Introduction
Head and neck squamous cell carcinoma (HNSCC) is the
sixth leading type of cancer globally, of which oral cavity
squamous cell carcinoma (OCSCC) accounts for ~50% of cases
(1). Etiologically, tobacco
smoking and alcohol consumption are two of the major risk factors
for OCSCC (2) Patients with OCSCC
often present with local invasion and lymph node metastases, but
usually without distant metastases. Oral tongue squamous cell
carcinoma (OTSCC) is a form of OCSCC that originates from the
anterior region of the tongue (3).
In addition to tobacco and alcohol, infection with human papilloma
virus (HPV), particularly HPV16, is strongly associated with the
occurrence of tongue cancer (4).
OCSCC, including OTSCC, is classified into stages I–IV according to
the tumor, node and metastasis (TNM) staging system of the American
Joint Committee on Cancer (AJCC) or the International Union for
Cancer Control (5). For the AJCC
stage I and II early OTSCC, the 5-year survival rates were reported
to be 67 and 51%, respectively, based on a population-based
database termed Surveillance, Epidemiology and End Results (SEER)
program (6). While, for stage III
and IV advanced OTSCC, the 5-year disease-specific survival rates
were 39 and 27%, respectively (7).
Oral carcinogenesis is a complex, diverse and successive process
that includes normal, dysplasia and carcinoma stages, and the tumor
microenvironment (TME) has an important role in malignant
transformation (8,9).
Microarrays based on gene expression are novel tools
that are employed to understand the multiple interactions and
identify the core networks of cancer (10) Publicly available microarray data
and innovative bioinformatics tools have improved the ability to
perform genome-wide expression studies independently. Various
genetic changes have been identified by computational statistical
methods in certain gene expression arrays involving HNSCC (11,12).
However, identification of the key genes or pathways involved in
the development of oral carcinogenesis is required, which will
contribute to the discovery of core mechanical interactions and the
development of targeted therapy for OTSCC.
It is hypothesized that the expression of certain
genes is dysregulated during oral cancer carcinogenesis. Wu et
al (13) investigated
interleukin (Il) 1β as the key gene in the TME during oral
carcinogenesis via bioinformatic network construction, which was
verified by immunohistochemical staining of human and rat samples.
Subsequently, chemoprevention targeting at Il1 was confirmed to
interrupt oral carcinogenesis in in vitro and in vivo
experiments. However, in carcinogenesis, modules composed of
functional genetic clusters usually cooperate together to fulfill
specific functions, rather than a single key gene.
The present study performed gene expression profile
analysis based on public microarray data from the Gene Expression
Omnibus (GEO). By using GSE42780, the current study compared
normal, dysplastic and carcinoma tissues in the TME. Differentially
expressed genes (DEGs) were identified and key pathways of DEGs
were enriched. In addition, a protein-protein interaction (PPI)
network was constructed and analyzed to identify the potential hub
modules of OTSCC.
Materials and methods
Microarray data
The gene expression data of the microarray GSE42780,
based on the platform of GPL1355 (Affymetrix Rat Genome 230 2.0
Array), was downloaded from the GEO (www.ncbi.nlm.nih.gov/geo) of the National Center of
Biotechnology Information. For GSE42780, following establishment of
the 4-nitroquinoline oxide-induced tongue cancer model, RNA samples
of rat tongue epithelia and submucosal fibroblasts were collected
for gene microarray scanning, including 3 from normal control, 3
from dysplasia and 3 from carcinoma, as described by Wu et
al (13).
Data processing
All of the data analysis in the present study was
conducted by R statistical software version 3.3 (www.r-project.org/) along with an open source software
for bioinformatics called Bioconductor (version 3.3; bioconductor.org/). Following robust multichip average
background correcting, quantile normalizing and calculation of
expression using the affy package (www.bioconductor.org/packages/3.3/bioc/html/affy.html;
version 1.50.0), the gene expression profile for all samples was
obtained. Subsequently, a linear model was fitted and empirical
Bayes statistics were computed by using the limma package (version
3.28.21; www.bioconductor.org/packages/3.3/bioc/html/limma.html).
As a result, the significant DEGs among groups of normal control,
dysplasia and carcinoma in tongue epithelia and submucosal
fibroblasts samples were identified using a Venn diagram webtool
(bioinformatics.psb.ugent.be/webtools/Venn/). Significant DEGs were
selected with criteria of P<0.01 and |log2 fold change (FC)|
≥1.5.
