group was excluded due to an
irreversible ventricular fibrillation during the stabilization
period. No significant difference in body or heart weight, areas at
risk or hemodynamic parameters between groups was observed
(Tables I and II).
 | Table I.Baseline and intraoperative
cardiodynamic data. |
Table I.
Baseline and intraoperative
cardiodynamic data.
| Variable | SO (n=10) | I/R (n=10) | EGCG (n=10) | EGCG +WOR (n=10) | WOR (n=9) |
|---|
| SAP, mmHg |
|
|
|
|
|
|
Baseline | 109±7 | 106±9 | 103±4 | 106±5 | 104±3 |
|
I-30 | 110±8 | 105±4 | 101±3 | 105±3 | 98±3 |
|
R-60 | 104±2 | 112±5 | 110±6 | 106±4 | 105±5 |
|
R-120 | 105±3 | 103±3 | 105±4 | 105±4 | 104±3 |
| MAP, mmHg |
|
|
|
|
|
|
Baseline | 99±6 | 95±6 | 94±4 | 91±6 | 95±5 |
|
I-30 | 98±7 | 92±5 | 88±5 | 92±4 | 89±4 |
|
R-60 | 96±5 | 94±4 | 94±4 | 91±6 | 92±7 |
|
R-120 | 97±3 | 94±6 | 92±6 | 94±7 | 91±6 |
| DAP, mmHg |
|
|
|
|
|
|
Baseline | 88±5 | 83±2 | 85±3 | 75±3 | 85±2 |
|
I-30 | 85±4 | 79±3 | 74±3 | 78±3 | 79±4 |
|
R-60 | 87±4 | 76±4 | 77±4 | 76±3 | 78±3 |
|
R-120 | 88±3 | 85±6 | 79±3 | 82±4 | 77±6 |
| HR, beats/min |
|
|
|
|
|
|
Baseline | 357±19 | 346±15 | 363±19 | 348±17 | 353±12 |
|
I-30 | 349±17 | 354±14 | 354±18 | 347±12 | 347±14 |
|
R-60 | 362±14 | 362±16 | 361±15 | 354±15 | 349±11 |
|
R-120 | 354±16 | 358±15 | 359±17 | 351±11 | 362±10 |
| Table II.Body weight, heart weight and
morphometric data. |
Table II.
Body weight, heart weight and
morphometric data.
| Variable | SO (n=10) | I/R (n=10) | EGCG (n=10) | EGCG + WOR
(n=10) | WOR (n=9) |
|---|
| Body weight, g |
236±13 |
233±12 |
227±16 |
241±13 |
239±15 |
| Heart weight,
g |
1.13±0.04 |
1.15±0.03 |
1.20±0.04 |
1.16±0.05 |
1.14±0.04 |
| LV volume,
cm3 |
0.47±0.04 |
0.48±0.04 |
0.49±0.04 |
0.50±0.04 |
0.46±0.05 |
| AR volume,
cm3 |
0.22±0.01 |
0.22±0.01 |
0.22±0.01 |
0.24±0.02 |
0.24±0.01 |
| AR/LV,
% |
48.4±5.5 |
45.3±5.6 |
46.0±4.7 |
49.4±3.2 |
49.3±2.0 |
| AI volume,
cm3 | 0 |
0.11±0.01 |
0.05±0.01a |
0.10±0.01b |
0.10±0.01b |
| Infarct size,
% | 0 |
50.0±3.2 |
22.5±4.2a |
40.2±4.4b |
43.2±6.0b |
Severity of I/R-induced ventricular
arrhythmia
The episodes and cumulative duration of the
premature ventricular contraction, ventricular tachycardia and
ventricular fibrillation were recorded. The arrhythmia scoring
system suggested by Miller et al (18) was used to assess the severity of
I/R-induced ventricular arrhythmia. As presented in Table III, ventricular arrhythmia scores
in the I/R, EGCG+WOR and WOR groups were significantly increased
compared with those in the SO and EGCG groups (P<0.05,
respectively).
| Table III.Incidence of ventricular
arrhythmia. |
Table III.
