Introduction
Myeloproliferative neoplasms (MPNs) are a
heterogeneous group of chronic disorders that are characterized by
increased proliferation of one or more of the myeloid lineages,
which are considered to arise from a mutated hematopoietic stem
cell (1). Primary myelofibrosis
(PMF) is one of the three classic types of MPN (2), and is characterized by megakaryocyte
hyperplasia, bone marrow fibrosis and abnormal stem cell
trafficking (3). The progression
of PMF from a precancerous neoplasm to an aggressive malignant
cancer, such as leukemia, is variable in speed and incidence rates,
but has a poor prognosis (4).
The underlying molecular pathogenesis of PMF is
partially understood by the identification of the relationships
between the disease and the functional mutations of Janus kinase 2
(5) and MPL proto-oncogene,
thrombopoietin receptor (6). In
addition, the mutational profiling of a number of genes, including
additional sex combs-like 1 transcriptional regulator, enhancer of
zeste 2 polycomb repressive complex 2, serine and arginine rich
splicing factor 2 and isocitrate dehydrogenase, was also identified
in patients with PMF at risk of premature death or leukemic
transformation (7). However,
calreticulin mutations in patients with PMF demonstrated a
favorable effect on overall survival (8). Although the mutational landscape of
PMF has been previously investigated, a comprehensive framework of
the molecular mechanisms of PMF pathogenesis has not been fully
elucidated.
Gene expression is highly regulated so that their
proper biological functions may be executed in a cell in response
to internal and/or external perturbations (9). Therefore, variations in gene
expression during disease deterioration processes may be causally
associated with phenotypic changes. That is, differentially
expressed genes (DEGs) may provide clues on the pathogenesis of
destructive diseases (10).
However, analysis of DEGs alone may lead to false-positive results,
as some genes may exhibit significant differences in expression
from certain stimuli that may not be related to the pathogenic
process (11). Disease
pathogenesis often involves a complex network of proteins and other
molecules (12). Therefore,
network-based systems biology approaches, such as protein-protein
interaction (PPI) networks, may provide insights into the
pathogenesis of a certain disease and may explain the underlying
molecular processes involved during the development and progression
of complex diseases (13).
Previously reported network-based methods have been widely used in
the identification of pathogenic genes in a number of diseases
(14–17). In addition, specific gene
expression data combined with gene networks and known pathogenic
genes may be effective in predicting unknown underlying pathogenic
genes for a certain disease (18).
To better understand the molecular pathogenesis of
PMF, the present study implemented a system-network approach to
predict the underlying pathogenic genes for PMF by integrating
protein interaction maps and gene expression data. These results
may aid in the identification of novel pathogenic genes and may
contribute to the clinical guidance for the treatment of PMF.
Materials and methods
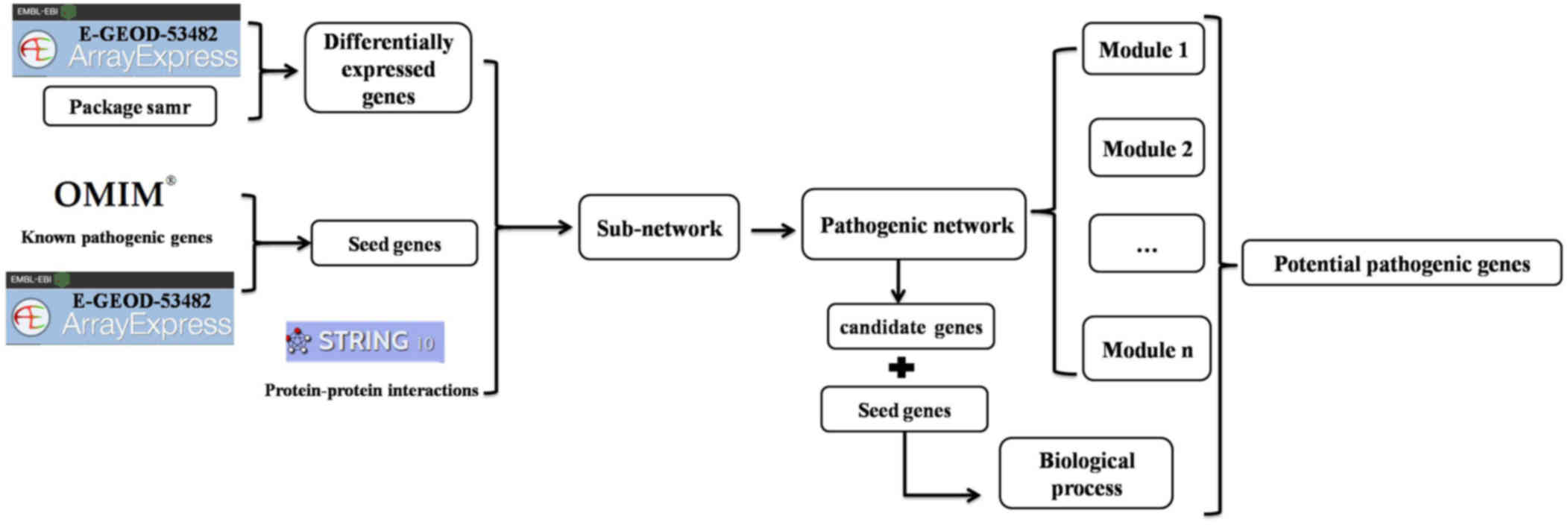
System-network approach
To identify potential pathogenic genes involved in
PMF, we performed a systemic analysis, as outlined in Fig. 1 and detailed in the following
sections.
