Introduction
Drug induced liver injury is the most frequent cause
of hepatic dysfunction. Drugs or their reactive metabolites are
known to induce distinct effects on gene expression and cellular
homeostasis in hepatocytes (1).
Indiscriminate usage of various drugs, chemicals, mycotoxins and
gamma radiation are potential threats to the integrity of the
liver. In vitro hepatotoxicity methods are routinely used to
evaluate the hepatotoxicity of drugs/chemicals to understand the
underlying mechanisms and to establish the associations with in
vivo hepatotoxicity (2).
Hepatic injury stimulated by D-galactosamine (GalN) is an
appropriate experimental model of human liver failure (3). GalN is an amino sugar metabolized by
the hepatocytes that induces liver damage and enhances the
production of reactive oxygen species (ROS) in hepatocytes
(4). GalN is an ideal hepatotoxic
model that resembles clinical hepatitis, in which oxidative stress
serves a major role (5). GalN
inhibits protein synthesis by depleting uridine triphosphate pool,
causing early generation of ROS and finally apoptosis (6).
ROS are also involved in cardiovascular diseases,
atherosclerosis, and various acute and chronic liver diseases
(7). ROS include superoxide
(O2•; an oxygen centered radical), thiols (a
sulphur-centered radical), trichloromethyl
(CCl3•; a carbon centered radical) or
nitricoxide (NO•). The other ROS continuously generated
in vivo are: O2•, hydroxyl radicals
(OH•) and H2O2. Continuous
interactions with biological systems by such free radicals either
formed endogenously, or exogenously via the inhalation/ingestion of
toxicants, chemicals or biological reactions, cause cumulative
damage to protein, lipid, DNA, carbohydrates and the membrane
(8).
Acetaminophen, the most widely used analgesic in the
United States, causes severe hepatic necrosis leading to acute
liver failure following suicidal overdoses (9). Platelets may contribute to
acetaminophen-induced liver injury via their interactions with
leukocytes to promote inflammation, as observed in various other
models of sterile inflammation (10). Platelets contain a host of
proinflammatory mediators, which potentially may serve a role in
acetaminophen-induced liver injury (10). In the last decade, the paradigm of
platelet function has expanded from primary hemostasis to
intravascular redox signaling and sterile inflammation. Oxidative
stress and the sterile immune response are considered to be
prominent hallmarks of hepatic injury (11); however, the role of platelets has
recently been considered in the context of liver injury. Khandoga
et al (12) postulated that
as activated platelets are able to generate ROS and NO, and release
proinflammatory mediators, they may exhibit the potential to induce
liver injury. Cyclooxygenase (COX) and lipoxygenase (LOX) products
derived from arachidonic acid (AA) are responsible for
microvasculature failure, and are implicated as pathogenic
mediators in endotoxemia (13).
Ito et al (14)
demonstrated that pretreated with a LOX inhibitor in mice
attenuated liver injury during endotoxemia. In addition, a
selective COX-2 inhibitor improved the survival rate in
endotoxin-challenged mice (15).
Based on the fact that GalN is the most established model of liver
disease, in which platelets contribute to the endotoxin-induced
liver injury, and ROS are involved in acute and chronic liver
diseases, the present study used electron spin resonance (ESR) and
spin-trapping methods to detect and identify the GalN induced free
radicals in human platelet suspension and mouse primary
hepatocytes. In addition, as the development of inhibitors of COX,
LOX and cytochrome P450 pathways may present novel insights into
the treatment of free radical mediated hepato-cardiac disorders,
the inhibitors were used to assess their role in regulating
GalN-induced free radicals.
Materials and methods
Chemicals and reagents
1-Aminobenzotriazole (ABT), AA861, AA, baicalein,
bovine serum albumin (BSA), collagen (type I, bovine achilles
tendon), dimethyl sulfoxide (DMSO), N-Acetyl-D-galactosamine,
heparin, indomethacin, L-glutamine, prostaglandin E1 (PGE1), sodium
citrate and thioacetamide were purchased from Sigma-Aldrich; Merck
KGaA (Darmstadt, Germany). 5,5-Dimethyl-1 pyrroline N-oxide (DMPO)
was purchased from Enzo Life Sciences, Inc. (Farmingdale, NY,
USA).