Incidence of ventricular
arrhythmia.
| Variable | SO (n=10) | I/R (n=10) | EGCG (n=10) | EGCG + WOR
(n=10) | WOR (n=9) |
|---|
| PVCs |
|
|
|
|
|
|
Episodes, n | 26.4±17.5 | 223.8±50.4 | 111.2±32.2 | 199.8±49.1 | 219.8±43.0 |
| VT |
|
|
|
|
|
|
Episodes, n | 0 | 27.2±7.5 | 14.8±3.6 | 21.8±5.2 | 28.0±7.8 |
| VF |
|
|
|
|
|
|
Episodes, n | 0 | 5.6±2.7 | 1.8±0.8 | 4.6±1.1 | 6.0±2.1 |
| Total duration,
sec | 0 | 40.2±18.8 | 6.2±3.3 | 38.0±16.2 | 39.2±15.1 |
| VAS | 1.4±0.3 |
8.3±1.0a |
5.1±0.8a,b |
8.0±0.6a,c |
8.4±0.8a,c |
EGCG protects against myocardial I/R
injury
The infarct size induced by an I/R injury was
significantly reduced by EGCG compared with the I/R group (22.5±4.2
vs. 50.0±3.2%, P<0.05). Administration of PI3K inhibitor (WOR)
eliminated the cardioprotective effects of EGCG on the infarct size
compared with the EGCG group (40.2±4.4 vs. 22.5±4.2%, P<0.05).
However, WOR alone did not affect the infarct size compared with
the I/R group (43.2±6.0 vs. 50.0±3.2%, P>0.05), suggesting that
the infarct reducing effects of EGCG involves the activation of the
PI3K-Akt signaling pathway (Table
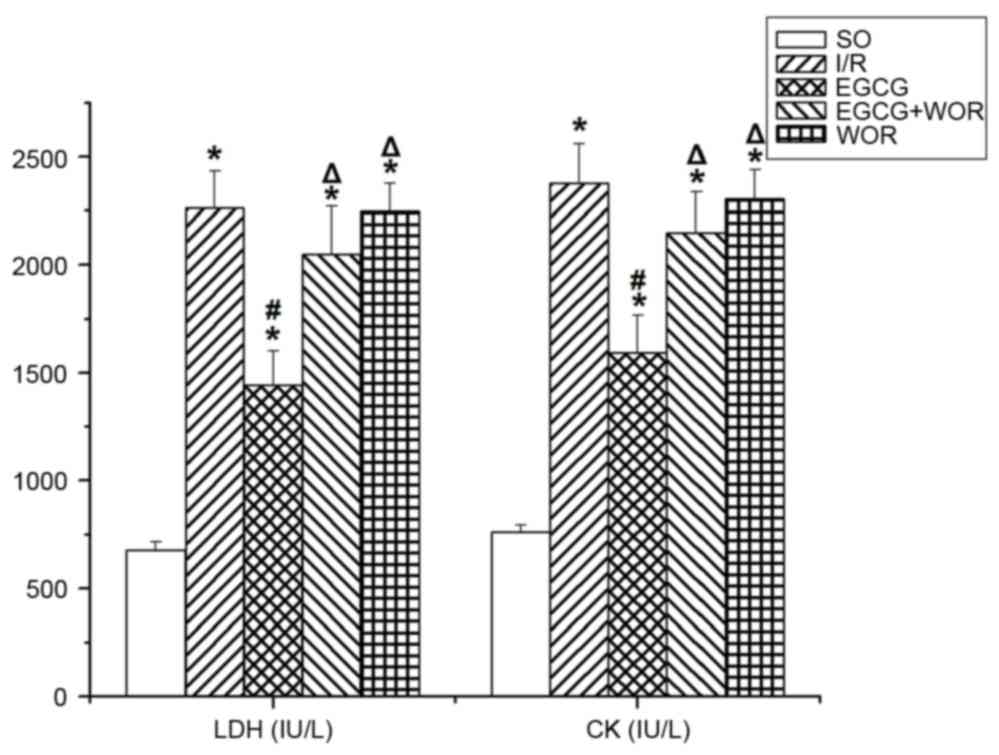
II). Following a 2 h reperfusion, LDH and CK levels in the I/R
group were significantly increased compared with the SO group
(P<0.05, respectively). However, a notable decrease of these
kinases was observed in the EGCG group (Fig. 2). No significant differences in LDH
and CK levels were observed between the WOR and EGCG+WOR group and
those in the I/R group.
