Introduction
Lung transplantation (LT) is performed as a
life-saving treatment for patients with end-stage lung disease
(1). Ischemia-reperfusion injury
(IRI) remains a significant contributor to morbidity and mortality
rates following LT (2). The
release of inflammatory mediators and reactive oxygen species (ROS)
promotes IRI, causing cellular injury, pneumocyte necrosis and
apoptosis (3).
Mitogen-activated protein kinases (MAPKs) are a
family of serine-threonine protein kinases, which are activated in
response to a variety of extracellular and intracellular stimuli,
including cytokines, oxidative stress and growth factors (4). Three major MAPK signaling pathways,
c-Jun NH2-terminal protein kinase (JNK), p38 MAPK (p38)
and extracellular signal-regulated protein kinase 1/2 (ERK 1/2),
regulate a variety of cellular activities, including proliferation,
differentiation, survival and death (5). MAPK activation by IRI in the heart,
liver and lungs has been reported in vitro and in
vivo (6–8). In addition, the inhibition of MAPKs
was shown to be pivotal in mediating the lung inflammatory response
and cell death induced by IRI in our previous studies (9,10).
Considering the significant cross-talk among these three signaling
pathways, how to obtain the optimal protective effects on lung IRI
by combined MAPK inhibition requires clarification.
Pulmonary microvascular endothelial cells (PMVECs)
provide a dynamic and semi-permeable barrier, which is critical for
lung gas exchange, regulation of fluids and solutes, passage of
macromolecules between the blood and interstitial compartments, and
adherence of circulating neutrophils during lung IRI (10–12).
PMVECs are also primary targets of reactive oxygen species (ROS)
and inflammatory mediators, for example, tumor necrosis factor-α,
which can stimulate PMVECs to express adhesion molecules, including
intercellular cell adhesion molecule-1 (10,13),
suggesting that PMVECs are appropriate for investigating IRI in the
pulmonary microcirculation endothelium. Therefore, the aim of the
present study was to evaluate the possible effects of the combined
inhibition of MAPKs in PMVECs of an IRI model of LT.
Materials and methods
Ethical approval
The protocol for the present study was approved by
Institutional Committee on Animal Care and Use of Harbin Medical
University (Harbin, China).
Isolation of rat PMVECS
Pathogen-free male Wistar rats, 4–5 weeks old and
weighing 60–80 g, were obtained from the Animal Experiment Center
of Harbin Medical University. Rats had free access to rodent chow
and water, and were maintained at 24±2°C with a 12 h light-dark
cycle. The rats were anesthetized with an intraperitoneal injection
of 350 mg/kg chloral hydrate (Sigma-Aldrich; Merck Millipore,
Darmstadt, Germany), and heparin (3,000 units) was injected
intraperitoneally. Subsequently, a tracheotomy was performed using
a sterile technique, and the lungs were perfused with medium 199
(M199; Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) and
harvested. The rats were then sacrificed by exsanguination.
The rat PMVECs were isolated according to the
protocols described in our previous study by Tan et al
(10). Briefly, following flushing
of the lungs with phosphate-buffered saline (PBS) at 4°C, the
visceral pleura were stripped from the lung parenchyma to preclude
mesothelial cells. The peripheral lung tissue was finely minced
into small sections (~1 mm3) and placed onto
25-mm2 culture flasks coated with 1% gelatin
(Sigma-Aldrich; Merck Millipore) containing M199 supplemented with
20% fetal calf serum (Invitrogen; Thermo Fisher Scientific, Inc.)
and 50 µg/ml endothelial cell growth supplement (BD Biosciences,
Franklin Lakes, NJ, USA) in a humidified atmosphere of 5%
CO2 at 37°C. Subsequently, 100 U/ml
penicillin-streptomycin (Beyotime Institute of Biotechnology,
Shanghai, China) and 8 µg/ml tylosin (Sigma-Aldrich; Merck
Millipore) were added to the culture medium. Following incubation
at 37°C for 60 h, the tissues were removed, and the M199 was
replaced to remove unattached cells. Contaminating cells were
removed by scraping and aspiration. The rat PMVECs were used for
subsequent experiments at passages 2–5.
Study design
The rat PMVECs were seeded at 1×105
cells/ml in 35-mm-diameter culture dishes 24 h prior to
transfection. The transfection was performed with 100 pmol of small
interfering RNAs (siRNAs) against JNK, p38 and ERK1/2 in the JNK
group, p38 group and ERK1/2 group, respectively. The PMVECs were
also cotransfected with 100 pmol each of the following siRNAs: JNK
siRNA and p38 siRNA in the J+p group, JNK siRNA and ERK1/2 siRNA in
the J+E group, p38 siRNA and ERK1/2 siRNA in the p+E group, and all
three in the J+p+E group. Non-targeting (NT) siRNA was used as a
control in the NT group. All siRNAs were prepared in Opti-MEM
(Invitrogen; Thermo Fisher Scientific, Inc.) and all transfection
procedures were performed using Lipofectamine RNAiMAX (Invitrogen;
Thermo Fisher Scientific, Inc.) for 48 h. The target sequences of
siRNAs were synthesized by Invitrogen; Thermo Fisher Scientific,
Inc., and were as follows: JNK, 5′-UCAAGGAAUAGUGUGUGCAGCUUAU-3′,
p38, 5′-GGACCUCCUUAUAGACGAAUU-3′, and ERK1/2,
5′-GACCGGAUGUUAACCUUUAUU-3′.
Simulated IR
Following transfection with the siRNAs, the PMVECs
were exposed in a sealed container to simulate the rapid
environmental changes during LT and were pre-ventilated with 95%
O2/5% CO2 at 1 l/min for 2 h, as described
previously (10).