Affymetrix microarray data
recruitment
The gene expression profile of PMF (accession no.
E-GEOD-53482) was obtained from the ArrayExpress database
(www.ebi.ac.uk/arrayexpress). This
data set was from an A-GEOD-13667-[HG-U219] Affymetrix Human Genome
U219 Array and the expression data were from mRNA expression
profiling in CD34+ cells from 42 patients with PMF and
31 healthy donors (19). The
microarray data and annotation files of PMF and healthy donors were
downloaded; the probe level gene expression profile was converted
into gene symbols and duplicated symbols were discarded. A total of
11,134 gene symbols were obtained for further analysis.
Identifying DEGs
The propensity of many diseases may be reflected in
the difference in gene expressions in particular cell types
(20). Therefore, differential
gene expression analysis between patients with PMF and healthy
controls was conducted on the gene expression profile. Significance
analysis of microarrays (SAM) (21) was performed using the samr package
(v2.0; cran.r-project.org/web/packages/samr/index.html) in R,
and was used to calculate gene expression values and identify DEGs
in PMF. Briefly, SAM allotted a score value to each gene based on
the change in expression relative to the standard deviation of
repeated measurements. Given a gene (i), the differential
expression level (Ci) was calculated as follows:
Ci=g(P,i)–g(H,i)sd(i)+sd0
Where g(P, i) and
g(H, i) represent the mean expression values
of gene i in PMF (P) and healthy (H) conditions, respectively;
sd(i) represents the standard deviation of repeated
measurements and sd0 was chosen to minimize variable
coefficient. Each gene was given a score value based on the gene
expression change compared with the standard deviation of repeated
measurements. Genes with a score greater than the threshold value
were considered to be potentially significant. The false discovery
rate (FDR) was used to estimate the percentage of genes identified
by chance (22). In the present
study, the threshold value of FDR <0.05 and a delta cut-off
value of >3.278 were used.
Identifying pathogenic networks
The Online Mendelian Inheritance in Man (OMIM)
database (www.omim.org) is a comprehensive
compilation of human genes and genetic phenotypes, with a focus on
the relationship between phenotype and genotype, and contains
information on all known Mendelian disorders and over 15,000 genes.
Prior to analysis, known pathogenic genes of PMF were retrieved
from OMIM. A total of 85 pathogenic genes were identified to be
associated with PMF in OMIM, of which 45 were in the E-GEOD-53482
gene expression profile, and these 45 genes were treated as seed
genes. In addition, a human PPI network was downloaded from the
Search Tool for the Retrieval of Interacting Genes/Proteins
(STRING) database (string-db.org).
If the genes that interacted with the seed genes
were potentially pathogenic, they may interact with the seed genes
to maintain essential biological processes. Therefore, the seed
genes and their adjacent DEGs within the PPI network were aligned
to the human PPI network, and a new network was extracted from the
PPI network. Subsequently, a smaller pathogenic gene network was
extracted from the new network and contained DEGs that interacted
with at least two seed genes. Genes in the pathogenic network were
considered to be candidate pathogenic genes and may be related to
PMF pathogenesis.
Candidate pathogenic genes
To improve confidence in the predicted pathogenic
genes, co-expression correlativity was analyzed between seed genes
and candidate pathogenic genes in pathogenic network. Co-expression
status was evaluated by Pearson's correlation coefficient between
the predicted pathogenic genes and seed genes, based on gene
expression level. Each gene was assigned a weight according to the
interactions and co-expression level with seed genes. If a gene was
co-expressed and interacted with several seed genes, the confidence
of it being a pathogenic gene increased. Based on Pearson
correlation coefficients, the weight value (w) for a gene (a) was
defined as follows:
w(a)=∑b∈DPC(a,b)XI(a,b)
Where D is the set of known pathogenic genes,
PC(a,b) was Pearson's correlation coefficient between
gene a and seed gene b and I(a,b) was an indication
function, where I(a,b) = ~1 if gene a interacted with
seed gene b and I(a,b) = ~0 if gene a did not interact
with seed gene b. The weight values instructed the correlations
between candidates and seed genes. Candidate pathogenic genes with
higher weight values had higher confidence as potentially involved
in pathogenic processes.
Cluster analysis of the pathogenic
network
Functionally related genes are often co-expressed in
many organisms, constituting conserved transcription modules
(23), where modules are groups of
genes with expression profiles that are highly correlated across
the samples (24). To further
investigate how the potential pathogenic genes respond to stimuli
in pathogenesis, cluster analysis was performed to mine the modules
from the pathogenic network using the Cytoscape (v3.3.0; www.cytoscape.org/) plugin ClusterONE (v1.0;
apps.cytoscape.org/apps/clusterone). A significance
score (SS) was utilized to measure the significance of the
predicted modules, which was defined as the geometric mean of
P-values accompanying the nodes in the module. The P-value for each
node was determined by the Mann-Whitney-Wilcoxon test based on gene
expression data under two conditions.