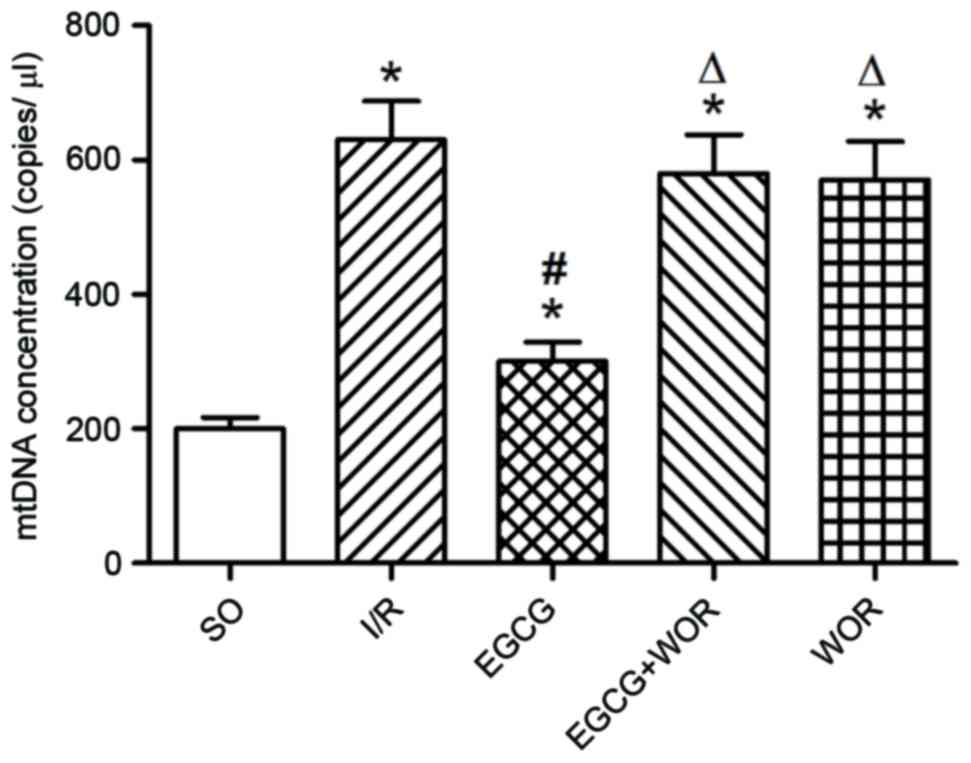
EGCG prevents mtDNA release caused by
I/R injury
mtDNA, as a pro-inflammatory agent, is released
following I/R injury to cause inflammatory damage to the heart. In
order to study the role of EGCG on mtDNA release, blood samples
were collected from all rats. As demonstrated in Fig. 3, EGCG significantly reduced the
plasma levels of mtDNA following I/R injury; 2.13-fold lower
compared with the I/R group (P<0.05). Additional treatment with
WOR is able to eliminate the mtDNA reducing effects of EGCG,
implying that EGCG may exert its inhibitory effect on mtDNA release
by activating PI3K-mediated signaling pathways.
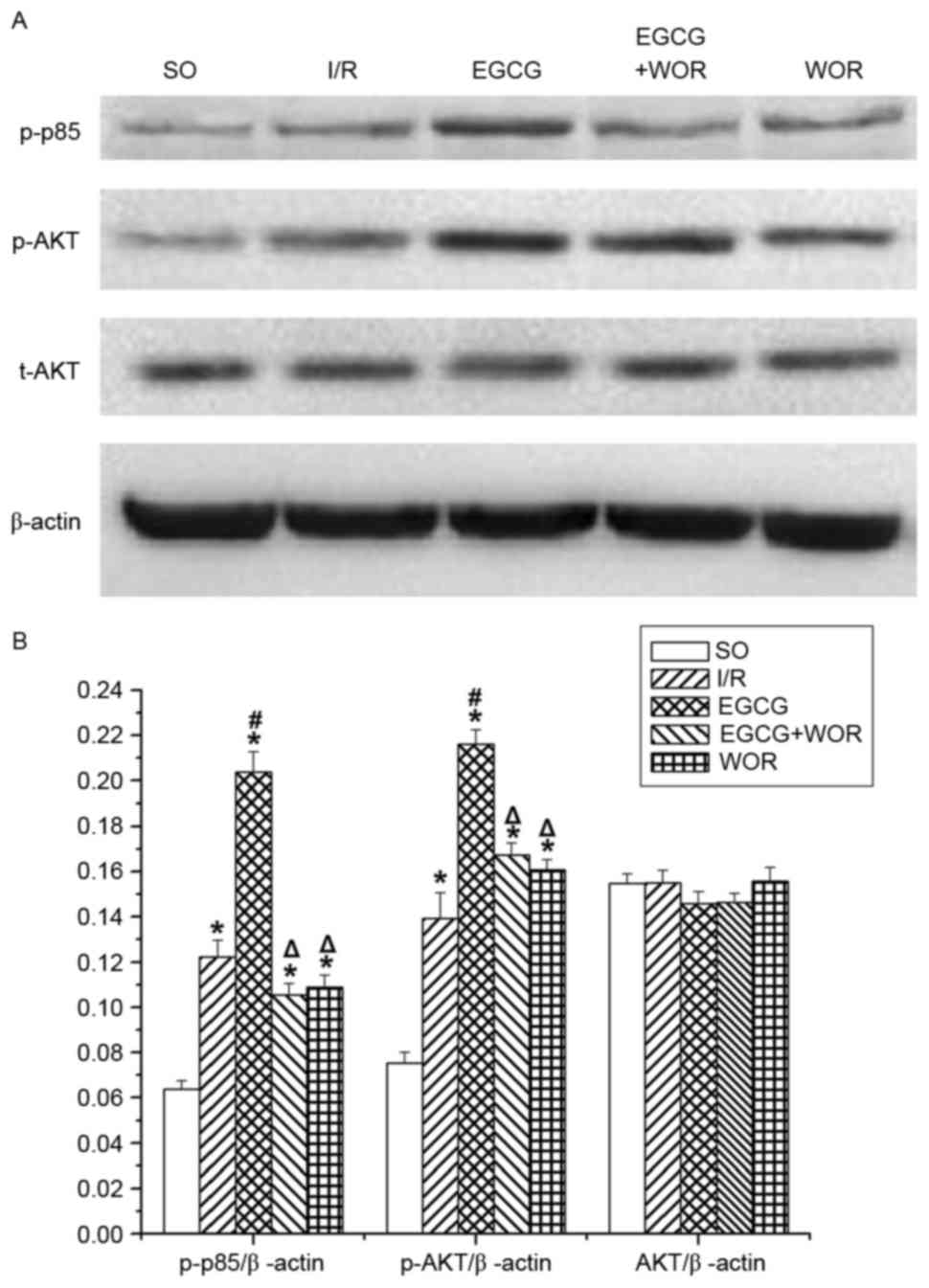
EGCG activates the PI3K/Akt signaling
pathway
To investigate the role of the PI3K/Akt signaling
pathway in the effect of EGCG, the expression of the p-p85 (an