Simulated cold storage
The container was placed in a refrigerator (4°C),
and M199 was immediately replaced with low-potassium dextran
solution (Vitrolife, Kungsbacka, Sweden) with gas insufflation
stoppage for 6 h.
Simulated implantation
The simulated implantation was performed by removing
the container from the refrigerator and allowing it to return room
temperature for 1 h.
Simulated reperfusion
Following the replacement of low-potassium dextran
solution immediately with M199 pre-heated to 37°C, the container
was ventilated with 50% O2/5% CO2/45%
N2 for 2 h. The culture dishes were then removed from
the container for detection. Gas concentrations in the container
were monitored with a gas analyzer (S/N 32590; Datex Ohmeda,
Helsinki, Finland).
Western blot analysis
The rat PMVECs in culture dishes were washed twice
with ice-cold PBS, and trypsinized and lysed in RIPA buffer
(Beyotime Institute of Biotechnology) supplemented with 1%
phenylmethanesulfonyl fluoride (Beyotime Institute of
Biotechnology). The PMVECs lysates were centrifuged at 13,201 × g
at 4°C for 20 min and the supernatants were collected. Protein
concentrations were determined using a BCA Protein Assay kit
(Beyotime Institute of Biotechnology) and proteins were boiled with
loading buffer at 95°C for 5 min. Protein samples (~20 µg) were
separated by 12% sodium dodecyl sulfate polyacrylamide gel
electrophoresis and transferred onto polyvinylidene fluoride
membranes. These membranes were blocked in 5% non-fat dried milk
for 2 h, and then incubated with primary polyclonal rabbit anti-rat
antibodies (1:1,000 dilution; Cell Signaling Technology, Inc.,
Danvers, MA, USA) against JNK (cat. no. 9252), p38 (cat. no. 9218),
ERK1 (cat. no. 4372) or ERK2 (cat. no. 9108) overnight at 4°C.
Following conjugation with a diluted horseradish peroxidase-labeled
secondary antibody (1:5,000; goat anti-rabbit IgG; cat. no. 7074;
Cell Signaling Technology, Inc.) at room temperature for 1 h. The
membranes were exposed to enhanced chemiluminescence (cat. no.
GIS2010; Tanon Science and Technology Co., Ltd., Shanghai, China),
and quantified using ImageJ 1.48v software (National Institutes of
Health, Bethesda, MD, USA). The expression levels of the measured
proteins were determined as the ratio of target proteins to that of
β-actin (1:1,000; cat. no. TA-09; Zhongshan Golden Bridge
Biotechnology, Beijing, China).
Measurements of inflammatory cytokines
and oxidation-reduction markers
Culture medium was collected and centrifuged at 573
× g at 4°C for 20 min. The culture medium concentrations of IL-1β
and IL-6 were assessed using an enzyme-linked immunosorbent assay
according to the manufacturer's protocol (R&D Systems, Inc.,
Minneapolis, MN, USA). The cells were homogenized in 100 µl of
ice-cold PBS and centrifuged at 13,000 × g at 4°C for 10 min. The
supernatants were collected and used to measure cellular levels of
malondialdehyde (MDA) and activity of superoxide dismutase (SOD)
using commercial kits (Nanjing Jiancheng Bioengineering Institute,
Nanjing, China).
Flow cytometry
An Annexin V-fluorescein isothiocyanate (FITC)
apoptosis detection kit (BD Biosciences) was used to detect
apoptosis. The cells were collected by trypsinization, washed twice
with ice-cold PBS and centrifuged at 297 × g at 4°C for 5 min.
Subsequently, the cells were resuspended in binding buffer and
adjusted to a density of 106 cells/ml. Equivalent
quantities of Annexin V-FITC and propidium iodide (PI) were added
to the cell suspension, followed by incubation in the dark at room
temperature for 10 min. Cell apoptosis was then determined using
flow cytometry (FACSort; BD Biosciences) according to the
manufacturer's protocol.
Statistical analysis
All data are expressed as the mean ± standard
deviation, and all experiments were repeated at least three times.
Differences between compared groups were determined using Tukey's
honest significant difference test. Statistical analysis was
performed using SPSS 22.0 software (IBM Corp., Armonk, NY, USA).
P<0.05 was considered to indicate a statistically significant
difference.