Gene Ontology (GO) enrichment and
Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway
analysis
GO (http://geneontology.org) offers a biological model
that classifies gene functions into biological process, molecular
function and cellular component (14). KEGG (www.genome/ad.jp/kegg/) is a database regarding
genomes, biological pathways, diseases, drugs and chemical
substances (15). The Database for
Annotation, Visualization and Integrated Discovery (DAVID;
https://david.ncifcrf.gov/), an
integration of biological data and analysis tools, provides
comprehensive functional annotation tools for a large number of
genes (16). In the current study,
GO annotation analysis and KEGG pathway enrichment analysis of DEGs
was performed using DAVID tools online. P<0.05 and gene counts
>2 were considered indicate significance.
PPI construction
The Search Tool for the Retrieval of Interacting
Genes/Proteins (STRING; string-db.org/) database provides a critical
assessment and integration of PPI, including direct (physical) and
indirect (functional) associations in a given organism (15). In the current study, the DEGs that
were identified previously were uploaded to the STRING database to
construct a PPI network, which provided improved comprehension of
the physical and functional organization of the proteome
systematically. Subsequently, the PPI network was reconstructed
with Cytoscape software (version 3.3.0; www.cytoscape.org/). As nodes with a high degree of
connectivity contribute more to the stability of the network, the
connectivity degree of each protein node in the PPI network was
calculated and the top five were identified as the hub nodes using
the Cytoscape plugin NetworkAnalyzer (release 2.7; med.bioinf.mpi-inf.mpg.de/netanalyzer/index.php).
Finally, all significant genes were clustered into several groups
to identify the important clusters using the Cytoscape plugin MCODE
(version 1.4.2; apps.cytoscape.org/apps/mcode).
Results
DEG analysis
For tongue epithelia samples, with the criteria of
P<0.01 and |log2(FC)| ≥1.5, 1,103 DEGs were identified when
comparing carcinoma with normal control, 251 DEGs when comparing
dysplasia with normal control and 515 DEGs when comparing carcinoma
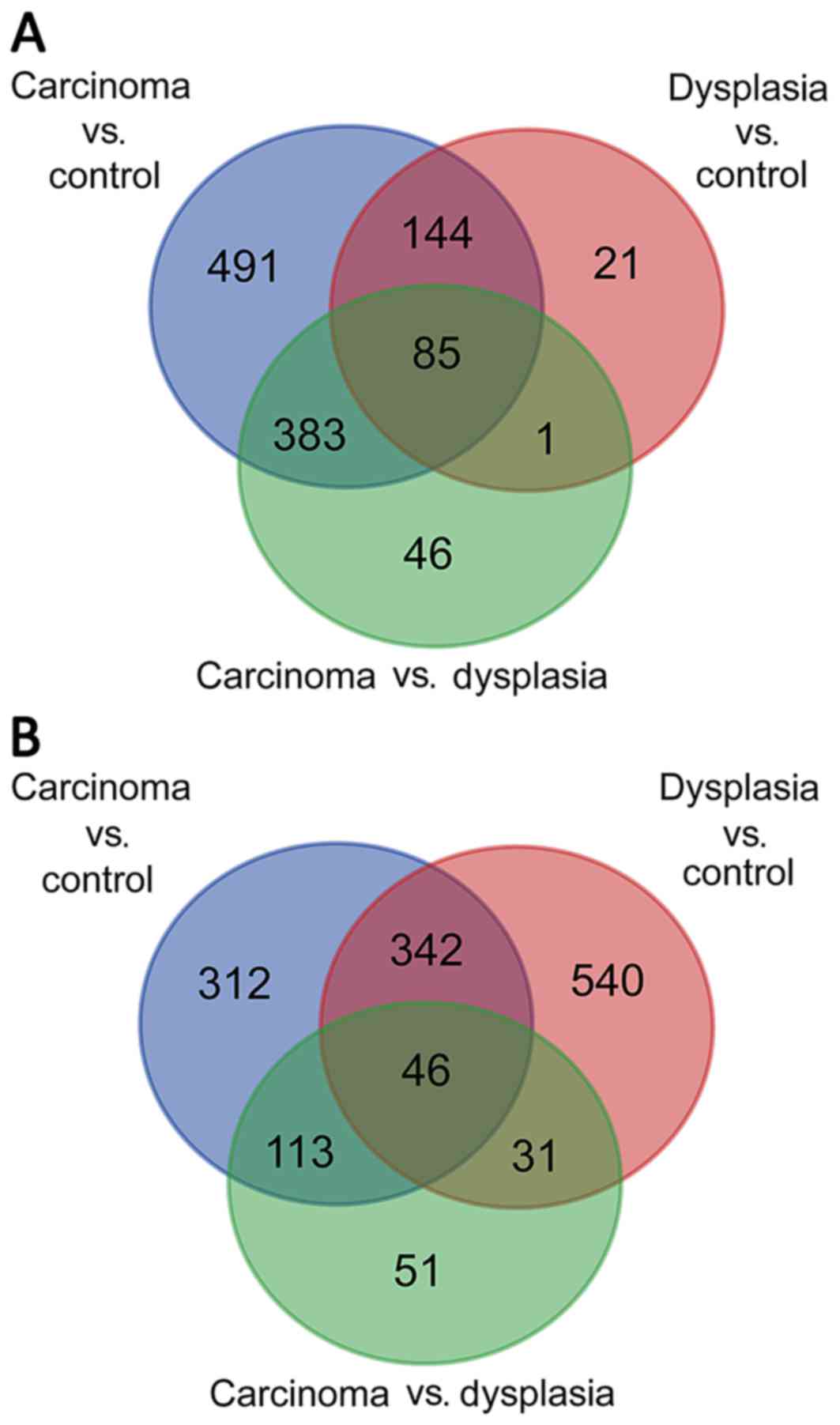
with dysplasia. As presented in Fig.
