Introduction
Neonatal hypoxic-ischemic (HI) encephalopathy
affects 2–6 out of 1,000 term births in the developed world,
associating with high mortality and lifelong chronic disabilities
(1,2). HI insult of human neonatal is a vital
cause of perinatal brain injury, which may cause cerebral palsy,
seizures, learning limitations and epilepsy (3,4).
Animal studies have demonstrated that the mechanisms leading to
injury in the neonatal brain are distinct from those involved in
adult brain injury (5). Apoptosis
appears to be prominent in neonatal HI, as this cascade is easily
engaged following brain insults during this developmental stage
(6,7). Thus, targeting apoptosis may be a
useful strategy for HI-induced brain injury.
Transforming growth factor (TGF)-β-activated kinase
1 (TAK1) belongs to the mitogen-activated protein kinases (MAPK)
kinase kinase (MAP3K) family (8),
and was first found to be activated by TGF-β and bone morphologic
proteins (9). TAK1 is essential to
activating the IκB kinase (IKK)/nuclear factor (NF)-κB signaling
pathways and the stress kinase [c-Jun N-terminal kinases (JNK) and
p38 MAPK] signaling pathways in response to various stressors
(10). Previous studies have
demonstrated that TAK1 is a central regulator of cell death and is
activated via a diverse set of intra- and extracellular stimuli
(8,11,12).
The TAK1-JNK signaling pathway has been demonstrated to be crucial
in cell apoptosis; For instance, this pathway is involved in
activated T-cell apoptosis in a model of lung and thyroid cancers
(11,13–15).
Thus, TAK1-JNK signaling pathways have been a widely used
therapeutic target in cancer and other types of disease (16–19).
However, the role of TAK1 in the neonatal brain
following HI is still unclear. Thus, the expression level and
distribution of phosphorylated (p)-TAK1 was investigated, at
various time-points following insult, by western blotting and
double immunofluorescence. The results demonstrated the presence of
p-TAK1, and that it localized with neurons and astrocytes. Further
study demonstrated that injection of NG25 prior to insult
significantly inhibited TAK1/JNK activity and markedly ameliorated
acute hypoxic-ischemic cerebral injury by inhibiting cell
apoptosis.
Materials and methods
HI rat model and treatment
The 7-day-ol d rat pups (20 pups, 4 pups in each
group, about 20 g) were purchased from the Animal Center of Sichuan
University (Chengdu, China) and on a 12-h night/12-h day cycle at a
room temperature of 22±2°C with free access to food and water. Each
group of four pups were kept in one feeding box. Then, the pups
were used for establishing the HI model. Briefly, the pups were
anesthetized with diethylether (0.1 mg/kg) and the body was
maintained at 37°C using a homoisothermy bench. A skin incision
(0.5 cm) was made in the midline of the neck and the right common
carotid artery (CCA) was permanently ligatured using 5–0 silk.
Following ligation of the CCA, the pups were returned to the dam
for 0.5 h to recover from anesthesia. The pups were placed in a
chamber at a constant 37°C for hypoxia (8% O2, 92%
N2) for 6, 12, 24 and 48 h. The sham group underwent a
neck dissection and the 5–0 silk was placed around the CCA, but was
not ligated. All animal procedures were approved by Sichuan
University Committee (Chengdu, China) on Animal Use and Care, and
all efforts were made to minimize animal suffering and the number
of animals sacrificed.
NG25 (MedChem Express, Monmouth Junction, NJ, USA),
a highly specific TAK1 inhibitor, was dissolved in Sigma-Aldrich
dimethyl sulfoxide (DMSO; Merck KGaA, Darmstadt, Germany) and
injected into the right cerebral hemisphere 30 min prior to HI
using a 30-gauge needle with a 5-µl Hamilton syringe (infusion
rate, 1 µl/min).
Immunofluorescence
At different designated time-points, the brains were
perfused and fixed in 4% paraformaldehyde for 48 h. Then the brains
were embedded in paraffin and sectioned (thickness, 4 mm). The
sections were probed with p-TAK1 primary antibody (cat no. 9339,
1:200; Cell Signaling Technology, Inc., Danvers, MA, USA) and mouse
monoclonal antibodies against NeuN (cat no. MAB377, 1:150) or glial
fibrillary acidic protein (GFAP; cat no. MAB360, 1:80) (both from
EMD Millipore, Billerica, MA, USA) and followed by incubation
(37°C) with a 1:120 dilution of a secondary antibody; either
fluorescein isothiocyanate- or tetramethylrhodamine-conjugated
anti-rabbit (cat no. sc-2012) or anti-mouse (cat no. sc-2010) IgG
(Santa Cruz Biotechnology, Inc., Dallas, TX, USA). The nucleus was
stained with 4′,6-diamidino-2-phenylindole (DAPI). Five sections
per rat were imaged by a fluorescence microscope (DTX500; Nikon
Corporation, Tokyo, Japan) and analyzed.
Western blot analysis
Cell extracts were prepared using a RIPA lysis
buffer (Beyotime Institute of Biotechnology, Haimen, China). At
different time-points after HI treatment, the cortex and
hippocampus from the right hemisphere were collected and dissected
(n=3 per group). The cortex extracts were prepared in the RIPA
lysis buffer containing a protease inhibitor cocktail (Merck KGaA).