Preparation of human platelet
suspensions
The present study was approved by the Taipei Medical
University Institutional Review Board (Taipei, Taiwan). All human
volunteers gave written informed consent. Human platelet
suspensions were prepared as previously described (16). Blood was collected from 20 male and
female (7:13) healthy human volunteers 20–30 years old; all
students from Taipei Medical University, Taiwan, who had taken no
medication during the preceding 2 weeks, between June-November
2016, and was mixed with acid citrate dextrose (ACD, 9:1).
Following centrifugation at 120 × g for 10 min at room temperature,
the supernatant platelet-rich plasma was supplemented with PGE1
(0.5 µM) and heparin (6.4 IU/ml), then incubated for 10 min at 37°C
and centrifuged at 500 × g for 10 min at the same temperature.
Freshly isolated platelets were suspended in 5 ml Tyrode's
solution, (pH 7.3; containing NaCl 137 mM, KCl 2.7 mM,
MgCl2 2.1 mM, NaH2PO4 0.4 mM,
NaHCO3 11.9 mM and glucose 11.1 nM). Then apyrase (1.0
U/ml), PGE1 (0.5 µM) and heparin (6.4 IU/ml) were added, and the
mixture was incubated for 10 min at 37°C and adjusted to
~4.5×108 platelets/ml.
Platelet aggregation
The turbidimetric method was applied to measure
platelet aggregation, using a Lumi-Aggregometer (Payton, Ontario,
Canada). Platelet suspensions (4.5×108 platelets/ml)
were preincubated at 37°C with various concentrations of GalN
(300–600 µM) or an isovolumetric solvent control (PBS) for 3 and
180 min prior to the addition of collagen (1 µg/ml). Additionally,
GalN (300–600 µM) was also preincubated for 3 min at 37°C prior to
the addition of a subthreshold concentration of collagen (0.5
µg/ml). The reaction was allowed to proceed for 6 min and the
extent of aggregation was expressed in light-transmission
units.
Isolation of primary mouse
hepatocytes
Ten male C57/BL6 mice (6–8 weeks and 25±5 g) were
purchased from BioLASCO (Taipei, Taiwan). All animal experiments
and care procedures were approved by the Institutional Animal Care
and Use Committee of Taipei Medical University. The mice were
housed in sterilized cages in a 12-h dark/light cycle at 20±1°C and
60±10% humidity with food and water ad libitum. Before undergoing
the experimental procedures, all animals were clinically normal and
free from apparent infection, inflammation, or neurologic
deficits.
Mouse hepatocyte isolation was performed with
collagenase perfusion as described by Sun et al (17). Specifically, the portal vein was
cannulated using a 22-gauge intravenous catheter and the liver was
perfused with calcium-free Krebs bicarbonate buffer followed by
collagenase [30 mg 494 IU/mg collagenase IV (Sigma-Aldrich; Merck
KGaA; C-5138)] in 280 ml Krebs bicarbonate containing 1.2 mM/l
CaCl2 and 1.8% BSA (Sigma-Aldrich; Merck KGaA; A-4503).
All solutions were maintained at 37°C and aerated using 95%
O2 and 5% CO2. The partially digested liver
was excised, passed over 60-µm nylon mesh and resuspended in Wilson
medium (Sigma-Aldrich; Merck KGaA; W-4125) with insulin
(Sigma-Aldrich; Merck KGaA; I-0516; 1 U/dl) and certified 10% fetal
bovine serum (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA,
USA; BRL 16000–044). Hepatocytes were purified by centrifugation
with the Wilson medium at 50 × g for 5 min at room temperature and
then resuspended in Wilson medium following a second centrifugation
at 80 × g for 5 min at room temperature in a gradient of 50%
Percoll-Wilson medium.
The viability of the hepatocytes, which were
maintained >85% confluency during the experiment, was determined
using trypan blue (0.008%) staining for 5 min at room temperature.
The cell concentration was adjusted to 3×105 cells/ml
with the isolated hepatocytes, then placed in a microtiter plate in
an incubator maintained at 5% CO2 at 37°C (18) and visualized using a light
microscope (Olympus Optical, CHT, Japan).