1A, following construction of a Venn diagram, 85 DEGs were
significantly differentially expressed among all three groups.
Regarding submucosal fibroblast samples, 813 DEGs
were identified when comparing carcinoma with normal control, 959
DEGs when comparing dysplasia with normal control and 241 DEGs when
comparing carcinoma with dysplasia. As demonstrated in Fig. 1B, the Venn diagram identified 46
DEGs that were significantly differentially expressed among all
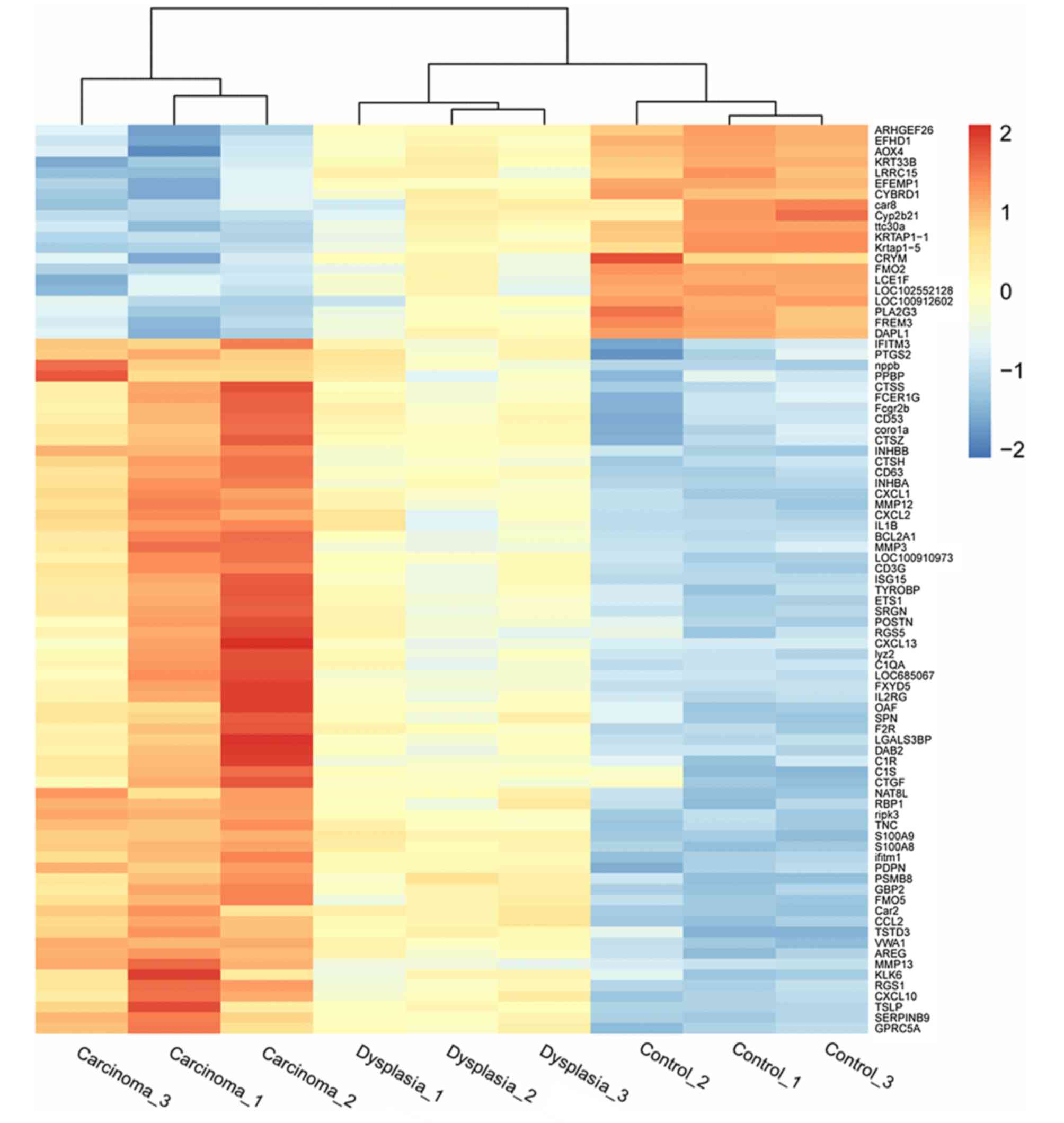
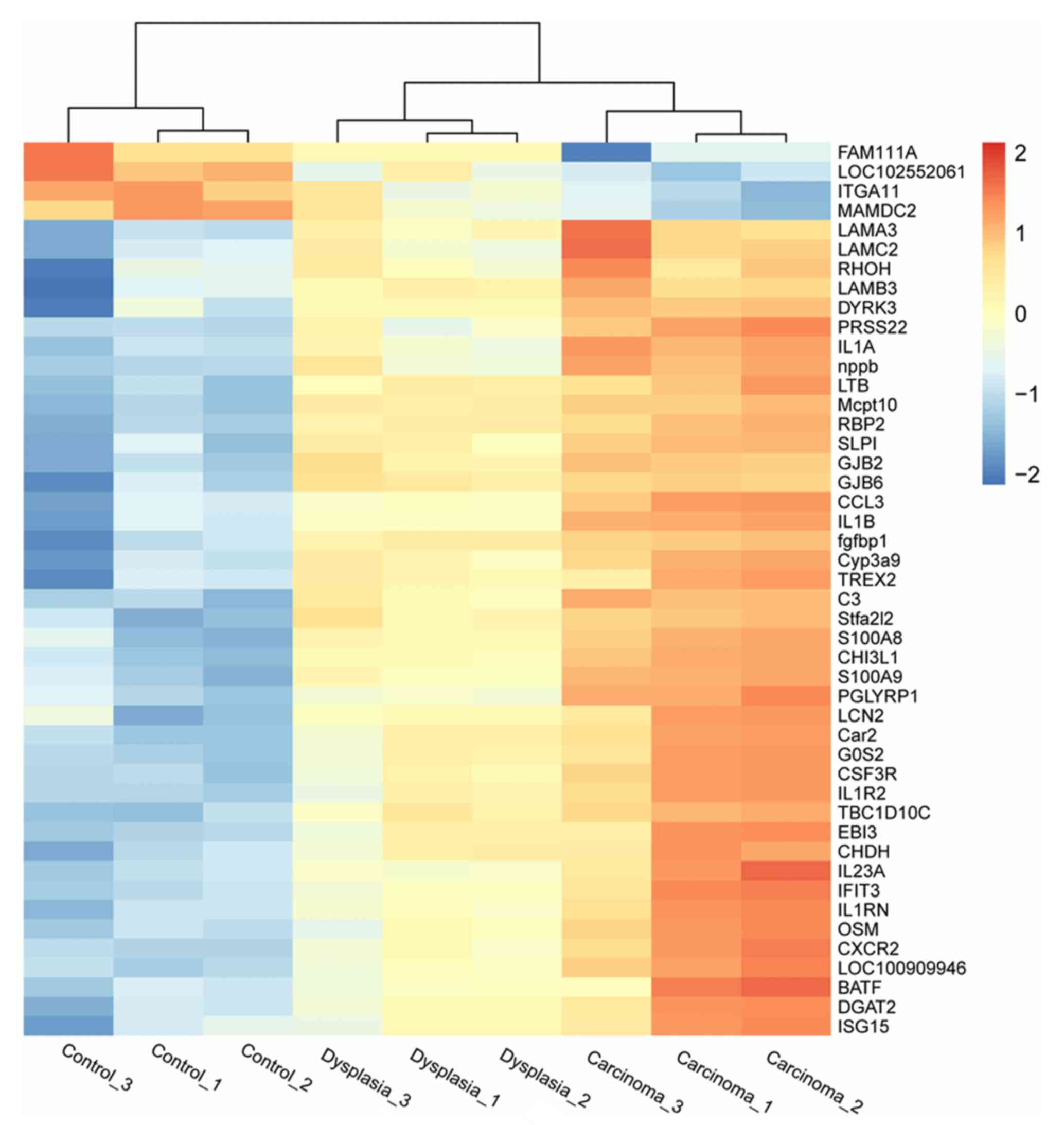
three groups. Heatmaps indicating differential expression of genes
among the three groups in epithelia and fibroblasts are presented
in Figs. 2 and 3, respectively.