Protein concentrations were determined using a BCA protein assay
kit (Beyotime Institute of Biotechnology) with bovine serum albumin
serving as the standard. Protein samples (20 µg per lane) were
resolved on 10% SDS-PAGE and transferred (100V, 50 min) to
polyvinylidene fluoride membranes (EMD Millipore). The membranes
were blocked in 5% nonfat milk in tris-buffered saline containing
0.05% Tween-20 for 1 h at room temperature and immunoblotted with
various antibodies as follows: p-TAK1 (cat no. 9339, 1:1,000);
rabbit anti-TAK1 monoclonal antibody (cat no. 5206, 1:800); rabbit
anti-p-JNK (Thr183/Tyr185) polyclonal antibody (cat no. 9255,
1:1,000); rabbit anti-p-c-Jun polyclonal antibody (cat no. 3270,
1:800) (all from Cell Signaling Technology, Inc.); rabbit anti-p53
polyclonal antibody (cat no. ab26, 1:200; Abcam, Cambridge, MA,
USA); and rabbit anti-caspase-3 polyclonal antibody (cat no. C9598,
1:100; Sigma-Aldrich; Merck KGaA) overnight at 4°C. A rabbit
anti-GAPDH polyclonal antibody (cat no. G9545, 1:2,000;
Sigma-Aldrich; Merck KGaA) served as an internal loading control.
The bound antibodies were visualized with horseradish
peroxidase-conjugated antibodies against rabbit or mouse IgG using
the electrochemiluminescence (ECL) or ECL advance western blotting
detection kit (Merck KGaA). Image-Pro Plus (version 6.0; Media
Cybernetics, Inc., Rockville, MD, USA) was used to quantify the
densities of the protein signals on X-ray films following
scanning.
TUNEL assay
To detect apoptotic cells in tumor tissue samples,
TUNEL assay using a DeadEnd™ Fluorometric TUNEL system
(Promega Corporation, Madison, WI, USA) was performed according to
the manufacturer's protocol. Cell nuclei with dark green
fluorescent staining were defined as apoptotic cells. To quantify
TUNEL-positive cells, the number of green fluorescence-positive
cells was imaged by a fluorescence microscope (DTX500; Nikon
Corporation, Tokyo, Japan) and counted in five random fields. Cell
nuclei were counterstained with DAPI.
Statistical analysis
Statistical analysis was performed using SPSS
version 19.0 (SPSS, Inc., Chicago, IL, USA). Numerical continuous
data are presented as the mean ± standard deviation and analyzed