Results
Rat PMVEC characteristics
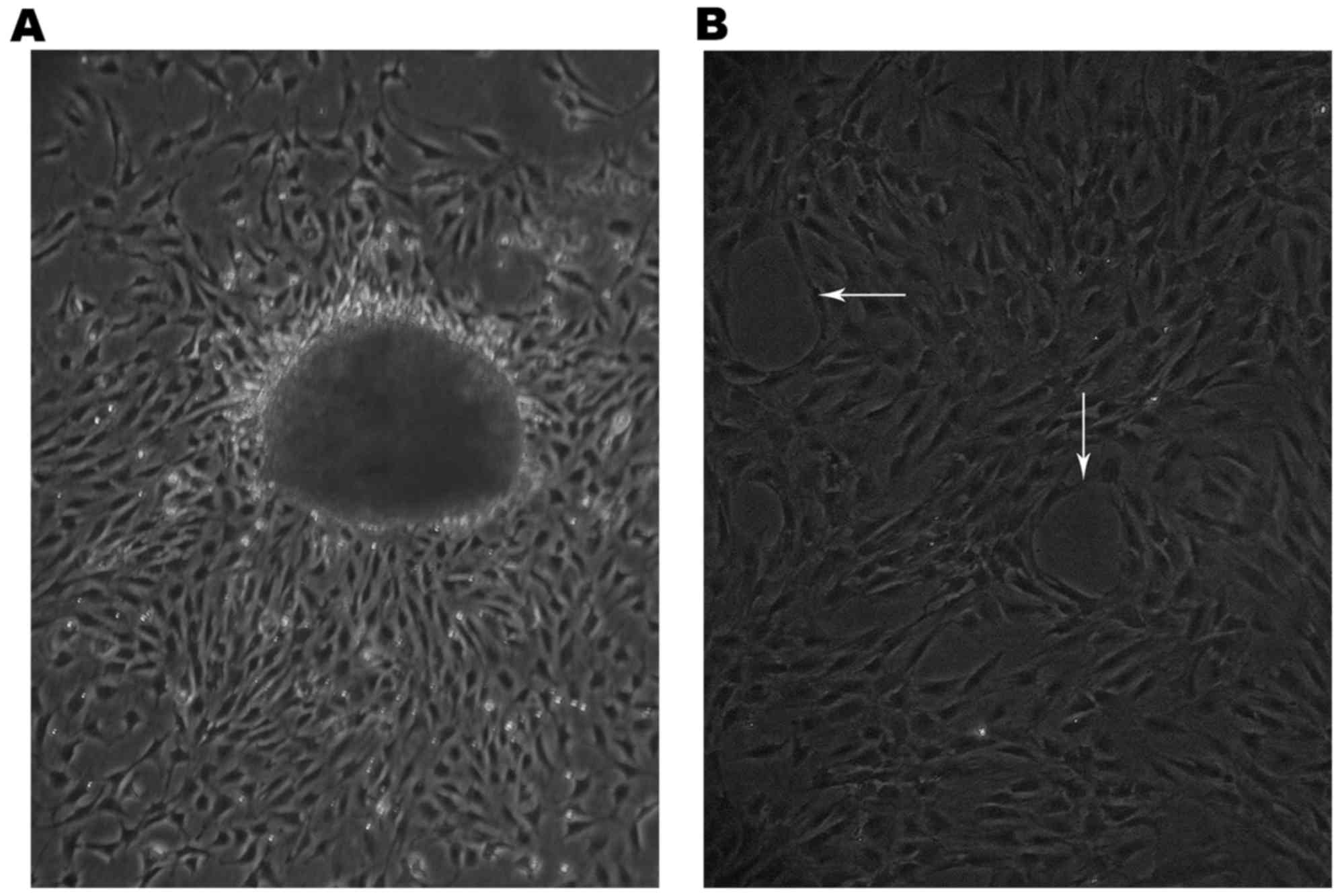
The cells had migrated from the edge of the small
lung tissues 60 h following tissue plating (Fig. 1A) and grew as capillary-like
structures on the gelatin (white arrows, Fig. 1B).
Protein expression of MAPKs
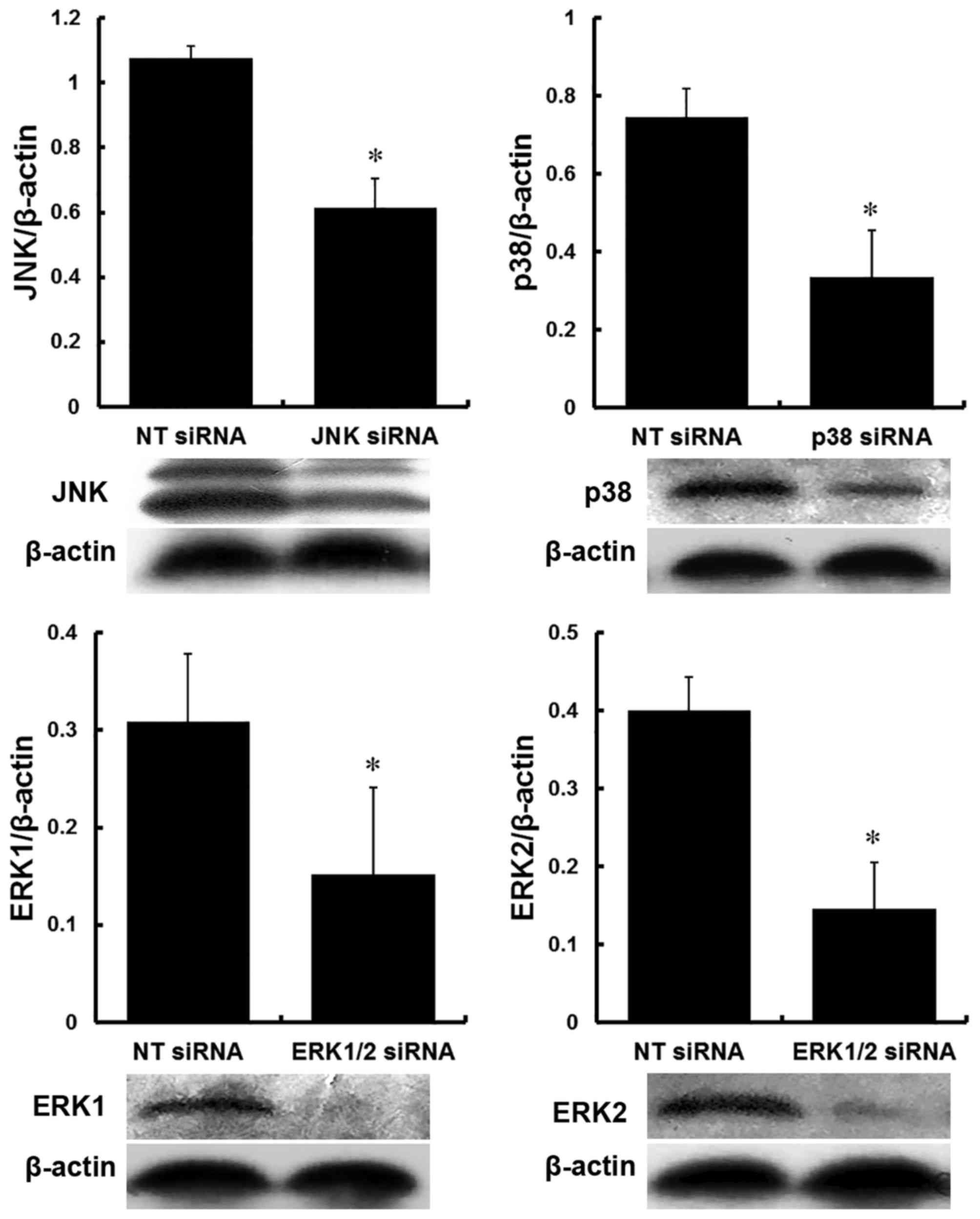
The protein expression levels of JNK, p38, ERK1 and
ERK2 in the PMVECs were reduced by >35% following transfection
with siRNAs against JNK, p38 and ERK1/2, compared with those in
cells transfected with NT siRNA. The protein inhibition ratio to
JNK was 42.37±9.80% following transfection with JNK siRNA
(P<0.05); inhibition to p38 was 55.70±12.90% following
transfection with p38 siRNA (P<0.05); inhibition ratios to ERK1
and ERK2 were 53.01±20.78 and 63.16±17.28%, respectively, following
transfection with ERK1/2 siRNA (P<0.05; Fig. 2).