Functional enrichment analyses
For tongue epithelia samples, the top five
significant GO biological process terms for DEGs were principally
associated with neutrophil chemotaxis, response to
lipopolysaccharide, inflammatory response, response to estradiol
and positive regulation of cell migration, as demonstrated in
Table I. The top five significant
KEGG pathways of DEGs were enriched in the tumor necrosis factor
(TNF) signaling pathway, cytokine-cytokine receptor interaction,
Staphylococcus aureus infection, chemokine signaling
pathway, and complement and coagulation cascades (Table II).
 | Table I.GO biological process enrichment
analyses of differentially expressed genes for epithelia. |
Table I.
GO biological process enrichment
analyses of differentially expressed genes for epithelia.
| GO term | Count | P-value |
|---|
| GO:0030593;
neutrophil chemotaxis lipopolysaccharide | 8 |
5.23×10−9 |
| GO:0032496; response
to | 12 |
2.92×10−8 |
| GO:0006954;
inflammatory response | 12 |
5.72×10−8 |
| GO:0032355; response
to estradiol | 10 |
1.91×10−7 |
| GO:0030335; positive
regulation of cell migration | 9 |
2.78×10−6 |
| Table II.KEGG enrichment analyses of
differentially expressed genes for epithelia. |
Table II.
KEGG enrichment analyses of
differentially expressed genes for epithelia.
| KEGG term | Count | P-value |
|---|
| rno04668; TNF
signaling pathway | 8 |
4.01×10−6 |
| rno04060;
cytokine-cytokine receptor interaction | 7 |
1.98×10−3 |
| rno05150;
Staphylococcus aureus infection | 4 |
4.39×10−3 |
| rno04062; chemokine
signaling pathway | 6 |
4.47×10−3 |
| rno04610; complement
and coagulation cascades | 4 |
9.76×10−3 |
For submucosal fibroblast samples, the top five
significant GO biological process terms of DEGs were principally
associated with neutrophil chemotaxis, response to
lipopolysaccharide, inflammatory response, cytokine-mediated
signaling pathway and fever generation (Table III). The top five significant
KEGG pathways of DEGs were enriched in cytokine-cytokine receptor
interaction, rheumatoid arthritis, amoebiasis, pertussis and
hematopoietic cell lineage (Table
IV).
| Table III.GO biological process enrichment
analyses of differentially expressed genes for fibroblasts. |
Table III.
GO biological process enrichment
analyses of differentially expressed genes for fibroblasts.
| GO term | Count | P-value |
|---|
| GO:0030593;
neutrophil chemotaxis | 6 |
3.01x10−7 |
| GO:0032496; response
to lipopolysaccharide | 8 |
4.97×10−6 |
| GO:0006954;
inflammatory response | 8 |
7.65×10−6 |
| GO:0019221;
cytokine-mediated signaling pathway | 6 |
2.20×10−5 |
| GO:0001660; fever
generation | 3 |
8.89×10−5 |
| Table IV.KEGG enrichment analyses of
differentially expressed genes for fibroblasts. |
Table IV.
KEGG enrichment analyses of