using one-way analysis of variance. P<0.05 was considered to
indicate a statistically significant difference.
Results
Induction of TAK1 phosphorylation in
the brain cortex of rats with HI
To determine the expression of p-TAK1 in the brain
cortex of rats with HI, the hemispheres of rats were collected at
different time-point post HI treatment and used for western
blotting. As presented in Fig. 1A and
B, a significant increase of p-TAK1 expression was observed in
the brain cortex 6 h after insult, followed by a 2-6-fold increase
in p-TAK1 protein expression in the brain of the HI model compared
with that of the sham group at 6, 12, 24 and 48 h. Subsequently,
double immunofluorescence was employed to detect the expression and
distribution of p-TAK1 in the rat cortex of sham control and
experimental group rats (24 h after HI). Furthermore, the
neuronal-specific marker, NeuN and the astrocyte-specific marker,
GFAP were used to indicate the neurons and astrocytes. As
demonstrated in Fig. 1C, compared
with the sham group, an increased expression level of p-TAK1 was
observed, which was localized with astrocytes at 24 h after insult.
NeuN and p-TAK1 double immunofluorescence indicate that a greater
expression level of p-TAK1 was observed and localized with neurons
at 24 h after insult, compared with the sham group (Fig. 1D). Thus, induction of p-TAK1 was
observed in the brain cortex of rats with HI, and p-TAK1 was
expressed by astrocytes and neurons.
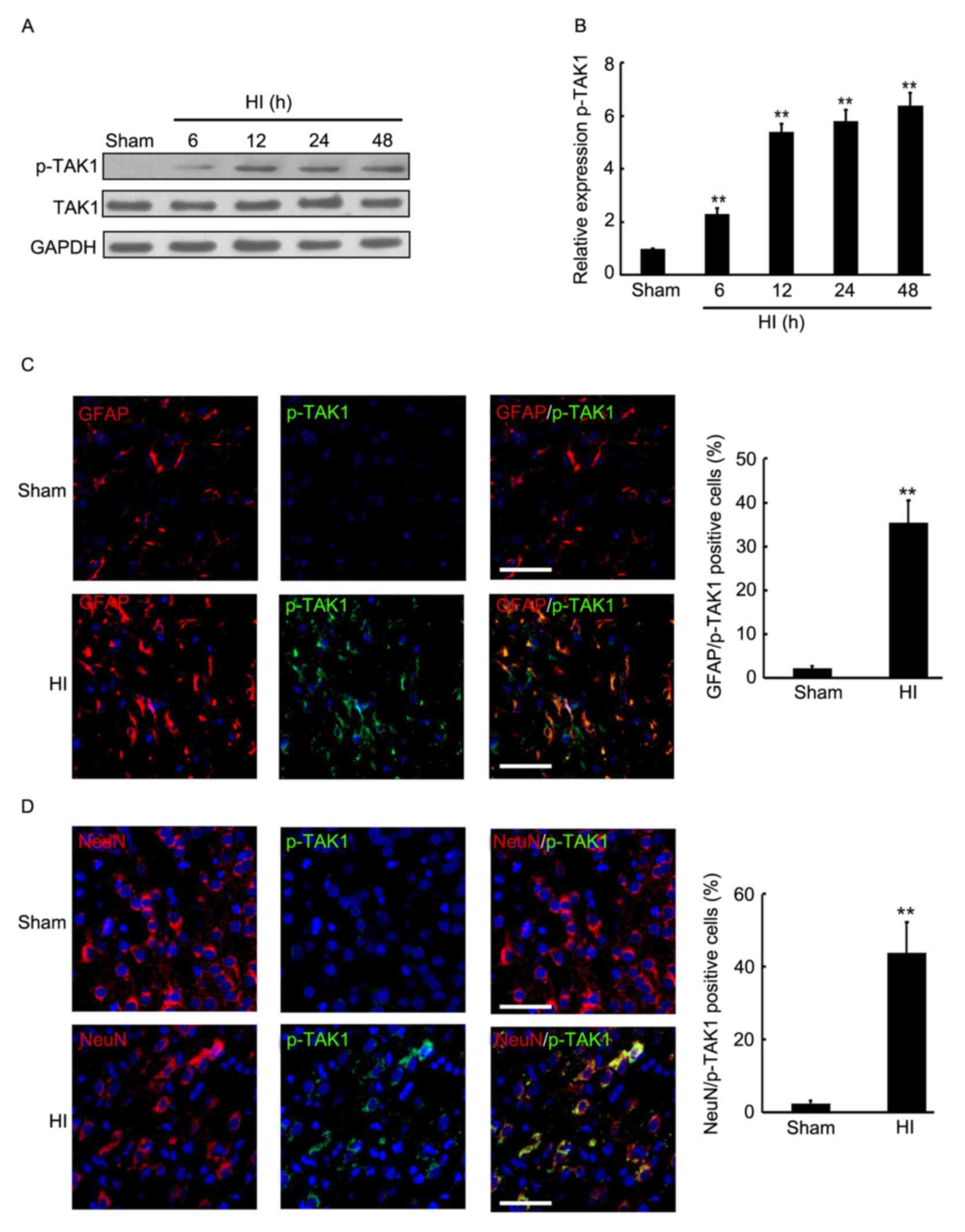 | Figure 1.Expression level and distribution of
TAK1 in the brain cortex of the rat HI model was determined by
western blotting and immunofluorescence. At 6, 12, 24 and 48 h
after brain insult, the brain cortex of rats was perfused and
sampled for western blotting and immunofluorescence analysis. Rats
without brain insult served as the sham controls. (A and B) Western
blot detection of p-TAK1 and TAK1 protein expression levels in the
sham group, and the HI model rats at 6, 12, 24 and 48 h after brain
insult (n=3). GAPDH served as the loading control. (C) Double
immunofluorescence GFAP (red) + p-TAK1 (green) were used in
paraffin-embedded sections sampled from the sham control rats, as