Measurement of free radicals in
platelet suspensions and primary mouse hepatocytes by ESR
spectrometry
The ESR spectrometry method was applied using a
Bruker EMX ESR spectrometer as described previously (19), with some minor modifications. The
culture medium was replaced with PBS solution prior to each
experiment. Each 150 µl platelet suspension (4.5×108
platelets/ml) and mouse hepatocytes (3×105 cells/ml)
were pre-warmed to 37°C for 2 min, and then the enzyme inhibitors
or other reagents [10 µM indomethacin (COX inhibitor), AA861 (LOX
inhibitor) and 30 µM ABT (a non-isoform specific cytochrome P450
inhibitor)] were added 3 min prior to the addition of GalN (600 µM)
and AA (100 µM). ESR spectra were recorded at room temperature
using a quartz flat cell designed for aqueous solutions. The dead
time of sample preparation and ESR analysis was exactly 30 sec
following the last addition. The conditions of ESR spectrometry
were as follows: 20 mW power at 9.78 GHz, with a scan range of 100
G and a receiver gain of 5×104. The modulation
amplitude, sweep time and time constant are given in the legends to
the figures. The ESR spectrum analysis was performed by using
WIN-EPR, version 921201 supplied by BRUKER-FRANZEN Analytik GmbH
(Bremen, Germany).
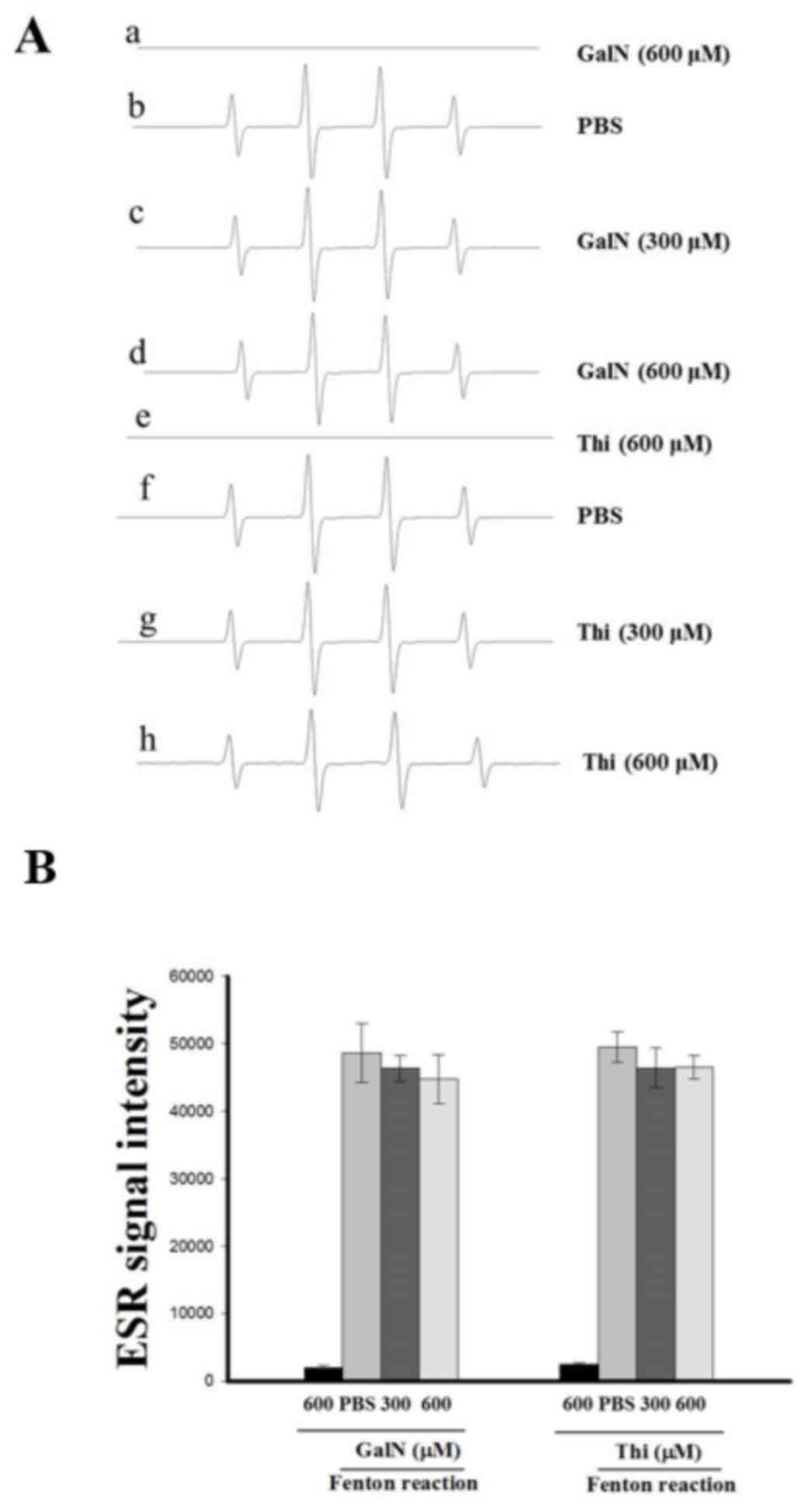
Measurement of Fenton reaction induced
OH• formation by ESR
The ESR method was used as described previously
(20). A Fenton reaction solution
(50 µM FeSO4 + 2 mM H2O2) was
pretreated with a solvent control (PBS) for 1 min with or without
GalN and thioacetamide (300–600 µM). The ESR spectrum analysis was
performed by using WIN-EPR, version 921201 supplied by
BRUKER-FRANZEN Analytik GmbH.