To measure the statistical significance of the
predicted modules, a P-value was calculated by an empirical
randomization test procedure for each module. The P-values of the
genes in a module were randomly shuffled, and each gene received a
new P-value. Subsequently, the SSs of the predicted modules were
recalculated following the shuffling of P-value labels and these
were regarded as the null distribution of the SSs. The
randomization was repeated 10,000 times. Finally, the P-value for a
module was defined as the probability that one module could be
detected in a randomization procedure with a smaller SS than that
of the predicted module.
Functional enrichment analysis of the
potential pathogenic genes
The Kyoto Encyclopedia of Genes and Genomes (KEGG)
pathway database is a comprehensive database that contains
information on numerous biochemical pathways (25). The KEGG database was used to
investigate pathway enrichment of the potential pathogenic genes in
PMF with the Database for Annotation, Visualization and Integrated
Discovery (DAVID) (26). Pathways
with P<0.05 and gene number >5 were considered as significant
pathways. If candidate pathogenic genes and seed genes were
involved in the same biological process, these candidate pathogenic
genes may be important for the pathogenesis of PMF.
Results
Identifying DEGs
Based on the obtained microarray data, SAM was used
to calculate gene expression values and to identify DEGs between
patients with PMF and healthy donors. With the threshold value of
FDR <0.05 and a delta cut-off value >3.278, a total of 845
DEGs were identified between PMF and healthy donors.
Identifying the pathogenic gene
network
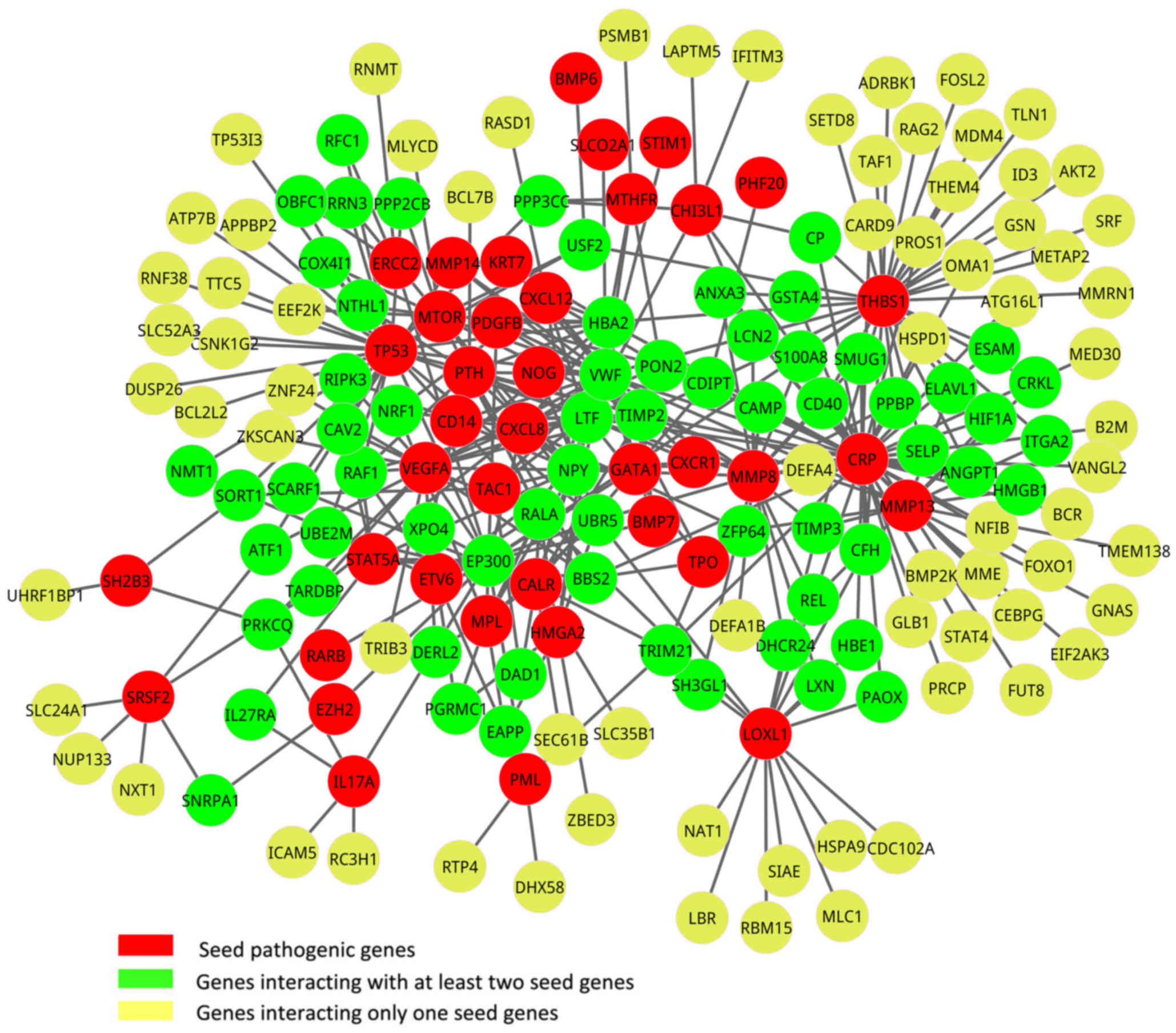
A total of 85 known pathogenic genes for PMF were
downloaded from OMIM, and 45 of these pathogenic genes were in the
gene expression profile and were considered as seed genes. Based on
the PPI data, the seed genes from OMIM and the DEGs, a new network
was constructed by aligning seed genes and DEGs to a human PPI
network, which contained a total of 39 seed genes and 139 DEGs
(Fig. 2). In addition, a
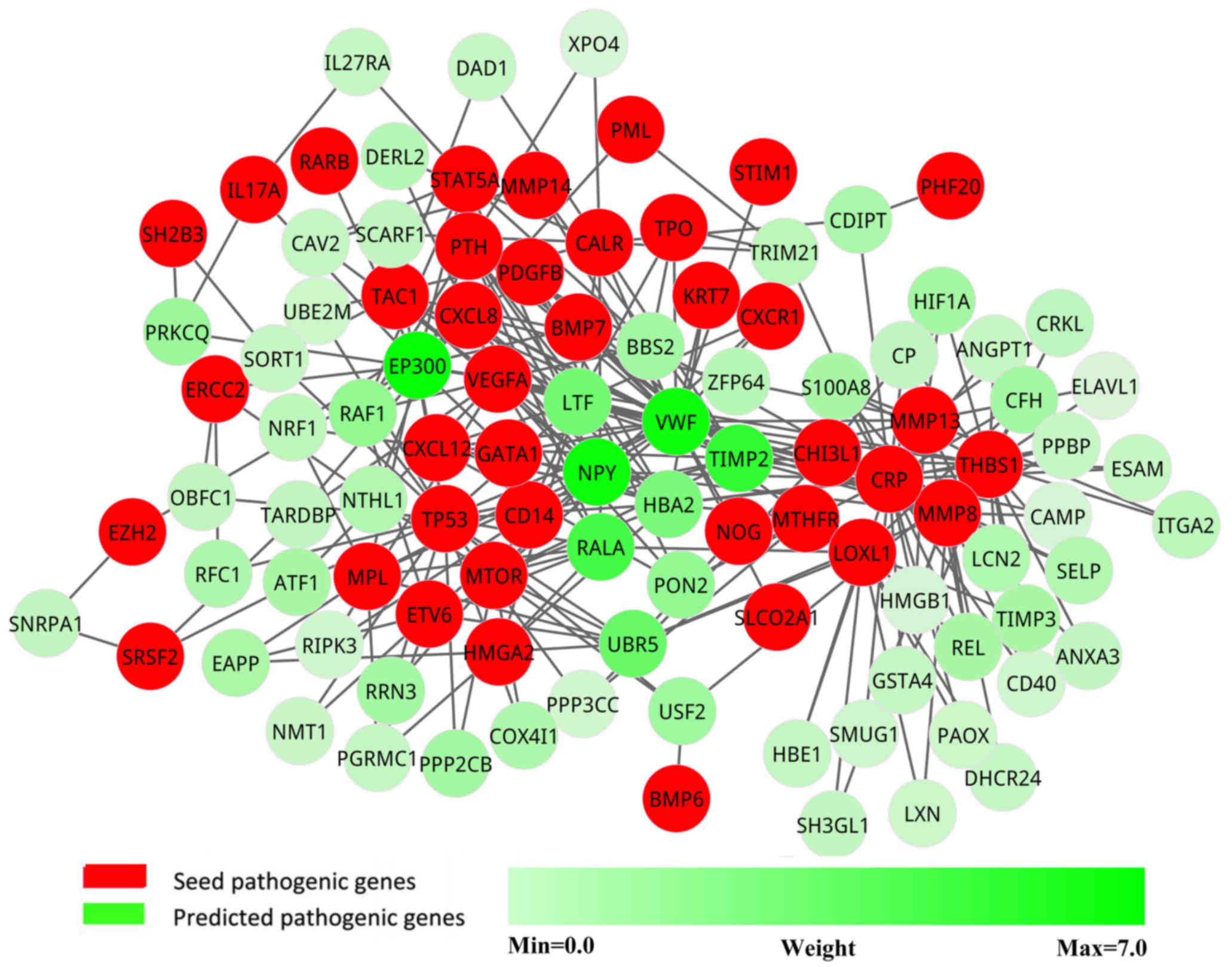
pathogenic network was extracted from the new network, in which the
genes interacted with at least two seed genes. This pathogenic
network comprised 103 nodes and 265 interactions (Fig. 3).
Candidate pathogenic genes
To increase the confidence in the predicted
pathogenic genes, each gene in the pathogenic network was assigned
a weight according to the co-expression level with seed genes. The
genes were ranked in descending order according to their weight
values. The top 10 candidate pathogenic genes (Table I) were more likely to be pathogenic
genes, as they had more interactions and higher correlations with
known pathogenic genes. These genes were considered as potential
pathogenic genes.
 | Table I.Top 10 candidate pathogenic genes of
primary myelofibrosis. |
Table I.