well as from the HI rats at 24 h after HI (each group, n=4). DAPI
staining demonstrated the cell nucleus. Scale bar, 100 µm. (D)
Double immunofluorescence NeuN (red) + p-TAK1 (green) were used in
paraffin-embedded sections sampled from the sham control rats, as
well as from the HI rats at 24 h after HI (each group, n=4). DAPI
staining demonstrated the cell nucleus. Scale bar, 100 µm.
**P<0.01 vs. sham. TAK1, transforming growth factor-β-activated
kinase 1; HI, hypoxia-ischemia; p, phosphorylated; GFAP, glial
fibrillary acidic protein; DAPI, 4′,6-diamidino-2-phenylindole. |
Induction of TAK1 downstream target
expression in the brain cortex of HI model rats
JNK is the direct target of TAK1, thus western
blotting was employed to determine the expression level of p-JNK at
different time-points in the rat cortex following HI. The current
results indicate that HI treatment induced p-JNK expression from
6–48 h after insult, compared with the sham control rats (Fig. 2A and B), whereas there were no
significant effects on total JNK expression (data not shown).
Furthermore, the JNK-associated downstream targets, p-c-Jun, p53
and caspase-3 expression levels were determined by western
blotting. The results demonstrate that p-c-Jun, p53 and caspase-3
expression levels were significantly increased from 6–48 h after
insult, compared with the sham control rats (Fig. 2C and D).
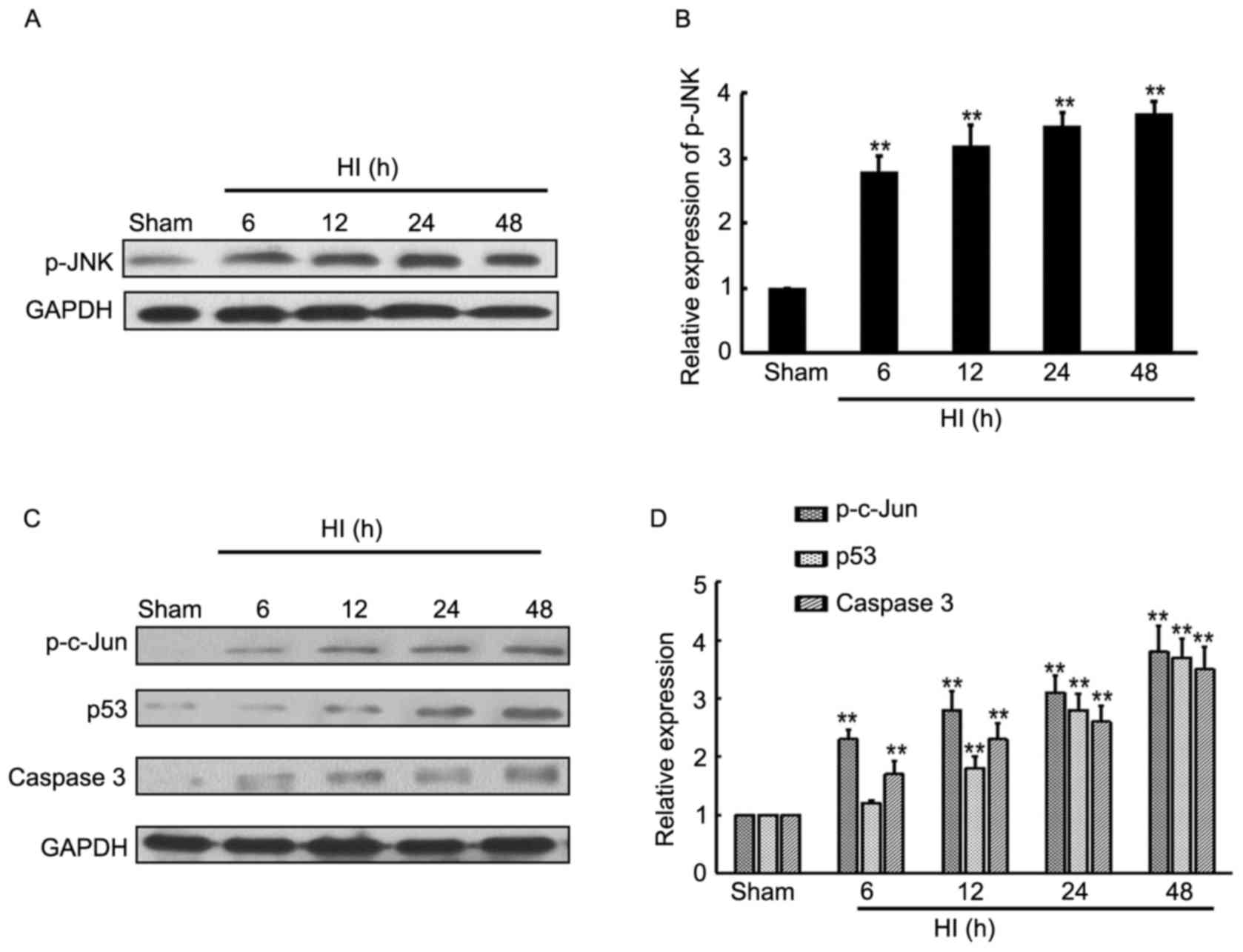 | Figure 2.Expression level of JNK and downstream
targets in brain cortex of rat HI model. (A and B) Western blotting
detection of p-JNK and JNK expression levels in the sham rats, and
HI model rats at 6, 12, 24 and 48 h after brain insult. GAPDH
served as a loading control (n=4). (C and D) Western blotting
detection of p-c-Jun, p53 and caspase-3 expression levels in sham
rats, and HI model rats at 6, 12, 24 and 48 h after brain insult.
GAPDH served as a loading control (n=4). **P<0.01 vs. sham. JNK,
c-Jun N-terminal kinase; HI, hypoxia-ischemia; p,
phosphorylated. |
NG25 inhibits p-JNK and downstream
target expression levels in the brain cortex of HI model rats
To further determine the potential neuroprotective
role of TAK1 silencing on HI-induced brain injury, NG25, an