activated regulatory subunit of PI3K), p-Akt and t-Akt were
examined. As demonstrated in Fig. 4A
and B, EGCG significantly increased the expression levels of
p-p85 (62.3±4.3% of the I/R group, P<0.05) and p-Akt (36.4±6.3%
of the I/R group, P<0.05). However, the upregulating effects of
EGCG on p-p85 and p-Akt expression were abolished by WOR.
EGCG-activates PI3K/Akt signaling
suppresses inflammatory cytokine expression
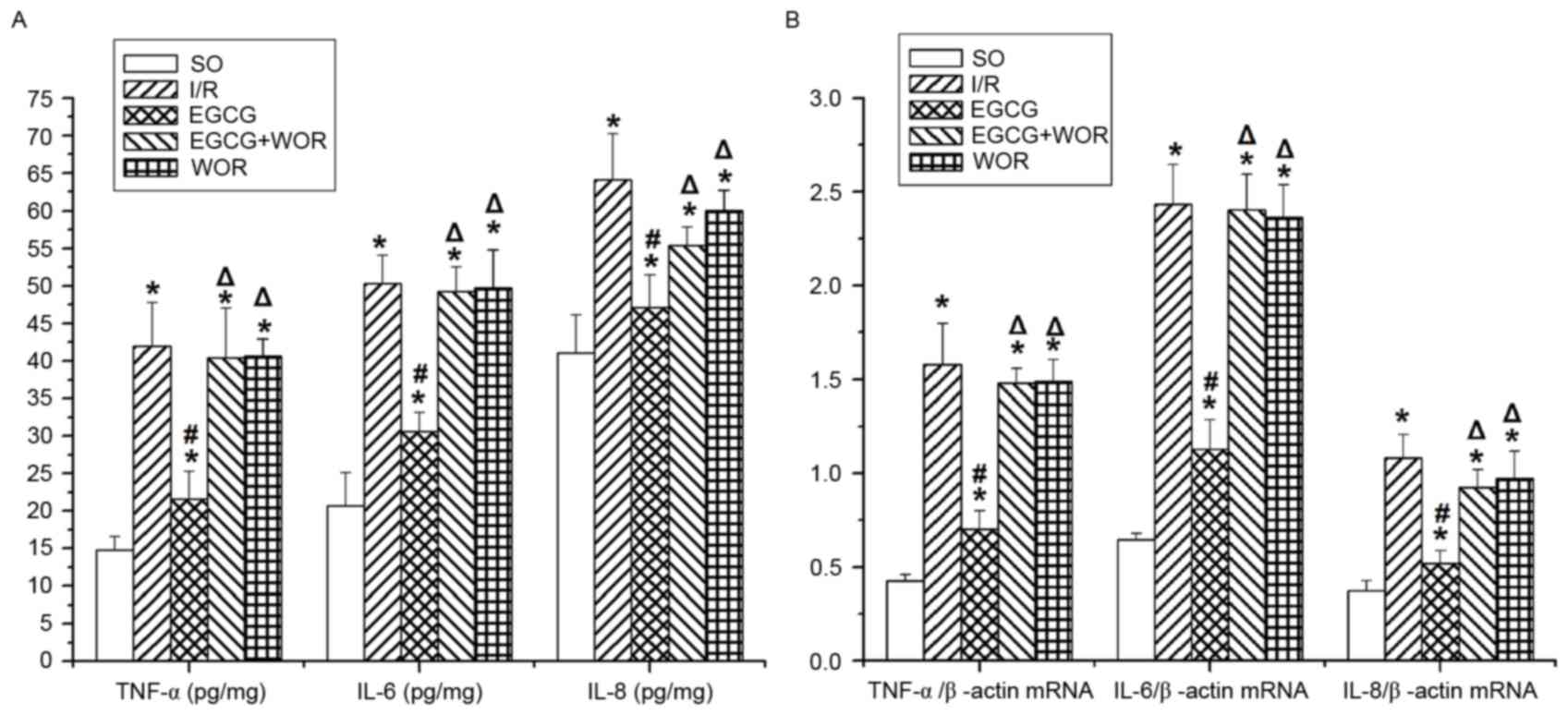
As demonstrated in Fig.
5A and B, the protein levels of TNF-α, IL-6 and IL-8 in heart
tissues were significantly higher in the I/R group than those in
the SO group. Consistently, an I/R injury increased the mRNA
expression of TNF-α by 3.68-fold, IL-6 by 3.75-fold and IL-8 by
2.89-fold compared with those in the SO group. It is noted that
EGCG markedly reduced the protein and mRNA levels of these
cytokines. However, the anti-inflammatory effect of EGCG could be
inhibited by WOR (P<0.05). Similarly, I/R-stimulated TNF-α, IL-6
and IL-8 overproduction remained unaffected in rat hearts treated
with WOR alone, suggesting that the EGCG-activated PI3K/Akt
signaling pathway may serve as a negative feedback mechanism in the
setting of I/R-triggered inflammatory responses.
Positive correlation between mtDNA
level and inflammatory cytokine expression
To further analyze the association between mtDNA and
inflammatory cytokines, a bivariate correlation study was performed
to examine these parameters in all groups with the exception of the
control group. As hypothesized, positive correlations were reached
between mtDNA level and TNF-α (r=0.765, P<0.01), IL-6 (r=0.665,
P<0.01) and IL-8 (r=0.652, P<0.01) expression levels
(Fig. 6A-C), implying that
increasing mtDNA level may contribute to inflammatory cytokine
expression levels.