differentially expressed genes for fibroblasts.
| KEGG term | Count | P-value |
|---|
| rno04060;
cytokine-cytokine receptor interaction | 9 |
4.63×10−8 |
| rno05323; rheumatoid
arthritis | 5 |
8.62×10−5 |
| rno05146;
amoebiasis | 5 |
2.02×10−4 |
| rno05133;
pertussis | 4 |
9.34×10−4 |
| rno04640;
hematopoietic cell lineage | 4 |
1.26×10−3 |
PPI analysis
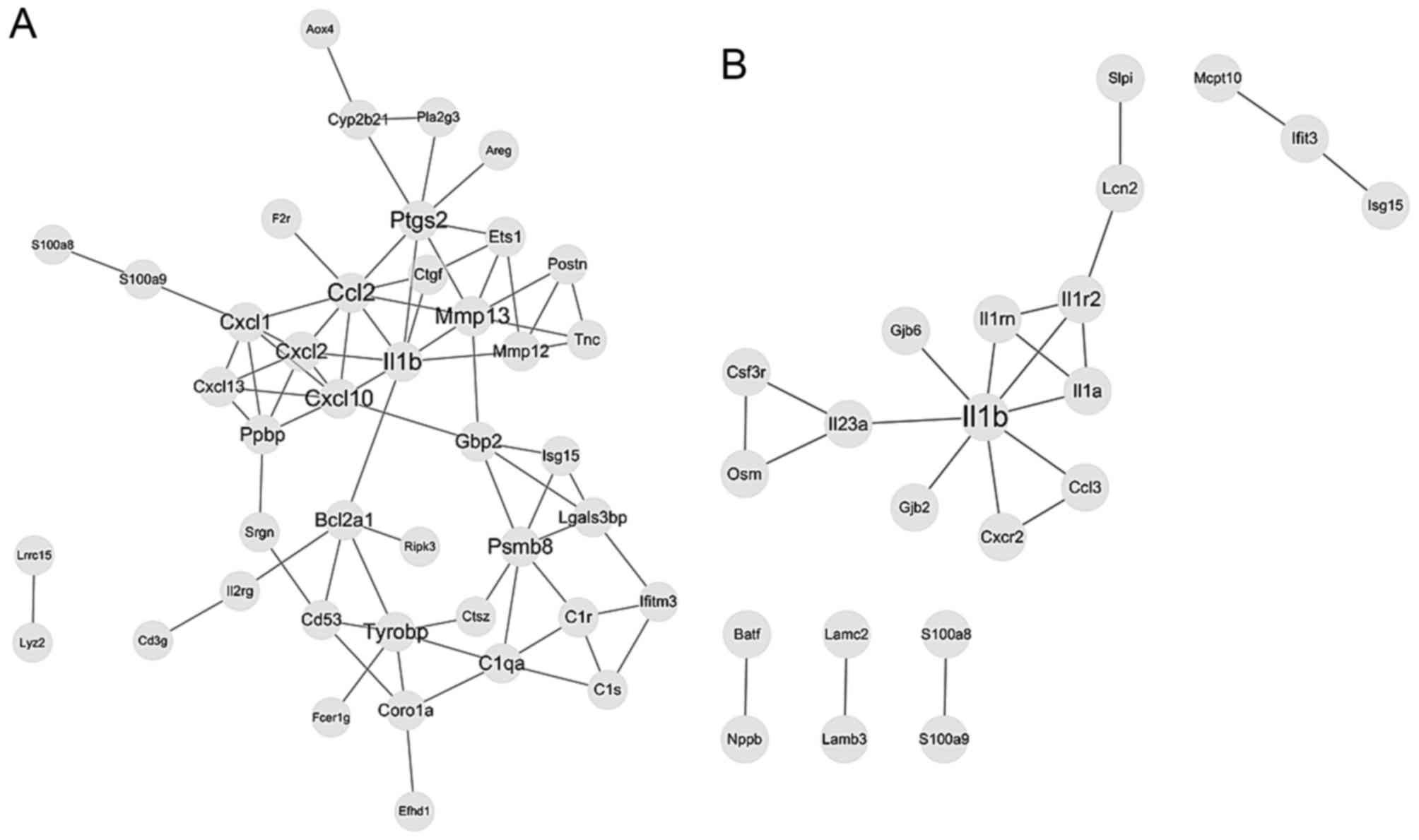
For DEGs of tongue epithelia samples, 42 nodes and
75 edges were included in the PPI network based on the STRING
database, as presented in Fig. 4A.
Topological analysis by plugin NetworkAnalyzer identified Il1β, C-C
motif chemokine ligand 2, matrix metallopeptidase 13,
prostaglandin-endoperoxide synthase 2 and C-X-C motif chemokine
ligand (Cxcl) 10 as hub nodes, which were considered to be key
proteins in the whole network. A module consisting of Cxcl1,
Cxcl10, Cxcl13, Cxcl2 and pro-platelet basic protein (Ppbp) was
recognized with a score >4 by plugin MCODE as a significant
cluster in the PPI network. DEGs of this module were significantly
associated with the TNF signaling pathway and cytokine-cytokine
receptor interaction.
Regarding submucosal fibroblast samples, 22 nodes
and 22 edges were included in the PPI network, as demonstrated in
Fig. 4B. Topological analysis
identified Il1β, Il1 receptor (Il1r) 2, Il1α, Il1r antagonist
(Il1rn) and Il23α as key proteins. A module consisting of Il1β,
Il1r2, Il1α and Il1rn was recognized as a significant cluster,
which was significantly associated with cytokine-cytokine receptor
interaction.
Discussion
The present study identified the DEGs when comparing
the normal, dysplasia and carcinoma expression profiles of tongue
cancer samples, which was followed by enrichment analysis and
network analysis. For the epithelial samples, 85 DEGs were
identified as being significantly differentially expressed among
the normal, dysplasia and carcinoma groups, which were associated
with neutrophil chemotaxis and inflammatory response GO biological
process terms, and associated with the KEGG terms TNF signaling
pathway and cytokine-cytokine receptor interaction. In the PPI
network, an important module consisting of Cxcl1, Cxcl10, Cxcl13,
Cxcl2 and Ppbp was significantly associated with the TNF signaling
pathway and cytokine-cytokine receptor interaction. Similarly, for
submucosal fibroblast samples, 46 DEGs were identified to be
significantly differentially expressed among the three groups,
which were associated with neutrophil chemotaxis and inflammatory
response GO biological process terms, and with cytokine-cytokine
receptor interaction from KEGG analysis. An important module
consisting of Il1b, Il1r2, Il1a and Il1rn was significantly
associated with cytokine-cytokine receptor interaction.