inhibitor of p-TAK1, was injected into the rat brains prior to HI
treatment. The results demonstrated that NG25 markedly inhibited
p-TAK1 expression in the brain cortex, compared with the HI and
DMSO groups (Fig. 3A and B).
Further immunofluorescence staining indicated that p-TAK1
expression was inhibited by NG25 in the brain cortex (Fig. 3C). Western blotting indicated that
p-JNK, p-c-Jun, p53 and caspase-3 expression levels were
significantly decreased by NG25 in the brain cortex, compared with
the HI and DMSO group (Fig. 3D and
E). Thus, the inhibitory role of NG25 on p-TAK1 and associated
downstream target expression levels was demonstrated.
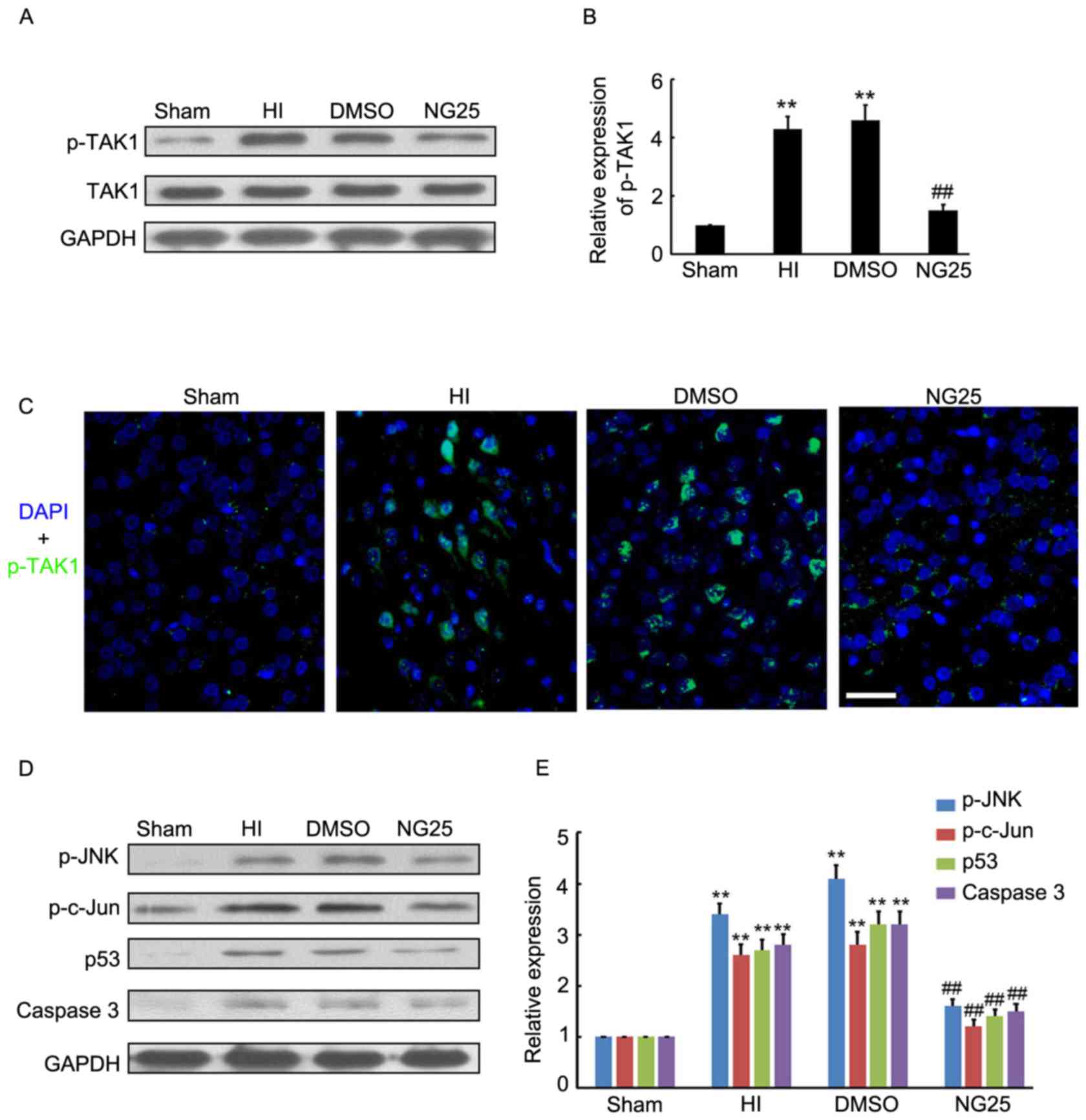 | Figure 3.Expression level of p-TAK1 and
downstream targets following NG25 treatment. (A and B) Western blot
detection of p-TAK1 and TAK1 expression levels in the sham group,
and samples from HI, DMSO-treated and NG25-treated rat brain
cortexes (n=4). (C) Immunofluorescence detection of p-TAK1 in the
sham group, and samples from HI, DMSO-treated and NG25-treated rat
brain cortexes (n=3). DAPI staining demonstrated the cell nucleus.
Scale bar, 50 µm. (D and E) Western blot detection of p-JNK,
p-c-Jun, p53 and caspase-3 expression levels in the sham group, and
samples from HI, DMSO-treated and NG25-treated rat brain cortexes
(n=4). **P<0.01 vs. sham controls; ##P<0.01 vs. HI
group. p, phosphorylated; TAK1, transforming growth
factor-β-activated kinase 1; HI, hypoxia-ischemia; DMSO, dimethyl
sulfoxide; DAPI, 4′,6-diamidino-2-phenylindole; JNK, c-Jun
N-terminal kinase. |
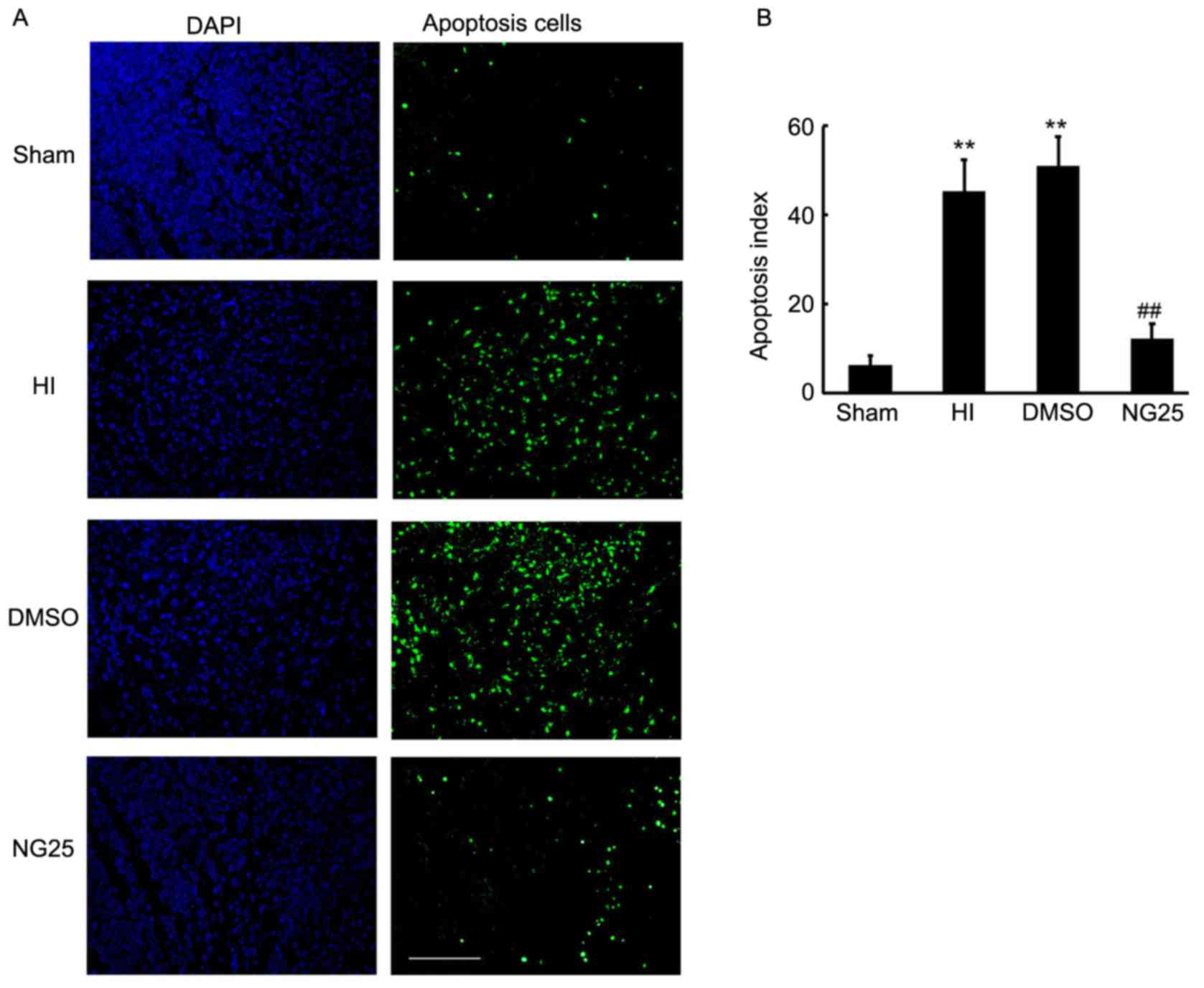
NG25 ameliorates neuronal apoptosis in
the brain cortex
Apoptosis is the major cause of brain injury
subsequent to HI. The TAK1-JNK signaling pathway is a well-known
inductor of apoptosis and has been widely researched in tumors
(13–15). Thus, in the current study, TUNEL
staining was employed to detect the apoptotic cells in the brain
cortex. As shown in Fig. 4A, an
increased number of apoptotic cells were observed in the HI and
DMSO-treated rats, when compared with the sham control rats
(Fig. 4B). Notably, less apoptosis
was observed in the NG25 injection group when compared with the HI
and DMSO-treated group (Fig. 4).