Top 10 candidate pathogenic genes of
primary myelofibrosis.
| Genes | Weight |
|---|
| EP300 | 6.84 |
| NPY | 6.61 |
| VWF | 6.49 |
| TIMP2 | 5.45 |
| RALA | 4.81 |
| UBR5 | 3.67 |
| LTF | 3.39 |
| HBA2 | 3.10 |
| RAF1 | 2.32 |
| PON2 | 2.30 |
Significance analysis of pathogenic
modules
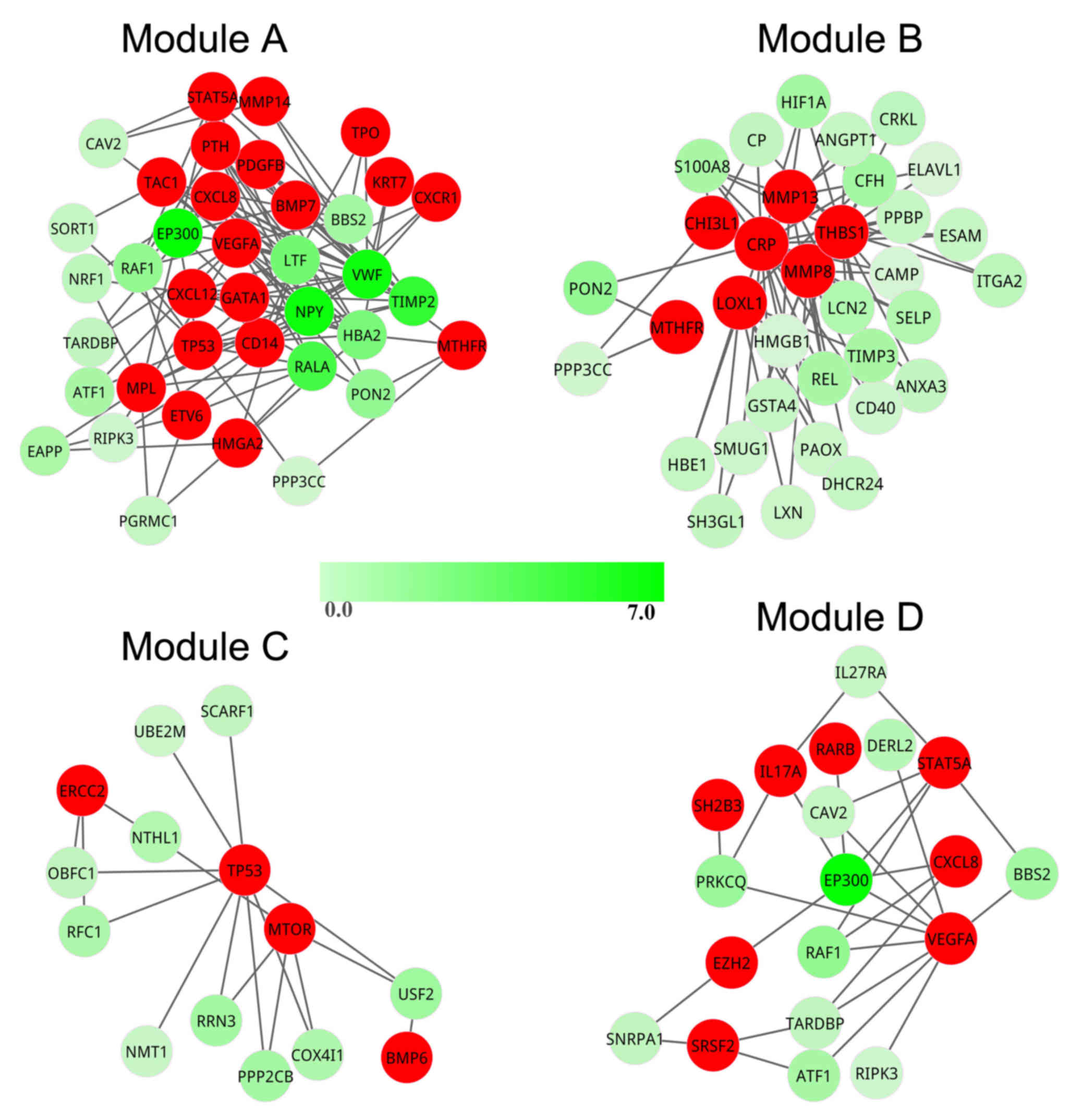
To investigate how the candidate pathogenic genes
responded to stimuli in the pathogenetic process, cluster analysis
was performed to mine the modules from the pathogenic network.
Using ClusterONE, four modules (modules A-D) were obtained from the
pathogenic network (Fig. 4). The
SS score was used to investigate whether a module could be detected
by chance. The SS values of modules A-D were 8.90×10−3,
8.20×10−3, 1.07×10−4 and
2.537×10−4, respectively.