Previous studies regarding bioinformatic analysis of
oral tongue cancer have been performed. A methylation profile
revealed that hypermethylation of microRNA (miRNA/miR)-10b with
downregulation of its target gene nuclear receptor subfamily 4
group A member 3 was associated with tongue tumor patient survival
(17). An additional study
employed single-nucleotide polymorphism microarrays with human
OTSCC samples to indicate that genetic changes, such as 20q11.2
gain encoding the E2F transcription factor 1 gene, may be acquired
through clonal evolution and may be responsible for metastasis
(18). miR-424 was reported to be
a marker specific for tongue tumorigenesis, which also may function
in the development of OTSCC (19).
However, the majority of studies have focused on the carcinoma
itself and ignored other components of the TME.
At present, it is established that the TME has
important roles in cancer progression, including primary growth,
invasion and metastasis. (8,9) The
TME refers to a complex network that primarily consists of cancer
cells, fibroblasts, endothelial cells and immune cells, as well as
cytokines, growth factors and blood vessels (20). Of these components,
cancer-associated fibroblasts have important effects on the
formation of the TME and the occurrence of tumorigenesis. In
addition, cytokines primarily produced by cancer cells and
fibroblasts establish the interaction between cancer cells and
other components of the extracellular matrix (21). The importance of both fibroblasts
and cytokines has been demonstrated in the current study. According
to analysis performed in the present study, neutrophil chemotaxis,
inflammatory response and cytokine-mediated signaling pathways were
enriched in GO analysis of the epithelial and fibroblast samples,
while cytokine-cytokine receptor interaction was enriched for KEGG
pathways in epithelial and fibroblast samples.
OTSCC is associated with a poor prognosis due to
local invasion and lymph node metastases (7). As a result, investigating the
potential key functional pathways and the key genes or proteins in
the TME is critical for the understanding of the underlying
mechanisms of the formation, invasion and metastasis of OTSCC. The
results of the present study indicate that the inflammatory immune
response and cytokine-mediated signaling interactions have
important functions in the tumor microenvironment. Il1b was
identified as the most important node in both epithelia and
fibroblast samples, which was consistent with results obtained by a
previous report (13). However, Wu
et al (13) only considered
FC, while the present study considered FC in addition to the
P-value. Wu et al (13)
demonstrated that Il1b acted as a paracrine signal to activate
associated pathways among different cells in the TME, which
subsequently established a cycle of reinforcement and generated a
comfortable TME for oral carcinogenesis. They subsequently
hypothesized that a chemoprevention strategy targeting Il1b in the
TME may interrupt oral malignant transformation.
The present study discovered a key module that
primarily consisted of chemokines, including Cxcl1, Cxcl10, Cxcl13
and Cxcl2, in epithelia samples, and a key module consisting of
Ils, including Il1b, Il1r2, Il1a and Il1rn, in fibroblast samples.
All of the secreted cytokines may recruit inflammatory cells, which
build up in the TME, and the subsequently release of growth
factors, cytokines, chemokines and enzymes may regulate tumor
growth, angiogenesis, invasion and metastasis (22). The balance between tumor immunity
and carcinogenesis may be broken when disequilibrium occurs among
secreted factors, which is a prospective antitumor immunity
strategy in the future (23).
The current studies aimed to perform gene expression
profile analysis of tongue cancer based on a microarray. In
addition, the majority of similar published bioinformatic analysis
studies were limited to single platforms, including mRNA, miRNA,
long non-coding RNA, methylation and mutation. However, a
comprehensive global analysis from the Cancer Genome Atlas
integrated multi-platform expression characterization from 279
patients with HNSCC. Therefore, studies focusing on integrated
multi-platform analysis are essential for the comprehension of
mechanisms underlying the TME of tongue tumors (19).
The identification of hub DEGs, enriched pathways
and important modules during oral cavity tongue carcinogenesis in
TME has the following promising applications: Improve understanding
of the potential molecular mechanical and cellular functional
changes of tongue malignant transformation of oral cancer;
demonstrate demands for integrated multi-platform analysis research
regarding the TME; and to reveal the importance of a stable
inflammatory immune network in the TME, which is useful for the
development of targeted therapy of OTSCC. Additional studies are
required to develop a more thorough understanding. The results of
the present study may provide insight into the translational
practice and future investigation.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant No. 81272892,
81471352).
References
|
1
|
Ferlay J, Shin HR, Bray F, Forman D,
Mathers C and Parkin DM: Estimates of worldwide burden of cancer in
2008: GLOBOCAN 2008. Int J Cancer. 127:2893–2917. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Blot WJ, Mclaughlin JK, Winn DM, Austin
DF, Greenberg RS, Preston-Martin S, Bernstein L, Schoenberg JB,
Stemhagen A and Fraumeni JF Jr: Smoking and drinking in relation to
oral and pharyngeal cancer. Cancer Res. 48:3282–3287.