Thus, our results indicated that knockdown of TAK1 by NG25 markedly
inhibited apoptosis in the brain cortex.
Discussion
The aim of the present study was to determine
whether, and how, TAK1 affects HI-induced brain injury in neonatal
rats. Thus, a HI model was established in 7-day-old rats, and the
expression and distribution of p-TAK1 was investigated at different
time-points following insult. p-TAK1 expression in the developing
brain was observed to be induced following HI, indicating that it
is involved in the development of HI-induced brain injury.
Furthermore, the inhibition of p-TAK1 expression with the specific
inhibitor, NG25 significantly alleviated the HI-induced brain
injury and apoptosis by preventing TAK1-JNK signaling pathway
activity. The results indicate that TAK1 is a potential therapeutic
target for neonatal HI-induced brain injury.
Various studies have indicated that apoptosis is
crucial during the development of HI-induced brain injury (20). An experimental study in neonatal
models has demonstrated that the extent of apoptosis-inducing
factor translocation to the nucleus correlates with the
morphological distribution of neuronal injury following
hypoxia-ischemia (21). In
addition, gene deletion of poly(ADP-ribose) polymerase-1 and
cyclophilin A significantly decreases apoptosis-inducing factor
translocation to the nucleus, which is accompanied by the reduction
of brain injury (22,23). Thus, apoptosis deficiency confers
considerable protection for neonatal HI-induced brain injury
(24,25). In the present study, the inhibitory
role of NG25 on brain apoptosis in neonatal HI was demonstrated and
the protective role of NG25 on HI-induced brain injury in neonatal
rats was further indicated.
The role of TAK1 inhibition on preventing neuronal
death following ischemia has been demonstrated in previous studies
(26–28). However, only the role of TAK1
inhibition on adult mice and rats was investigated (26–28).
Notably, neonatal HI encephalopathy affects 2–6 out of 1,000 term
births in the developed world, which is associated with high
mortality and lifelong chronic disabilities (1,2).
Thus, further work is required to develop novel strategies for
neonatal HI therapy. In the current study, the expression and
function of TAK1 in neonatal HI rats was the focus, and 7-day-old
pups were used to establish the HI model and undergo treatment. The
study demonstrated that inhibition of TAK1 efficiency ameliorated
neuronal apoptosis in the neonatal HI rats.
JNKs are important stress responsive kinases that
are activated by various forms of insults, including oxidative
stress and ischemia. JNK activation precedes cell death by
apoptosis and inflammation in many cell types (29). It has been shown that JNK
hyperactivation in neurons, microglia and vascular endothelial
cells is important in overweight-aggravated HI injury in the
neonatal brain (30). A previous
study demonstrated that the JNK/forkhead box O3/Bim signaling
pathway is involved in neuronal apoptosis in the developing rat
brain following HI and that agents targeting JNK may potentially
protect neurons from HI-induced damage (31). In another study, the JNK/activator
protein 1 signaling pathway participated in neuronal apoptosis in
the neonatal rat brain following HI, and inhibition of JNK with
D-JNKi or TAT-JBD efficiently protects the neonatal brain against
ischemic brain damage and subsequent cognitive and motor impairment
(32,33). Thus, JNK is a widely used
therapeutic target for neonatal HI-induced brain injury. In the
present study, JNK hyperactivation was observed in the neurons
following HI, which is consistent with previous studies (30–33).
As predicted, knockdown of p-TAK1 with NG25 significantly inhibited
p-JNK expression levels and the associated downstream target
expression. NG25, widely used in recent studies (34,35),
was injected with a lower dose into rats in the current study, as
compared with the 5Z-7-oxozeaenol that was used in the previous
study (28); a lower dose of
molecular compound causes fewer side effects. The current study
indicates that NG25 serves the neuroprotective role in the
developing brain following insult by inhibiting the activity of JNK
and the associated brain apoptosis.