To determine the statistical significance of the
four predicted modules, a P-value was determined for each using an
empirical randomization test procedure. The P-values of modules A-D
were 1.13×10−2, 1.16×10−2,
1.00×10−4 and 1.00×10−4, respectively, which
demonstrated that these four modules were statistically
significant. In module A, a total of 38 genes formed a tightly
connected module with 19 seed genes. In modules B-D, there were 34,
14 and 19 genes, covering 7, 4 and 8 seed genes, respectively. In
addition, there were nine potential pathogenic genes contained in
module A [E1A-binding protein p300 (EP300), neuropeptide Y
(NPY), von Willebrand factor (VWF), TIMP
metallopeptidase inhibitor 2 (TIMP2), RAS-like
proto-oncogene A (RALA), lactotransferrin (LTF),
hemoglobin α2 (HBA2), RAF-1 proto-oncogene, serine/threonine
kinase (RAF1) and paraoxonase 2 (PON2)], however,
there was only one potential pathogenic gene in modules B
(PON2) and D (EP300), and no potential pathogenic
genes were identified in module C. Therefore, module A contained
the greatest number of seed genes and potential pathogenic genes;
thus, it was regarded as the most important module in PMF.
Functional enrichment analysis
Functional enrichment analysis demonstrated that the
known and candidate pathogenic genes in PMF participated in four
pathways, including pathways in cancer, cytokine-cytokine receptor
interaction, TGF-β signaling pathway and focal adhesion (Table II). Candidate pathogenic genes
EP300, RALA and RAF1 were enriched in pathways
in cancer, candidate pathogenic gene EP300 was enriched in
TGF-β signaling pathway and candidate pathogenic genes VWF
and RAF1 were enriched in focal adhesion pathway.
| Table II.Enriched Kyoto Encyclopedia of Genes
and Genomes pathways of the seed genes and candidate pathogenic
genes in primary myelofibrosis. |
Table II.
Enriched Kyoto Encyclopedia of Genes
and Genomes pathways of the seed genes and candidate pathogenic
genes in primary myelofibrosis.
| ID | Term | n | P-value | Genes |
|---|
| hsa05200 | Pathways in
cancer | 10 |
1.67×10−4 | EP300, PDGFB,
STAT5A, VEGFA, PML, TP53, RALA, RAF1, RARB and MTOR |
| hsa04060 | Cytokine-cytokine
receptor interaction | 8 |
1.20×10−3 | IL17A, PDGFB,
VEGFA, CXCR1, TPO, MPL, BMP7 and CXCL12 |
| hsa04350 | TGF-β signaling
pathway | 5 |
2.24×10−3 | NOG, EP300,
BMP7, THBS1 and BMP6 |
| hsa04510 | Focal adhesion | 5 |
3.96×10−2 | VWF, PDGFB,
VEGFA, RAF1 and THBS1 |
Discussion
In the present study, a system-network approach was
performed to predict candidate pathogenic genes for PMF by
integrating protein interaction map and gene expression data. A
total of 10 potential pathogenic genes were identified for PMF, all
of which were involved in pathogenic module A, except for ubiquitin
protein ligase E3 component n-recognin 5 (UBR5). In
addition, the candidate pathogenic genes mainly participated in
pathways in cancer, TGF-β signaling pathway and focal adhesion
pathway.
Among the 10 candidate pathogenic genes,
EP300 exhibited the highest weight value in the pathogenic
network and participated in two signaling pathways, which indicated
an important role for this gene in the development of PMF. EP300 is
a histone acetyltransferase that regulates transcription through
chromatin remodeling and is an important factor in cell
proliferation and differentiation (27). It was previously reported that
EP300 may be a target for adenoviral E1A oncoprotein, and the
binding of these proteins has been associated with malignant
transformations, which suggested that EP300 may function as
a tumor suppressor gene (28).
Additional evidence for a role of EP300 in tumorigenesis was
provided by another study that demonstrated that EP300 was
fused with the mixed-lineage leukemia gene in leukemia (29). Mutations in EP300 were
detected in >5% of patients with PMF (30). Results from a previous study
indicated that EP300 was an attractive candidate gene for
human myeloproliferative disorders, and point mutations in the
coding region of EP300 may be the cause of myeloproliferation
(31). The present results
indicated that EP300 had the most interactions and the
highest correlations with known pathogenic genes; further analysis
may be able to determine the specific role of EP300 in the
pathogenesis of PMF.
Three other candidate pathogenic genes, RALA,
RAF1 and VWF, were selected by functional analysis.
RALA is one of two proteins in the Ral protein family, a subfamily
in the RAS super-family of small GTPases, and acts as a downstream
effector of RAS (32). It has been
reported that RALA was involved in a number of complex
diseases, such as lung cancer (33), bladder cancer (34), pancreatic cancer (35), colorectal cancer (36) and ovarian cancer. RAF1 is a
proto-oncogene that functions downstream of the RAS sub-family of
membrane associated GTPases (37).
RAF1 oncogenic signaling was linked to activation of the
mesenchymal-to-epithelial transition pathway in metastatic breast
cancer cells (38). In addition,
RAF1 has been considered as a therapeutic target in disease
treatment (39). VWF
encodes a multimeric glycoprotein that mediates the adhesion of
platelets to the subendothelial matrix and to endothelial surfaces,
and is a carrier for coagulation factor VIII in the circulation
(40). Colon cancer cell adherence
to human umbilical vein endothelial cells was demonstrate to be
enhanced by the addition of VWF to the media in a co-culture system
and adherence was blocked by the addition of VWF antibodies and the
platelet inhibitor ticlopidne (41). VWF deficiency may lead to
bleeding diathesis of the skin and mucous membrane. In the present
study, these three candidate pathogenic genes were identified as
being involved in the same biological process with the seed genes,
and may exert influence in PMF development by interacting with
known pathogenic genes.