Inflammatory cytokines and
oxidation-reduction markers
Compared with the NT group, significant decreases in
the levels of IL-1β were observed in all other groups following
simulation of IRI (P<0.05). In addition, there were significant
reductions in the J+p and J+p+E groups, compared with the rest of
the groups (P<0.05). No significant difference was observed
between the J+p and J+p+E groups (P>0.05; Fig. 3A).
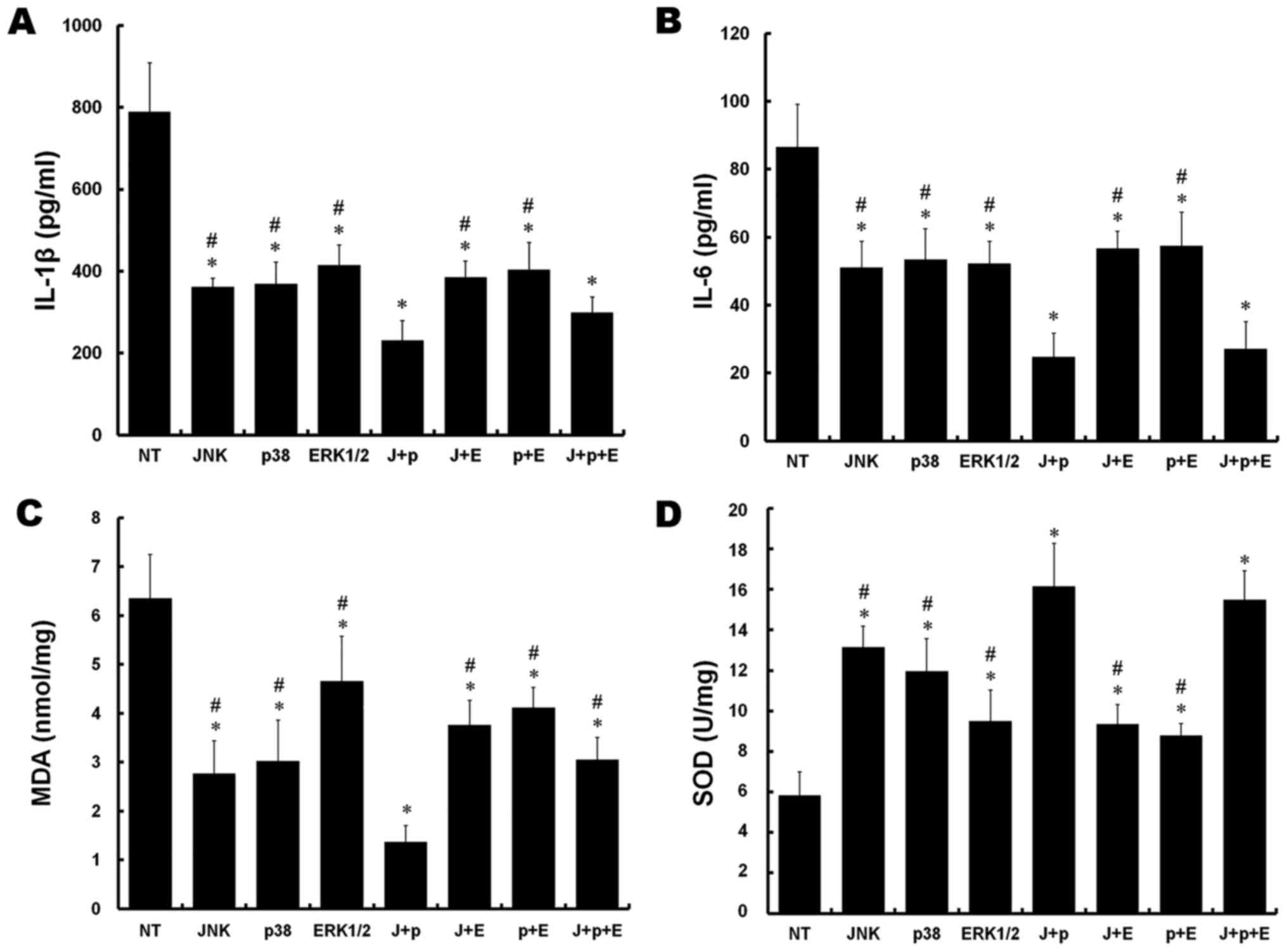 | Figure 3.Effect of silencing of MAPKs on
pro-inflammatory cytokines and oxidation-reduction markers in rat
PMVECs. (A) Culture medium concentrations of IL-1β. (B) Culture
medium concentrations of IL-6. (C) MDA levels. (D) SOD activity.