In conclusion, the present results indicate that
TAK1 is involved in the development of neonatal HI-induced brain
injury. To the best of our knowledge, the present study is the
first to demonstrate that knockdown of p-TAK1, via
intracerebroventricular injection of NG25 (a specific inhibitor of
TAK1), prior to insult significantly attenuates acute HI cerebral
injury by inhibiting JNK signaling pathway activity and the
associated brain apoptosis. Thus, TAK1 is proposed as a potential
therapeutic target for the treatment of neonatal HI-induced brain
injury. Further study are needed to determine the long term
validity and the potential toxicity of NG25.
Acknowledgements
The present study was supported by the National
Science Foundation of China (grant no. 81401239).
References
|
1
|
Wachtel EV and Hendricks-Muñoz KD: Current
management of the infant who presents with neonatal encephalopathy.
Curr Probl Pediatr Adolesc Health Care. 41:132–153. 2011.
View Article : Google Scholar
|
|
2
|
Selway LD: State of the science: Hypoxic
ischemic encephalopathy and hypothermic intervention for neonates.
Adv Neonatal Care. 10:60–68. 2010. View Article : Google Scholar
|
|
3
|
Robertson CM, Finer NN and Grace MG:
School performance of survivors of neonatal encephalopathy
associated with birth asphyxia at term. J Pediatr. 114:753–760.
1989. View Article : Google Scholar
|
|
4
|
Marlow N, Rose AS, Rands CE and Draper ES:
Neuropsychological and educational problems at school age
associated with neonatal encephalopathy. Arch Dis Child Fetal
Neonatal Ed. 90:F380–F387. 2005. View Article : Google Scholar :
|
|
5
|
Back SA, Han BH, Luo NL, Chricton CA,
Xanthoudakis S, Tam J, Arvin KL and Holtzman DM: Selective
vulnerability of late oligodendrocyte progenitors to
hypoxia-ischemia. J Neurosci. 22:455–463. 2002.
|
|
6
|
Li L, Klebe D, Doycheva D, McBride DW,
Krafft PR, Flores J, Zhou C, Zhang JH and Tang J: G-CSF ameliorates
neuronal apoptosis through GSK-3β inhibition in neonatal
hypoxia-ischemia in rats. Exp Neurol. 263:141–149. 2015. View Article : Google Scholar
|
|
7
|
Xiao Q, Ford AL, Xu J, Yan P, Lee KY,
Gonzales E, West T, Holtzman DM and Lee JM: Bcl-x pre-mRNA splicing
regulates brain injury after neonatal hypoxia-ischemia. J Neurosci.
32:13587–13596. 2012. View Article : Google Scholar :
|
|
8
|
Mihaly SR, Ninomiya-Tsuji J and Morioka S:
TAK1 control of cell death. Cell Death Differ. 21:1667–1676. 2014.
View Article : Google Scholar :
|
|
9
|
Yamaguchi K, Shirakabe K, Shibuya H, Irie
K, Oishi I, Ueno N, Taniguchi T, Nishida E and Matsumoto K:
Identification of a member of the MAPKKK family as a potential
mediator of TGF-beta signal transduction. Science. 270:2008–2011.
1995. View Article : Google Scholar
|
|
10
|
Sato S, Sanjo H, Takeda K, Ninomiya-Tsuji
J, Yamamoto M, Kawai T, Matsumoto K, Takeuchi O and Akira S:
Essential function for the kinase TAK1 in innate and adaptive
immune responses. Nat Immunol. 6:1087–1095. 2005. View Article : Google Scholar
|
|
11
|
Hirata Y, Sugie A, Matsuda A, Matsuda S
and Koyasu S: TAK1-JNK axis mediates survival signal through Mcl1
stabilization in activated T cells. J Immunol. 190:4621–4626.
2013.
|
|
12
|
Wang JS, Wu D, Huang DY and Lin WW: TAK1
inhibition-induced RIP1-dependent apoptosis in murine macrophages
relies on constitutive TNF-α signaling and ROS production. J Biomed
Sci. 22:762015. View Article : Google Scholar :
|
|
13
|
Augeri DJ, Langenfeld E, Castle M,
Gilleran JA and Langenfeld J: Inhibition of BMP and of TGFβ
receptors downregulates expression of XIAP and TAK1 leading to lung
cancer cell death. Mol Cancer. 15:272016. View Article : Google Scholar :
|
|
14
|
Lin P, Niu W, Peng C, Zhang Z and Niu J:
The role of TAK1 expression in thyroid cancer. Int J Clin Exp
Pathol. 8:14449–14456. 2015.
|
|
15
|
Okada M, Matsuzawa A, Yoshimura A and
Ichijo H: The lysosome rupture-activated TAK1-JNK pathway regulates
NLRP3 inflammasome activation. J Biol Chem. 289:32926–32936. 2014.
View Article : Google Scholar :
|
|
16
|
Bao D, Lu D, Liu N, Dong W, Lu YD, Qin C
and Zhang LF: Tomoregulin-1 prevents cardiac hypertrophy after
pressure overload in mice by inhibiting TAK1-JNK pathways. Dis
Model Mech. 8:795–804. 2015. View Article : Google Scholar :
|
|
17
|
Fan Y, Cheng J, Vasudevan SA, Patel RH,
Liang L, Xu X, Zhao Y, Jia W, Lu F, Zhang H, et al: TAK1 inhibitor
5Z-7-oxozeaenol sensitizes neuroblastoma to chemotherapy.
Apoptosis. 18:1224–1234. 2013. View Article : Google Scholar :
|
|
18
|
Hayakawa Y, Hirata Y, Kinoshita H,
Sakitani K, Nakagawa H, Nakata W, Takahashi R, Sakamoto K, Maeda S
and Koike K: Differential roles of ASK1 and TAK1 in Helicobacter
pylori-induced cellular responses. Infect Immun. 81:4551–4560.