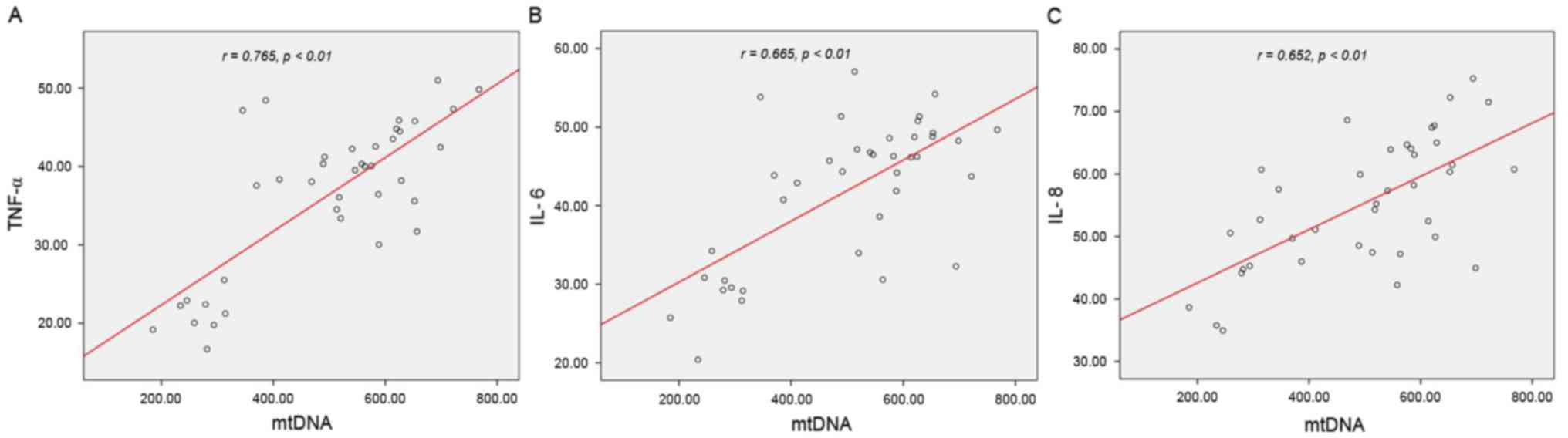 | Figure 6.Positive correlations between mtDNA
level and inflammatory cytokine expression levels. Bivariate
correlation was performed and identified positive correlations
between mtDNA level and (A) TNF-α (r=0.765, P<0.01, (B) IL-6
(r=0.665, P<0.01, and (C) IL-8 (r=0.652, P<0.01, in I/R,
EGCG, EGCG+WOR, WOR groups. mtDNA, mitochondrial DNA; TNF, tumor
necrosis factor; IL, interleukin; I/R, ischemia and reperfusion
group; EGCG, epigallocatechin-3-gallate/epigallocatechin-3-gallate
group; EGCG+WOR, EGCG plus wortmannin group; WOR, wortmannin-only
group. |
Discussion
The present study revealed the beneficial effects of
EGCG postconditioning on preventing myocardial I/R injury in a rat
model. EGCG was demonstrated to limit infarct size and reduced the
severity of myocardial injury and ventricular arrhythmia
effectively. It was also identified that EGCG was able to down
regulate plasma mtDNA levels, and the expression levels of TNF-α,
IL-6 and IL-8 in myocardial tissue following an I/R injury. In
addition, the cardioprotective and anti-inflammatory effects of
EGCG were blocked by a specific PI3K inhibitor. Therefore, the
results suggested that EGCG-induced anti-inflammatory action may
attenuate myocardial I/R injury by reducing mtDNA release under the
activation of the PI3K/Akt signaling pathway.
Existing evidence indicated that I/R is able to
trigger a vigorous inflammatory response. A series of cytokines
generated by infiltrated leukocytes and regional myocytes may
further augment the inflammatory cascades and exacerbate myocardial
injury (3–5). Consistent with previous studies
(8–11), an association was observed between
an increased level of classical pro-inflammatory cytokines (TNF-α,
IL-6 and IL-8) and a more severe reperfusion-induced myocardial
injury in terms of higher ventricular arrhythmia scores, myocardial
kinases levels and larger infarct size. The data suggested a
contributory role for inflammation in the pathogenesis of
myocardial I/R injury. Additionally, the results implied a
promising potential in designing therapeutic strategies aimed at
suppressing inflammatory responses at the time of reperfusion for
an improved cardiovascular outcome.
The cardioprotective effects of green tea have been
ascribed consistently to the antioxidant activity of EGCG, its
major polyphenolic constituent. Indeed, multiple studies have
demonstrated that EGCG could prevent endothelial dysfunction and
reduce myocardial I/R injury by inhibiting the release of reactive
oxygen species (12,15,19,20).
However, several other studies have also identified that EGCG
actually serves as a pro-oxidant in intact cells (21,22),
suggesting that the beneficial actions of EGCG may involve
mechanisms unassociated with its antioxidant capabilities. The
present study identified that EGCG significantly reduced plasma
mtDNA levels and the expression levels of TNF-α, IL-6 and IL-8 in
the myocardial tissue following an I/R injury. Notably, this
reduction was associated with limited infarct size and decreased
myocardial kinases levels. These findings indicated the
effectiveness of EGCG-induced anti-inflammatory action in
protecting hearts against I/R injury.
mtDNA serves as a pro-inflammatory agent, released
following cell injury to induce inflammatory responses, featuring
high expression levels of TNF-α, IL-6 and IL-8 (16,23,24).