Statistical analysis
Experimental results are expressed as the mean ±
standard error of the mean and are accompanied by the number (n) of
observations. Data were assessed using an analysis of variance
followed by the Newman-Keuls post hoc test for multiple
comparisons. All statistical tests were carried out using SigmaPlot
version 10 software (Systat Software Inc., San Jose, USA).
P<0.05 was considered to indicate a statistically significant
difference.
Results
GalN stimulated hydroxyl radical
production in human platelet suspension
The ESR spin-trapping technique was employed to
detect free radical production during the reaction between the
platelet suspension and GalN. In the absence of GalN-induced
stimulation, only weak spin trapping of the hydroxyl radical
(OH•) was detected with DMPO (Fig. 1A). By contrast, a typical
concentration dependent 4-line OH•signal
(aN=aH=14.8 G) was detected when GalN
(150–600 µM) was reacted with the human platelet suspension in the
presence of 100 mM DMPO (Fig.
1Ab-Ad). DMSO, an amphiphilic compound, which acted as a free
radical scavenger (Fig. 1Ae),
effectively scavenged these radicals. In addition, similar radical
signals were observed in thioacetamide treated platelet suspension
to that detected in PBS treated platelets (Fig. 1Af), which indicated that
OH• signals produced in the platelet suspension are
dependent upon the type of inducer used in the spin trapping
reaction. Fig. 1B illustrates the
statistical analysis of results presented in 1A, in which GalN
significantly induced a 4-line OH• signal in a
dose-dependent manner (150–600 µM). Moreover, GalN at a
concentration of 600 µM, induced the highest significant
OH• signal compared to PBS (P<0.001), but this signal
was significantly reversed by DMSO (P<0.001).
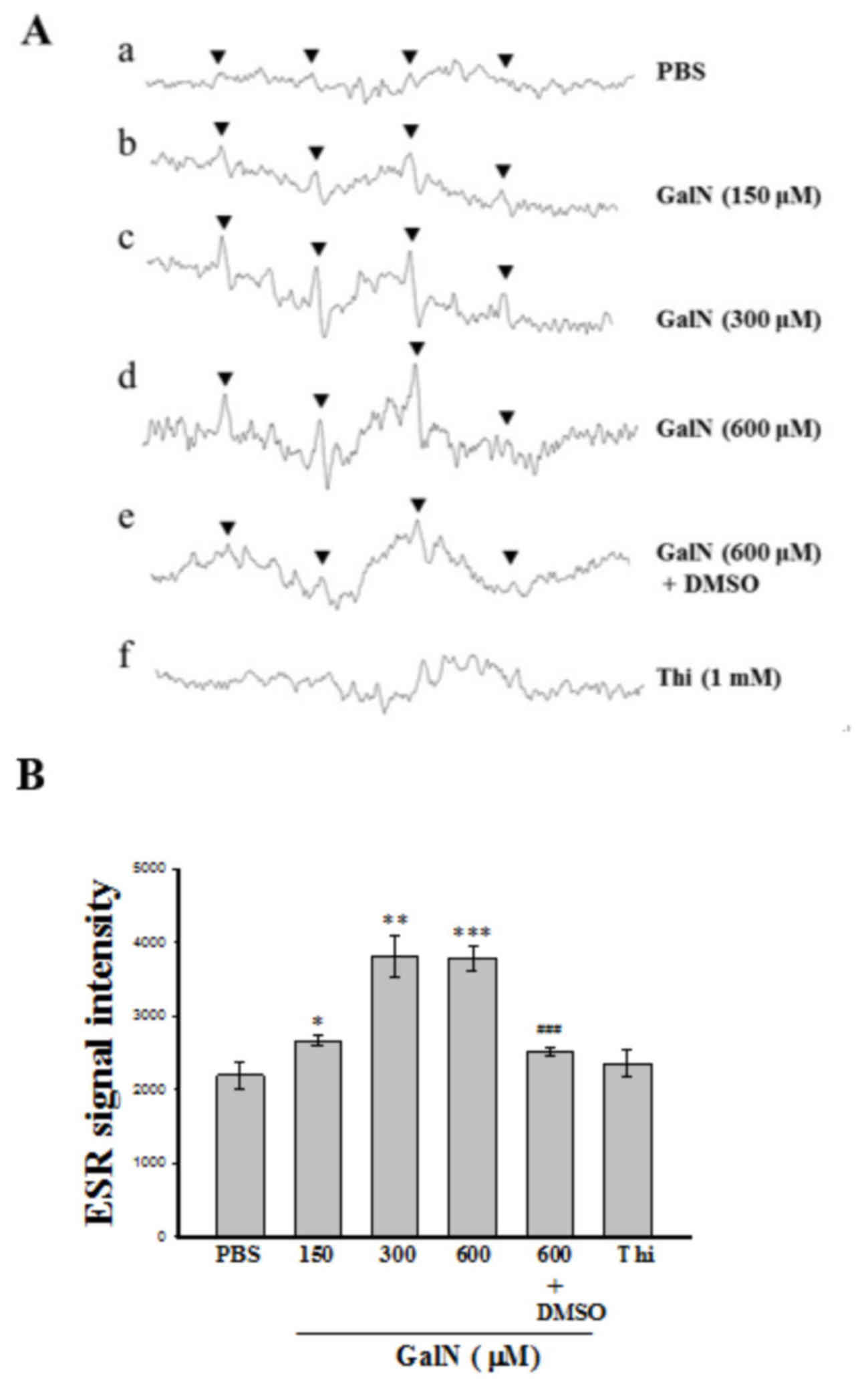 | Figure 1.Analysis of ESR spectra. (A) ESR