*P<0.05, vs. NT group; #P<0.05, vs. J+P group.
J+p, JNK and p38; J+E, JNK and ERK1/2; p+E, p38 and ERK1/2; and
J+p+E, JNK, p38 and ERK1/2. PMVECs, pulmonary microvascular
endothelial cells; MAPKs, mitogen-activated protein kinases; IL,
interleukin; MDA, malondialdehyde; SOD, superoxide dismutase; JNK,
c-Jun NH2-terminal protein kinase; ERK, extracellular
signal-regulated kinase; NT, non-targeting. |
The levels of IL-6 were decreased in all groups,
compared with that in the NT group (P<0.05). In addition, the
levels of IL-6 were decreased in the J+p and J+p+E groups, compared
with the other groups (P<0.05). No significant difference was
observed between the J+p and J+p+E groups (P>0.05, Fig. 3B).
The levels of MDA were substantially decreased in
all groups transfected or cotransfected with target siRNAs,
compared with that in the NT group (P<0.05). In addition, the
level of MDA in the J+p group was significantly decreased, compared
with the levels in the other groups (P<0.05; Fig. 3C).
Compared with the NT group, increased SOD activity
was observed in all other groups (P<0.05). The activities of SOD
in the J+p and J+p+E groups increased significantly, compared with
those in the other groups (P<0.05), however, no significant
differences were observed between the J+p and J+p+E groups
(P>0.05; Fig. 3D).
Early apoptosis
Annexin V− and PI− cells were
used as controls, and Annexin V+ and PI−
cells are representative of early-apoptotic cells. The presence of
Annexin V+ and PI+ cells is considered as a
sign of late apoptosis, whereas Annexin V− and
PI+ cells are considered necrotic. In the NT group,
numerous early-apoptotic cells were observed. Compared with the NT
group, the percentages of early-apoptotic cells were decreased in
the JNK, p38, ERK, J+p, J+E, and J+p+E groups (P<0.05). In
addition, the percentages of early-apoptotic cells in the J+p and
J+p+E groups were decreased significantly, compared with those in
the other groups (P<0.05). No significant differences were
observed between the J+p and J+p+E groups (P>0.05; Fig. 4).
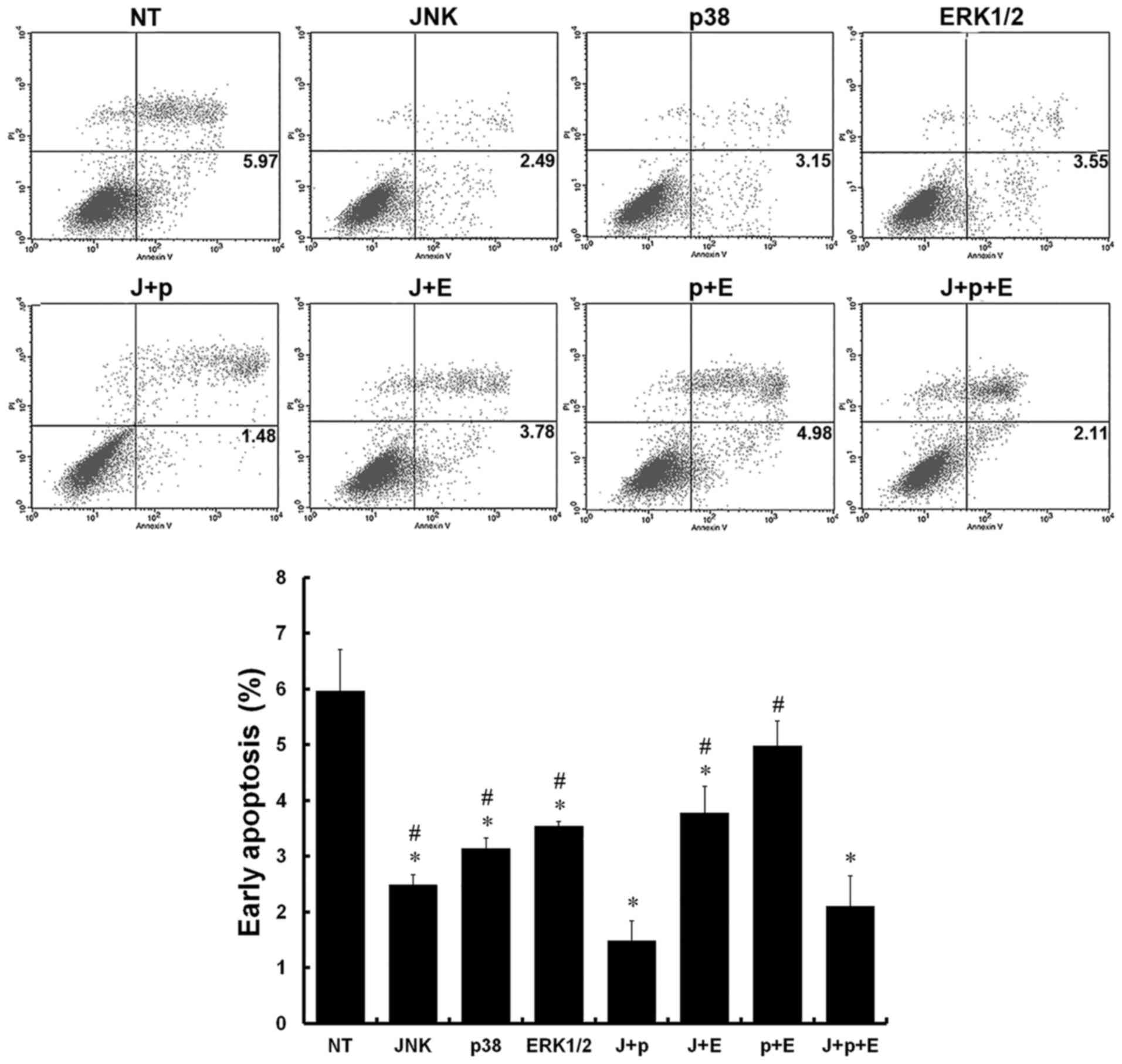 | Figure 4.Effect of silencing MAPKs on early
apoptosis. Stages of apoptosis were detected using flow cytometry
in rat PMVECs. *P<0.05, vs. NT group; #P<0.05, vs.