In conclusion, by using a system-network approach
that combined protein interaction network and gene expression data
with known pathogenic genes, the present study predicted 10
candidate pathogenic genes and several signaling pathways that may
be related to the pathogenesis of PMF. These predictions may shed
light on the pathogenesis of PMF and may provide guidelines for
future experimental verification.
Acknowledgements
We thank all members of the Department of Oncology
and Hematology, Hubei Provincial Hospital of Integrated Chinese and
Western Medicine.
References
|
1
|
Jamieson CH, Barroga CF and Vainchenker
WP: Miscreant myeloproliferative disorder stem cells. Leukemia.
22:2011–2019. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tefferi A and Vardiman JW: Classification
and diagnosis of myeloproliferative neoplasms: The 2008 World
Health Organization criteria and point-of-care diagnostic
algorithms. Leukemia. 22:14–22. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Vannucchi AM, Guglielmelli P and Tefferi
A: Advances in understanding and management of myeloproliferative
neoplasms. CA Cancer J Clin. 59:171–191. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Triviai I, Ziegler M, Bergholz U, Oler AJ,
Stübig T, Prassolov V, Fehse B, Kozak CA, Kröger N and Stocking C:
Endogenous retrovirus induces leukemia in a xenograft mouse model
for primary myelofibrosis. Proc Natl Acad Sci USA. 111:8595–8600.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Levine RL, Wadleigh M, Cools J, Ebert BL,
Wernig G, Huntly BJ, Boggon TJ, Wlodarska I, Clark JJ, Moore S, et
al: Activating mutation in the tyrosine kinase JAK2 in polycythemia
vera, essential thrombocythemia, and myeloid metaplasia with
myelofibrosis. Cancer Cell. 7:387–397. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pardanani AD, Levine RL, Lasho T, Pikman
Y, Mesa RA, Wadleigh M, Steensma DP, Elliott MA, Wolanskyj AP,
Hogan WJ, et al: MPL515 mutations in myeloproliferative and other
myeloid disorders: A study of 1182 patients. Blood. 108:3472–3476.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Vannucchi AM, Lasho TL, Guglielmelli P,
Biamonte F, Pardanani A, Pereira A, Finke C, Score J, Gangat N,
Mannarelli C, et al: Mutations and prognosis in primary
myelofibrosis. Leukemia. 27:1861–1869. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tefferi A, Lasho TL, Finke CM, Knudson RA,
Ketterling R, Hanson CH, Maffioli M, Caramazza D, Passamonti F and
Pardanani A: CALR vs JAK2 vs MPL-mutated or triple-negative
myelofibrosis: Clinical, cytogenetic and molecular comparisons.
Leukemia. 28:1472–1477. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kostka D and Spang R: Finding disease
specific alterations in the co-expression of genes. Bioinformatics.
20 Suppl 1:i194–i199. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liu Y, Chen SH, Jin X and Li YM: Analysis
of differentially expressed genes and microRNAs in alcoholic liver
disease. Int J Mol Med. 31:547–554. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
MacFarlane RC and Singh U: Identification
of differentially expressed genes in virulent and nonvirulent
Entamoeba species: Potential implications for amebic
pathogenesis. Infect Immun. 74:340–351. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lim J, Hao T, Shaw C, Patel AJ, Szabó G,
Rual JF, Fisk CJ, Li N, Smolyar A, Hill DE, et al: A
protein-protein interaction network for human inherited ataxias and
disorders of Purkinje cell degeneration. Cell. 125:801–814. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu ZP, Wang Y, Zhang XS and Chen L:
Network-based analysis of complex diseases. IET Syst Biol. 6:22–33.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jia Y, Nie K, Li J, Liang X and Zhang X:
Identification of therapeutic targets for Alzheimer's disease via
differentially expressed gene and weighted gene co-expression
network analyses. Mol Med Rep. 14:4844–4848. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Horvath S, Zhang B, Carlson M, Lu KV, Zhu
S, Felciano RM, Laurance MF, Zhao W, Qi S, Chen Z, et al: Analysis
of oncogenic signaling networks in glioblastoma identifies ASPM as
a molecular target. Proc Natl Acad Sci USA. 103:17402–17407. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jiang W, Li X, Rao S, Wang L, Du L, Li C,
Wu C, Wang H, Wang Y and Yang B: Constructing disease-specific gene
networks using pair-wise relevance metric: Application to colon
cancer identifies interleukin 8, desmin and enolase 1 as the
central elements. BMC Syst Biol. 2:722008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pe'er D and Hacohen N: Principles and
strategies for developing network models in cancer. Cell.
144:864–873. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu X, Tang WH, Zhao XM and Chen L: A
network approach to predict pathogenic genes for Fusarium
graminearum. PLoS One. 5:e130212010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Norfo R, Zini R, Pennucci V, Bianchi E,
Salati S, Guglielmelli P, Bogani C, Fanelli T, Mannarelli C, Rosti
V, et al: miRNA-mRNA integrative analysis in primary myelofibrosis
CD34+ cells: Role of miR-155/JARID2 axis in abnormal
megakaryopoiesis. Blood. 124:e21–e32. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhao J, Yang TH, Huang Y and Holme P:
Ranking candidate disease genes from gene expression and protein
interaction: A Katz-centrality based approach. PLoS One.