spectra detected from the reaction of human platelet suspensions
with GalN in the presence of DMPO. (a) Human platelets
(4.5×108 platelets/ml, 150 µl) were preincubated with
DMPO (100 mM) followed by the addition of GalN at (b) 150, (c) 300
and (d) 600 µM. (e) 600 µM GalN plus 1% DMSO. (f) Human platelets
(4.5×108 platelets/ml, 150 µl) preincubated with DMPO
and then 1 mM thio was added. Instrument parameters were as
follows: Modulation amplitude, 1 G; time constant, 164 msec;
scanning for 42 sec with 10 scans accumulated. The ESR spectra are
labeled to show their components: DMPO-hydroxyl radical adducts
(▼). (B) Statistical data are presented as the mean ± standard
error of the mean (n=4). *P<0.05; **P<0.01 and ***P<0.001,
vs. the reaction of PBS treated platelets;
###P<0.001, vs. the reaction of GalN (600 µM)
incubated platelets. GalN, D-galactosamine; ESR, electron spin
resonance; DMPO, 5,5-dimethyl-1-pyrroline N-oxide; Thi,
thioacetamide. |
Role of COX/LOX in GalN-induced
OH• in human platelets
Since COX and LOX products derived from AA, such as
prostaglandins and leukotrienes, are associated with pathogenic
mediators in endotoxemia, the inhibitors of these enzymes were
employed in order to determine whether they serve a role in
GalN-induced OH• production in human platelets.
Platelets were incubated with 10 µM indomethacin (COX inhibitor),
AA861 (LOX inhibitor) and 30 µM ABT (a non-isoform specific
cytochrome P450 inhibitor) 3 min prior to the addition of GalN. The
results demonstrated that in the presence of indomethacin, GalN
generated a condensed (84.3±1.7%) ESR signal intensity, however,
AA861 and ABT demonstrated no significant effect (8.7±9.3 and
24.8±14.8%, respectively) in this reaction system (Fig. 2A). These observations suggested a
role for COX enzyme in the generation of GalN-induced
OH• in human platelets.