1988.PubMed/NCBI
|
|
3
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Castellsagué X, Alemany L, Quer M, Halec
G, Quirós B, Tous S, Clavero O, Alòs L, Biegner T, Szafarowski T,
et al: HPV involvement in head and neck cancers: Comprehensive
assessment of biomarkers in 3680 patients. J Natl Cancer Inst.
108:djv4032016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Greene FL, Page DL, Fleming ID, Fritz A,
Balch CM, Haller DG and Morrow M: American Joint Committee on
Cancer: AJCC Cancer Staging Manual. 6th edition. Springer; New
York, NY: pp. 157–164. 2002
|
|
6
|
Rusthoven K, Ballonoff A, Raben D and Chen
C: Poor prognosis in patients with stage I and II oral tongue
squamous cell carcinoma. Cancer. 112:345–351. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sessions DG, Lenox J, Spector GJ, Chao C
and Chaudry OA: Analysis of treatment results for base of tongue
cancer. Laryngoscope. 113:1252–1261. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hanahan D and Coussens LM: Accessories to
the crime: Functions of cells recruited to the tumor
microenvironment. Cancer Cell. 21:309–322. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Quail DF and Joyce JA: Microenvironmental
regulation of tumor progression and metastasis. Nat Med.
19:1423–1437. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bullinger L, Döhner K, Bair E, Fröhling S,
Schlenk RF, Tibshirani R, Döhner H and Pollack JR: Use of
gene-expression profiling to identify prognostic subclasses in
adult acute myeloid leukemia. N Engl J Med. 350:1605–1616. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chung CH, Ely K, McGavran L,
Varella-Garcia M, Parker J, Parker N, Jarrett C, Carter J, Murphy
BA, Netterville J, et al: Increased epidermal growth factor
receptor gene copy number is associated with poor prognosis in head
and neck squamous cell carcinomas. J Clin Oncol. 24:4170–4176.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chung CH, Parker JS, Karaca G, Wu J,
Funkhouser WK, Moore D, Butterfoss D, Xiang D, Zanation A, Yin X,
et al: Molecular classification of head and neck squamous cell
carcinomas using patterns of gene expression. Cancer Cell.
5:489–500. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wu T, Hong Y, Jia L, Wu J, Xia J, Wang J,
Hu Q and Cheng B: Modulation of IL-1β reprogrammes the tumor
microenvironment to interrupt oral carcinogenesis. Sci Rep.
6:202082016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ashburner M, Ball CA, Blake JA, Botstein
D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT,
et al: Gene ontology: Tool for the unification of biology. the gene
ontology consortium. Nat Genet. 25:25–29. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kanehisa M and Goto S: KEGG: Kyoto
encyclopedia of genes and genomes. Nucleic Acids Res. 28:27–30.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dennis G Jr, Sherman BT, Hosack DA, Yang
J, Gao W, Lane HC and Lempicki RA: DAVID: Database for annotation,
visualization and integrated discovery. Genome Biol. 4:P32003.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Krishnan NM, Dhas K, Nair J, Palve V,
Bagwan J, Siddappa G, Suresh A, Kekatpure VD, Kuriakose MA and
Panda B: A minimal DNA methylation signature in oral tongue
squamous cell carcinoma links altered methylation with tumor
attributes. Mol Cancer Res. 14:805–819. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Morita T, Uzawa N, Mogushi K, Sumino J,
Michikawa C, Takahashi KI, Myo K, Izumo T and Harada K:
Characterizing genetic transitions of copy number alterations and
allelic imbalances in oral tongue carcinoma metastasis. Genes
Chromosomes Cancer. 55:975–986. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Boldrup L, Coates PJ, Laurell G, Wilms T,
Fahraeus R and Nylander K: Downregulation of miRNA-424: A sign of
field cancerisation in clinically normal tongue adjacent to
squamous cell carcinoma. Br J Cancer. 112:1760–1765. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Joyce JA and Pollard JW:
Microenvironmental regulation of metastasis. Nat Rev Cancer.
9:239–252. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Polyak K, Haviv I and Campbell IG:
Co-evolution of tumor cells and their microenvironment. Trends
Genet. 25:30–38. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lewis CE and Pollard JW: Distinct role of
macrophages in different tumor microenvironments. Cancer Res.
66:605–612. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kortylewski M, Xin H, Kujawski M, Lee H,
Liu Y, Harris T, Drake C, Pardoll D and Yu H: Regulation of the
IL-23 and IL-12 balance by Stat3 signaling in the tumor
microenvironment. Cancer Cell. 15:114–123. 2009. View Article : Google Scholar : PubMed/NCBI
|