It has been reported that abnormally high mtDNA release from
oxidative mitochondria is responsible for triggering subsequent
inflammatory responses (25). Yao
et al (26) identified that
mtDNA escaped from damaged mitochondria functioned as a type of
DAMP to stimulate inflammation through the TL9-RAGE pathway. The
data from the present study also demonstrated a significant
positive association between the serum level of mtDNA and the
expression levels of inflammatory cytokines in all four experiment
groups. Given the antioxidant characteristic of EGCG, the results
indicated that EGCG could efficiently prevent the release of mtDNA
from damaged myocardium, which would further reduce the expression
levels of several inflammatory cytokines and to function as a
protective agent against I/R injury.
PI3K/Akt is an intracellular signaling pathway and
is generally regarded as the primary pro-survival mechanism in the
ischemic-reperfused myocardium (6,7,9,10). A
series of in vitro and in vivo studies have proposed
that activation of the PI3K/Akt signaling by procedures including
ischemic preconditioning or postconditioning, or by administration
of pharmacological agents, is critical for protecting the
myocardium from lethal I/R-induced cell apoptosis (6,7,12,13,15,19,20,22).
Consistent with previous studies, the present study recorded a
significant reduction in myocardial kinases and infarct size in rat
hearts through the PI3K/Akt-dependent signaling mechanism. The data
clearly demonstrate that there is a causal association between EGCG
administration and upregulation of p-p85 and p-Akt, suggesting the
active involvement of PI3K/Akt signaling pathway in EGCG-induced
anti-inflammatory and cardioprotective effects.
There are several limitations in the present study.
The reperfusion duration is relatively short compared with several
other studies (5,11). As sustained activation of the
PI3K/Akt signaling pathway may lead to myocardial fibrosis and
hypertrophy (10,22), in turn compromising the blood and
oxygen supply of the viable myocytes within the risk zone, the role
of EGCG-activated PI3K/Akt signaling in the long-term effects of
cardioprotection remains to be elucidated. In addition,
preconditioning and postconditioning may provide effective
protection against myocardial I/R injury. However, the present
study adopted EGCG postconditioning rather than preconditioning.
The reason for this was that the postconditioning procedures may be
more applicable in clinical practices.
In conclusion, the present study demonstrated that
the anti-inflammatory and cardioprotective effects of EGCG
postconditioning in vivo appear to involve the prevention of
mtDNA release. Therefore, EGCG and related compounds may provide a
novel therapeutic strategy for attenuating myocardial I/R
injury.
Acknowledgementss
The present study was supported in part by the
National Natural Science Foundation of China (grant nos. 81300155
and 81670327).
References
|
1
|
Go AS, Mozaffarian D, Roger VL, Benjamin
EJ, Berry JD, Blaha MJ, Dai S, Ford ES, Fox CS, Franco S, et al:
Heart disease and stroke statistics-2014 update: A report from the
American Heart Association. Circulation. 129:e28–e292. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Carrick D and Berry C: Prognostic
importance of myocardial infarct characteristics. Eur Heart J
Cardiovasc Imaging. 14:313–315. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Minamino T: Cardioprotection from
ischemia/reperfusion injury: Basic and translational research. Circ
J. 76:1074–1082. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hausenloy DJ and Yellon DM: Myocardial
ischemia-reperfusion injury: A neglected therapeutic target. J Clin
Invest. 123:92–100. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sanada S, Komuro I and Kitakaze M:
Pathophysiology of myocardial reperfusion injury: Preconditioning,
postconditioning, and translational aspects of protective measures.
Am J Physiol Heart Circ Physiol. 301:H1723–H1741. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal
T, Junger W, Brohi K, Itagaki K and Hauser CJ: Circulating
mitochondrial DAMPs cause inflammatory responses to injury. Nature.
464:104–107. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yue R, Xia X, Jiang J, Yang D, Han Y, Chen
X, Cai Y, Li L, Wang WE and Zeng C: Mitochondrial DNA oxidative
damage contributes to cardiomyocyte ischemia/reperfusion-injury in
rats: Cardioprotective role of lycopene. J Cell Physiol.
230:2128–2141. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang JZ, Liu Z, Liu J, Ren JX and Sun TS:
Mitochondrial DNA induces inflammation and increases TLR9/NF-κB
expression in lung tissue. Int J Mol Med. 33:817–824. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Williams DL, Ozment-Skelton T and Li C:
Modulation of the phosphoinositide 3-kinase signaling pathway
alters host response to sepsis, inflammation, and
ischemia/reperfusion injury. Shock. 25:432–439. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hu G, Huang X, Zhang K, Jiang H and Hu X:
Anti-inflammatory effect of B-Type natriuretic peptide
postconditioning during myocardial ischemia-reperfusion:
Involvement of PI3K/Akt signaling pathway. Inflammation.