J+p group. J+p, JNK and p38; J+E, JNK and ERK1/2; p+E, p38 and
ERK1/2; and J+p+E, JNK, p38 and ERK1/2. PMVECs, pulmonary
microvascular endothelial cells; MAPKs, mitogen-activated protein
kinases; IL, interleukin; MDA, malondialdehyde; SOD, superoxide
dismutase; JNK, c-Jun NH2-terminal protein kinase; ERK,
extracellular signal-regulated kinase; NT, non-targeting; PI,
propidium iodide. |
Discussion
The major finding of the present study was that the
dual inhibition of JNK and p38 decreased the levels of IL-1β, IL-6
and MDA, and percentage of early-apoptotic cells, and increased the
activity of SOD in PMVECs in an IRI model of LT. In addition, the
inhibition of ERK1/2 had a marginal effect on the levels of IL-1β,
IL-6 and SOD, and on early apoptosis, compared with the effects of
the dual inhibition of JNK and p38.
Due to their ease of harvesting and being relatively
inexpensive, several cell types have been used to investigate lung
IRI previously, including pulmonary artery endothelial cells
(PAECs) and human umbilical vein endothelial cells (HUVECs)
(14,15). PMVECs were used as the cell model
to mimic lung IRI in the present study for several reasons. PMVECs
are the early target cells of IRI and important in the initiation
and development of pulmonary inflammation (16). Significant differences between
PMVECs and PAECs have also been confirmed, including in morphology,
proliferation, endothelial function, and endothelial barrier
integrity (17,18). Previously, the replacement of
PMVECs with HUVECs to investigate lung IRI showed that they differ
from PMVECs in terms of biological properties and immune
recognition (19). These
discrepancies suggest that PMVECs may be more appropriate for
investigating IRI in the pulmonary microcirculation
endothelium.
Activation of the immune system is important in lung
IRI (20). When the immune
response is activated, pattern recognition molecules, including
Toll-like receptors, are activated, triggering the MAPK signaling
pathways (21). Ultimately, MAPKs
induce the production of pro-inflammatory cytokines and chemokines,
which contribute significantly to lung IRI (22,23).
As MAPKs are important in aggravating lung IRI, inhibiting the MAPK
pathways may be an effective way to ameliorate lung IRI. Therefore,
the present study inhibited MAPKs to identify a method to alleviate
lung IRI and investigate the role of MAPKs, and examine the effects
on lung IRI.
siRNAs are a novel class of RNA inhibitors, which
specifically degrade target RNAs via the RNA-induced silencing
complex (24). They have become
increasingly popular as a potential technique for silencing
specific genes due to their higher specificity, higher
effectiveness, lower dose and fewer side effects, compared with
inhibitors used in previous studies (25,26).
In the present study, due to the transfection with siRNAs, the
protein expression levels of JNK, p38, ERK1 and ERK2 in the PMVECs
were reduced by >40%, which indicated that the MAPK pathways
were inhibited significantly and provided a suitable system for
investigating the role of MAPKs in lung IRI.
Previous studies have investigated the functions of
JNK, p38 and ERK1/2 alone and in combination; however, comparison
of their therapeutic efficacies has not been evaluated. Due to the
substantial cross-talk among MAPKs, circumscribed investigations or
speculative hypotheses may ignore potential possibilities. In the
present study, a comprehensive method was used to investigate the
functions of MAPKs in IRI. JNK, p38 and ERK1/2 were silenced, and
the independent function of each pathway during IRI was confirmed.
In addition, to observe the effect of multiple inhibitions on IRI,
gene silencing of any two of the MAPKs or all three was performed.
This enabled determination of a preferred therapeutic strategy
through the inhibition of MAPKs to ameliorate IRI, using reliable
data rather than theoretical possibilities.
Cargnello and Roux (4) and Murayama et al (27) found that JNK and p38 are important
components involved in inflammation and apoptosis. The results of
the present study showed that the inhibition of JNK or p38
decreased the levels of IL-1β and IL-6, and decreased the
percentage of early-apoptotic cells. Therefore, the activation of
JNK and p38 was confirmed to be pro-inflammatory and pro-apoptotic
in the model, which was similar to the results reported Zhang et
al and Liou et al (28,29).
ERK1/2 has been suggested to be pro-inflammatory and pro-apoptotic
(10,30), anti-apoptotic (31), or not involved in inflammation or
apoptosis (32). However, the true
effects of the ERK1/2 pathway differ depending on the stimuli and
cell types (33). In the present
study, the inhibition of ERK1/2 led to reductions in inflammation
and apoptosis. The data indicated that ERK1/2 was a
pro-inflammatory and pro-apoptotic pathway in the IRI model. It has
been reported that MAPK proteins can be activated by oxidative
stress (34,35), whereas JNK mediates the increase of
ROS production during stress (36), and the inhibition of p38 and ERK1/2
pathways has been shown to be involved in resistance to oxidative
stress during renal IRI (37). The
data obtained in the present study suggested that the silencing of
JNK, p38 or ERK1/2 attenuated oxidative stress to different
extents. Therefore, the inhibition of JNK, p38 or ERK1/2 may
ameliorate lung IRI via anti-inflammatory, anti-apoptotic and
anti-oxidative mechanisms.