6:e243062011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tusher VG, Tibshirani R and Chu G:
Significance analysis of microarrays applied to the ionizing
radiation response. Proc Natl Acad Sci USA. 98:5116–5121. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Reiner A, Yekutieli D and Benjamini Y:
Identifying differentially expressed genes using false discovery
rate controlling procedures. Bioinformatics. 19:368–375. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Choi JK, Yu U, Yoo OJ and Kim S:
Differential coexpression analysis using microarray data and its
application to human cancer. Bioinformatics. 21:4348–4355. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ravasz E, Somera AL, Mongru DA, Oltvai ZN
and Barabási AL: Hierarchical organization of modularity in
metabolic networks. Science. 297:1551–1555. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kanehisa M and Goto S: KEGG: Kyoto
Encyclopedia of Genes and Genomes. Nucleic Acids Res. 28:27–30.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Huang DW, Sherman BT, Tan Q, Collins JR,
Alvord WG, Roayaei J, Stephens R, Baseler MW, Lane HC and Lempicki
RA: The DAVID gene functional classification tool: A novel
biological module-centric algorithm to functionally analyze large
gene lists. Genome Biol. 8:R1832007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gayther SA, Batley SJ, Linger L, Bannister
A, Thorpe K, Chin SF, Daigo Y, Russell P, Wilson A, Sowter HM, et
al: Mutations truncating the EP300 acetylase in human cancers. Nat
Genet. 24:300–303. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bryan EJ, Jokubaitis VJ, Chamberlain NL,
Baxter SW, Dawson E, Choong DY and Campbell IG: Mutation analysis
of EP300 in colon, breast and ovarian carcinomas. Int J Cancer.
102:137–141. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ida K, Kitabayashi I, Taki T, Taniwaki M,
Noro K, Yamamoto M, Ohki M and Hayashi Y: Adenoviral E1A-associated
protein p300 is involved in acute myeloid leukemia with
t(11;22)(q23;q13). Blood. 90:4699–4704. 1997.PubMed/NCBI
|
|
30
|
Li B, Gale RP, Xu Z, Qin T, Song Z, Zhang
P, Bai J, Zhang L, Zhang Y, Liu J, et al: Non-driver mutations in
myeloproliferative neoplasm-associated myelofibrosis. J Hematol
Oncol. 10:992017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Steensma DP, Pardanani A, Stevenson WS,
Hoyt R, Kiu H, Grigg AP, Szer J, Juneja S, Hilton DJ, Alexander WS
and Roberts AW: More on Myb in myelofibrosis: Molecular analyses of
MYB and EP300 in 55 patients with myeloproliferative disorders.
Blood. 107:1733–1735. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bodemann BO and White MA: Ral GTPases and
cancer: Linchpin support of the tumorigenic platform. Nat Rev
Cancer. 8:133–140. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Male H, Patel V, Jacob MA, Borrego-Diaz E,
Wang K, Young DA, Wise AL, Huang C, Van Veldhuizen P,
O'Brien-Ladner A, et al: Inhibition of RalA signaling pathway in
treatment of non-small cell lung cancer. Lung Cancer. 77:252–259.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Oxford G and Theodorescu D: The role of
Ras superfamily proteins in bladder cancer progression. J Urol.
170:1987–1993. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Neel NF, Stratford JK, Shinde V, Ecsedy
JA, Martin TD, Der CJ and Yeh JJ: Response to MLN8237 in pancreatic
cancer is not dependent on RalA phosphorylation. Mol Cancer Ther.
13:122–133. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Győrffy B, Stelniec-Klotz I, Sigler C,
Kasack K, Redmer T, Qian Y and Schäfer R: Effects of RAL signal
transduction in KRAS- and BRAF-mutated cells and prognostic
potential of the RAL signature in colorectal cancer. Oncotarget.
6:13334–13346. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Varga A and Baccarini M: RAF-1
(C-RAF)Encyclopedia of Signaling Molecules. Springer; New York: pp.
1562–1570. 2012
|
|
38
|
Leontovich AA, Zhang S, Quatraro C, Iankov
I, Veroux PF, Gambino MW, Degnim A, McCubrey J, Ingle J, Galanis E
and D'Assoro AB: Raf-1 oncogenic signaling is linked to activation
of mesenchymal to epithelial transition pathway in metastatic
breast cancer cells. Int J Oncol. 40:1858–1864. 2012.PubMed/NCBI
|
|
39
|
Maurer G, Tarkowski B and Baccarini M: Raf
kinases in cancer-roles and therapeutic opportunities. Oncogene.
30:3477–3488. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Franchini M, Frattini F, Crestani S,
Bonfanti C and Lippi G: von Willebrand factor and cancer: A renewed
interest. Thromb Res. 131:290–292. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Morganti M, Carpi A, Amo-Takyi B,
Sagripanti A, Nicolini A, Giardino R and Mittermayer C: Von
Willebrand's factor mediates the adherence of human tumoral cells
to human endothelial cells and ticlopidine interferes with this
effect. Biomed Pharmacother. 54:431–436. 2000. View Article : Google Scholar : PubMed/NCBI
|