2013. View Article : Google Scholar :
|
|
19
|
Ikeda Y, Morioka S, Matsumoto K and
Ninomiya-Tsuji J: TAK1 binding protein 2 is essential for liver
protection from stressors. PLoS One. 9:e880372014. View Article : Google Scholar :
|
|
20
|
Hagberg H, Mallard C, Rousset CI and
Xiaoyang W: Apoptotic mechanisms in the immature brain: Involvement
of mitochondria. J Child Neurol. 24:1141–1146. 2009. View Article : Google Scholar :
|
|
21
|
Zhu C, Qiu L, Wang X, Hallin U, Candé C,
Kroemer G, Hagberg H and Blomgren K: Involvement of
apoptosis-inducing factor in neuronal death after hypoxia-ischemia
in the neonatal rat brain. J Neurochem. 86:306–317. 2003.
View Article : Google Scholar
|
|
22
|
Hagberg H, Wilson MA, Matsushita H, Zhu C,
Lange M, Gustavsson M, Poitras MF, Dawson TM, Dawson VL,
Northington F and Johnston MV: PARP-1 gene disruption in mice
preferentially protects males from perinatal brain. J Neurochem.
90:1068–1075. 2004. View Article : Google Scholar
|
|
23
|
Zhu C, Wang X, Deinum J, Huang Z, Gao J,
Modjtahedi N, Neagu MR, Nilsson M, Eriksson PS, Hagberg H, et al:
Cyclophilin A participates in the nuclear translocation of
apoptosis-inducing factor in neurons after cerebra l
hypoxia-ischemia. J Exp Med. 204:1741–1748. 2007. View Article : Google Scholar :
|
|
24
|
Gu Y, Zhang Y, Bi Y, Liu J, Tan B, Gong M,
Li T and Chen J: Mesenchymal stem cells suppress neuronal apoptosis
and decrease IL-10 release via the TLR2/NFkB pathway in rats with
hypoxic-ischemic brain damage. Mol Brain. 8:652015. View Article : Google Scholar :
|
|
25
|
Sun MY, Cui KJ, Yu MM, Zhang H, Peng XL
and Jiang H: Bax inhibiting peptide reduces apoptosis in neonatal
rat hypoxic-ischemic brain damage. Int J Clin Exp Pathol.
8:14701–14708. 2015.
|
|
26
|
Neubert M, Ridder DA, Bargiotas P, Akira S
and Schwaninger M: Acute inhibition of TAK1 protects against
neuronal death in cerebral ischemia. Cell Death Differ.
18:1521–1530. 2011. View Article : Google Scholar :
|
|
27
|
White BJ, Tarabishy S, Venna VR, Manwani
B, Benashski S, McCullough LD and Li J: Protection from cerebral
ischemia by inhibition of TGFβ-activated kinase. Exp Neurol.
237:238–245. 2012. View Article : Google Scholar :
|
|
28
|
Zhang D, Hu Y, Sun Q, Zhao J, Cong Z, Liu
H, Zhou M, Li K and Hang C: Inhibition of transforming growth
factor beta-activated kinase 1 confers neuroprotection after
traumatic brain injury in rats. Neuroscience. 238:209–217. 2013.
View Article : Google Scholar
|
|
29
|
Cao J, Semenova MM, Solovyan VT, Han J,
Coffey ET and Courtney MJ: Distinct requirements for p38alpha and
c-Jun N-terminal kinase stress-activated protein kinases in
different forms of apoptotic neuronal death. J Biol Chem.
279:35903–35913. 2004. View Article : Google Scholar
|
|
30
|
Tu YF, Tsai YS, Wang LW, Wu HC, Huang CC
and Ho CJ: Overweight worsens apoptosis, neuroinflammation and
blood-brain barrier damage after hypoxic ischemia in neonatal brain
through JNK hyperactivation. J Neuroinflammation. 8:402011.
View Article : Google Scholar :
|
|
31
|
Li D, Li X, Wu J, Li J, Zhang L, Xiong T,
Tang J, Qu Y and Mu D: Involvement of the JNK/FOXO3a/Bim pathway in
neuronal apoptosis after Hypoxic-Ischemic brain damage in neonatal
rats. PLoS One. 10:e01329982015. View Article : Google Scholar :
|
|
32
|
Nijboer CH, Bonestroo HJ, Zijlstra J,
Kavelaars A and Heijnen CJ: Mitochondrial JNK phosphorylation as a
novel therapeutic target to inhibit neuroinflammation and apoptosis
after neonatal ischemic brain damage. Neurobiol Dis. 54:432–444.
2013. View Article : Google Scholar
|
|
33
|
Nijboer CH, van der Kooij MA, van Bel F,
Ohl F, Heijnen CJ and Kavelaars A: Inhibition of the JNK/AP-1
pathway reduces neuronal death and improves behavioral outcome
after neonatal hypoxic-ischemic brain injury. Brain Behav Immun.
24:812–821. 2010. View Article : Google Scholar
|
|
34
|
Tan L, Nomanbhoy T, Gurbani D, Patricelli
M, Hunter J, Geng J, Herhaus L, Zhang J, Pauls E, Ham Y, et al:
Discovery of type II inhibitors of TGFβ-activated kinase 1 (TAK1)
and mitogen-activated protein kinase kinase kinase kinase 2
(MAP4K2). J Med Chem. 58:183–196. 2015. View Article : Google Scholar
|
|
35
|
Wang Z and Zhang H, Shi M, Yu Y, Wang H,
Cao WM, Zhao Y and Zhang H: TAK1 inhibitor NG25 enhances
doxorubicin-mediated apoptosis in breast cancer cells. Sci Rep.
6:327372016. View Article : Google Scholar :
|