The results of the present study showed that the
inhibition of JNK or p38 alone decreased the levels of IL-1β, IL-6
and MDA, and the percentage of early-apoptotic cells, and increased
the activity of SOD. The dual inhibition of these two kinases
further increased this effect. In addition, the dual inhibition of
JNK and p38 was the most effective technique for attenuating
inflammation, apoptosis and oxidative stress in the IRI model of
the present study. There are several possible reasons for these
results. JNK interacts with and shares components with p38. These
respond to common upstream activators and phosphorylate common
downstream targets (28). JNK and
p38 phosphorylate pro-apoptotic protein B-cell lymphoma
2-interacting modulator of cell death at the same site to initiate
apoptosis and also activate effector caspases, including caspase 3,
cooperatively (31,32). They can also regulate cytokine
expression by modulating transcription factors, including nuclear
factor-κB (4). Owing to the
significant cross-talk, the silencing of either JNK or p38 alone is
not sufficient to ameliorate apoptosis, inflammation or oxidative
stress, therefore, the dual inhibition of the two kinases is
important to obtain substantial amelioration. The gene silencing of
JNK, p38 and ERK1/2 simultaneously had a notable effect on
attenuating IRI. However, compared with the dual inhibition of JNK
and p38, the additional inhibition of ERK1/2 had no increased
positive effect on IRI, only resulting economic loss and waste of
resources.
A limitation of the present study was that the
mechanism underlying the interactions of MAPKs in IRI were not
precisely determined. For example, the common downstream signaling
pathways of JNK and p38, which can enhance the protective effect on
IRI, remain to be elucidated. In addition, artificial cell model
cannot completely mimic the actual physiological processes during
LT, including alloimmunity and lung compliance.
In conclusion, the dual inhibition of JNK and p38
led to maximal amelioration of lung IRI via anti-inflammatory,
anti-oxidative and anti-apoptotic mechanisms. These results
demonstrated an optimal protective measure in MAPK pathways during
lung IRI, and provide a therapeutic strategy against lung IRI
induced by transplantation for further animal experiments and
clinical applications.
Acknowledgements
This study was supported by the Nature Science
Foundation of Heilongjiang Province (Youth, grant no. QC2015125)
and the Research Foundation of the Second Affiliated Hospital of
Harbin Medical University (grant no. KYBS201505).
References
|
1
|
What is lung transplantation? Am J Respir
Crit Care Med. 192:P7–P8. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen F and Date H: Update on
ischemia-reperfusion injury in lung transplantation. Curr Opin
Organ Transplant. 20:515–520. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hu R, Chen ZF, Yan J, Li QF, Huang Y, Xu
H, Zhang XP and Jiang H: Endoplasmic reticulum stress of
neutrophils is required for ischemia/reperfusion-induced acute lung
injury. J Immunol. 195:4802–4809. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cargnello M and Roux PP: Activation and
function of the MAPKs and their substrates, the MAPK-activated
protein kinases. Microbiol Mol Biol Rev. 75:50–83. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kim EK and Choi EJ: Pathological roles of
MAPK signaling pathways in human diseases. Biochim Biophys Acta.
1802:396–405. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yue TL, Wang C, Gu JL, Ma XL, Kumar S, Lee
JC, Feuerstein GZ, Thomas H, Maleeff B and Ohlstein EH: Inhibition
of extracellular signal-regulated kinase enhances
Ischemia/Reoxygenation-induced apoptosis in cultured cardiac
myocytes and exaggerates reperfusion injury in isolated perfused
heart. Circ Res. 86:692–699. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li J, Wang F, Xia Y, Dai W, Chen K, Li S,
Liu T, Zheng Y, Wang J, Lu W, et al: Astaxanthin pretreatment
attenuates hepatic ischemia reperfusion-induced apoptosis and
autophagy via the ROS/MAPK pathway in mice. Mar Drugs.
13:3368–3387. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sakiyama S, Hamilton J, Han B, Jiao Y,
Shen-Tu G, de Perrot M, Keshavjee S and Liu M: Activation of
mitogen-activated protein kinases during human lung
transplantation. J Heart Lung Transplant. 24:2079–2085. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lv X, Tan J, Liu D, Wu P and Cui X:
Intratracheal administration of p38α short-hairpin RNA plasmid
ameliorates lung ischemia-reperfusion injury in rats. J Heart Lung
Transplant. 31:655–662. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tan J, Liu D, Lv X, Wang L, Zhao C, Che Y,
Xie Q and Cui X: MAPK mediates inflammatory response and cell death
in rat pulmonary microvascular endothelial cells in an
ischemia-reperfusion model of lung transplantation. J Heart Lung
Transplant. 32:823–831. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang N, Zhang D, Sun G, Zhang H, You Q,
Shao M and Yue Y: Lipopolysaccharide-induced caveolin-1
phosphorylation-dependent increase in transcellular permeability
precedes the increase in paracellular permeability. Drug Des Devel
Ther. 9:4965–4977. 2015.PubMed/NCBI
|
|
12
|
Audia JP, Lindsey AS, Housley NA, Ochoa
CR, Zhou C, Toba M, Oka M, Annamdevula NS, Fitzgerald MS, Frank DW
and Alvarez DF: In the absence of effector proteins, the
Pseudomonas aeruginosa type three secretion system needle tip
complex contributes to lung injury and systemic inflammatory
responses. PLoS One. 8:e817922013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yuan Q, Jiang YW, Ma TT, Fang QH and Pan
L: Attenuating effect of Ginsenoside Rb1 on LPS-induced lung injury
in rats. J Inflamm. 11:402014. View Article : Google Scholar
|
|
14
|
McCourtie AS, Merry HE, Farivar AS, Goss
CH and Mulligan MS: Alveolar macrophage secretory products augment
the response of rat pulmonary artery endothelial cells to hypoxia
and reoxygenation. Ann Thorac Surg. 85:1056–1060. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Casiraghi M, Tatreau JR, Abano JB,
Blackwell JW, Watson L, Burridge K, Randell SH and Egan TM: In
vitro modeling of nonhypoxic cold ischemia-reperfusion simulating
lung transplantation. J Thorac Cardiovasc Surg. 138:760–767. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yuan Q, Jiang YW and Fang QH: Improving
effect of Sivelestat on lipopolysaccharide-induced lung injury in
rats. APMIS. 122:810–817. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Parra-Bonilla G, Alvarez DF, Al-Mehdi AB,
Alexeyev M and Stevens T: Critical role for lactate dehydrogenase A
in aerobic glycolysis that sustains pulmonary microvascular
endothelial cell proliferation. Am J Physiol Lung Cell Mol Physiol.