37:1669–1674. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wynne AM, Mocanu MM and Yellon DM:
Pioglitazone mimics preconditioning in the isolated perfused rat
heart: A role for the prosurvival kinases PI3K and P42/44MAPK. J
Cardiovasc Pharmacol. 46:817–822. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Boucher M, Pesant S, Falcao S, de Montigny
C, Schampaert E, Cardinal R and Rousseau G: Post-ischemic
cardioprotection by A2A adenosine receptors: Dependent of
phosphatidylinositol 3-kinase pathway. J Cardiovasc Pharmacol.
43:416–422. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Giakoustidis DE, Giakoustidis AE, Iliadis
S, Koliakou K, Antoniadis N, Kontos N, Papanikolaou V, Papageorgiou
G, Kaldrimidou E and Takoudas D: Attenuation of liver
ischemia/reperfusion induced apoptosis by
epigallocatechin-3-gallate via down-regulation of NF-kappaB and
c-Jun expression. J Surg Res. 159:720–728. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Al-Maghrebi M, Renno WM and Al-Ajmi N:
Epigallocatechin-3-gallate inhibits apoptosis and protects
testicular seminiferous tubules from ischemia/reperfusion-induced
inflammation. Biochem Biophys Res Commun. 420:434–439. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kim HS, Quon MJ and Kim JA: New insights
into the mechanisms of polyphenols beyond antioxidant properties;
lessons from the green tea polyphenol, epigallocatechin 3-gallate.
Redox Biol. 2:187–195. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Qin C, Liu R, Gu J, Li Y, Qian H, Shi Y
and Meng W: Variation of perioperative plasma mitochondrial DNA
correlate with peak inflammatory cytokines caused by cardiac
surgery with cardiopulmonary bypass. J Cardiothorac Surg.
10:852015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Miller LE, Hosick PA, Wrieden J, Hoyt E
and Quindry JC: Evaluation of arrhythmia scoring systems and
exercise-induced cardioprotection. Med Sci Sports Exerc.
44:435–441. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kim JA, Formoso G, Li Y, Potenza MA,
Marasciulo FL, Montagnani M and Quon MJ: Epigallocatechin gallate,
a green tea polyphenol, mediates NO-dependent vasodilation using
signaling pathways in vascular endothelium requiring reactive
oxygen species and Fyn. J Biol Chem. 282:13736–13745. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Elbling L, Weiss RM, Teufelhofer O, Uhl M,
Knasmueller S, Schulte-Hermann R, Berger W and Micksche M: Green
tea extract and (−)-epigallocatechin-3-gallate, the major tea
catechin, exert oxidant but lack antioxidant activities. FASEB J.
19:807–809. 2005.PubMed/NCBI
|
|
21
|
Potenza MA, Marasciulo FL, Tarquinio M,
Tiravanti E, Colantuono G, Federici A, Kim JA, Quon MJ and
Montagnani M: EGCG, a green tea polyphenol, improves endothelial
function and insulin sensitivity, reduces blood pressure, and
protects against myocardial I/R injury in SHR. Am J Physiol
Endocrinol Metab. 292:E1378–E1387. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ai W, Zhang Y, Tang QZ, Yan L, Bian ZY,
Liu C, Huang H, Bai X, Yin L and Li H: Silibinin attenuates cardiac
hypertrophy and fibrosis through blocking EGFR-dependent signaling.
J Cell Biochem. 110:1111–1122. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cao H, Ye H, Sun Z, Shen X, Song Z, Wu X,
He W, Dai C and Yang J: Circulatory mitochondrial DNA is a
pro-inflammatory agent in maintenance hemodialysis patients. PLoS
One. 9:e1131792014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pinti M, Cevenini E, Nasi M, De Biasi S,
Salvioli S, Monti D, Benatti S, Gibellini L, Cotichini R, Stazi MA,
et al: Circulating mitochondrial DNA increases with age and is a
familiar trait: Implications for ‘inflamm-aging’. Eur J Immunol.
44:1552–1562. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhou R, Yazdi AS, Menu P and Tschopp J: A
role for mitochondria in NLRP3 inflammasome activation. Nature.
469:221–225. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yao X, Carlson D, Sun Y, Ma L, Wolf SE,
Minei JP and Zang QS: Mitochondrial ROS induces cardiac
inflammation via a pathway through mtDNA damage in a
pneumonia-related sepsis model. PLoS One. 10:e01394162015.
View Article : Google Scholar : PubMed/NCBI
|