299:L513–L522. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Obiako B, Calchary W, Xu N, Kunstadt R,
Richardson B, Nix J and Sayner SL: Bicarbonate disruption of the
pulmonary endothelial barrier via activation of endogenous soluble
adenylyl cyclase, isoform 10. Am J Physiol Lung Cell Mol Physiol.
305:L185–L192. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dib H, Chafey P, Clary G, Federici C, Le
Gall M, Dwyer J, Gavard J, Tamas N, Bussone G, Broussard C, et al:
Proteomes of umbilical vein and microvascular endothelial cells
reflect distinct biological properties and influence immune
recognition. Proteomics. 12:2547–2555. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Carroll MC and Holers VM: Innate
autoimmunity. Adv Immunol. 86:137–157. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen GY and Nuñez G: Sterile inflammation:
Sensing and reacting to damage. Nat Rev Immunol. 10:826–837. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Weyker PD, Webb CA, Kiamanesh D and Flynn
BC: Lung ischemia reperfusion injury: A bench-to-bedside review.
Semin Cardiothorac Vasc Anesth. 17:28–43. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ferrari RS and Andrade CF: Oxidative
stress and lung ischemia-reperfusion injury. Oxid Med Cell Longev.
2015:5909872015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Aljuffali IA, Lin YK and Fang JY:
Noninvasive approach for enhancing small interfering RNA delivery
percutaneously. Expert Opin Drug Deliv. 13:265–280. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kesharwani P, Gajbhiye V and Jain NK: A
review of nanocarriers for the delivery of small interfering RNA.
Biomaterials. 33:7138–7150. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chopra P, Kanoje V, Semwal A and Ray A:
Therapeutic potential of inhaled p38 mitogen-activated protein
kinase inhibitors for inflammatory pulmonary diseases. Expert Opin
Investig Drugs. 17:1411–1425. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Murayama T, Tanabe M, Matsuda S, Shimazu
M, Kamei S, Wakabayashi G, Kawachi S, Matsumoto K, Yamazaki K,
Matsumoto K, et al: JNK (c-Jun NH2 terminal kinase) and p38 during
ischemia reperfusion injury in the small intestine.
Transplantation. 81:1325–1330. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang X, Bedard EL, Potter R, Zhong R,
Alam J, Choi AM and Lee PJ: Mitogen-activated protein kinases
regulate HO-1 gene transcription after ischemia-reperfusion lung
injury. Am J Physiol Lung Cell Mol Physiol. 283:L815–L829. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Liou SF, Ke HJ, Hsu JH, Liang JC, Lin HH,
Chen IJ and Yeh JL: San-Huang-Xie-Xin-Tang prevents rat hearts from
ischemia/reperfusion-induced apoptosis through eNOS and MAPK
pathways. Evid Based Complement Alternat Med. 2011:9150512011.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Arthur JS and Ley SC: Mitogen-activated
protein kinases in innate immunity. Nat Rev Immunol. 13:679–692.
2013. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Darling NJ and Cook SJ: The role of MAPK
signalling pathways in the response to endoplasmic reticulum
stress. Biochim Biophys Acta. 1843:2150–2163. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Engelbrecht AM, Niesler C, Page C and
Lochner A: p38 and JNK have distinct regulatory functions on the
development of apoptosis during simulated ischaemia and reperfusion
in neonatal cardiomyocytes. Basic Res Cardiol. 99:338–350. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Dhanasekaran DN and Reddy EP: JNK
signaling in apoptosis. Oncogene. 27:6245–6251. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Garcia-Fernandez LF, Losada A, Alcaide V,
Alvarez AM, Cuadrado A, González L, Nakayama K, Nakayama KI,
Fernández-Sousa JM, Muñoz A, et al: Aplidin induces the
mitochondrial apoptotic pathway via oxidative stress-mediated JNK
and p38 activation and protein kinase C delta. Oncogene.
21:7533–7544. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wei H, Li Z, Hu S, Chen X and Cong X:
Apoptosis of mesenchymal stem cells induced by hydrogen peroxide
concerns both endoplasmic reticulum stress and mitochondrial death
pathway through regulation of caspases, p38 and JNK. J Cell
Biochem. 111:967–978. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sehgal V and Ram PT: Network motifs in JNK
signaling. Genes Cancer. 4:409–413. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ka SO, Hwang HP, Jang JH, Hyuk Bang I, Bae
UJ, Yu HC, Cho BH and Park BH: The protein kinase 2 inhibitor
tetrabromobenzotriazole protects against renal ischemia reperfusion
injury. Sci Rep. 5:148162015. View Article : Google Scholar : PubMed/NCBI
|