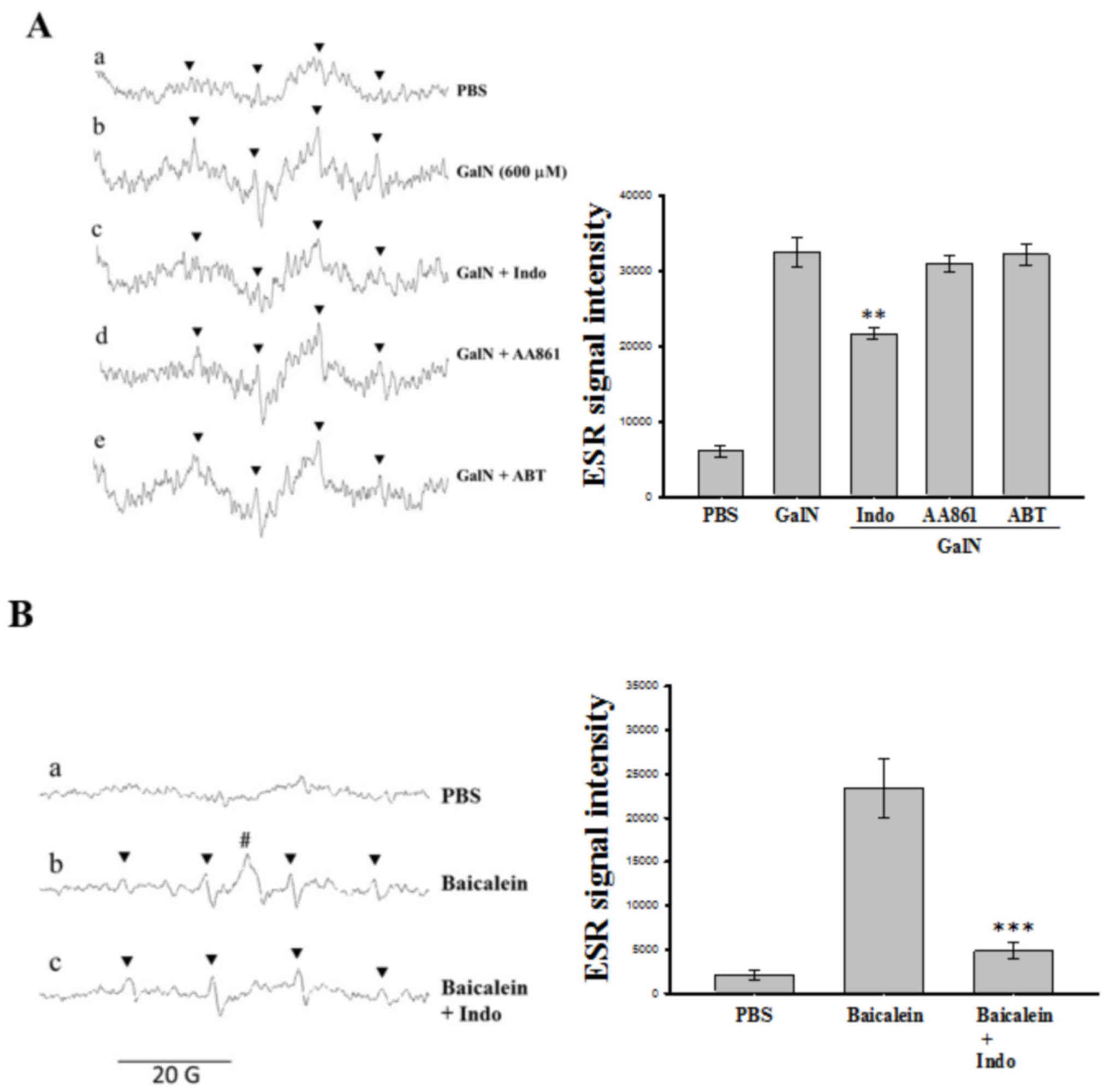 | Figure 2.Effects of various inhibitors on
GalN-induced hydroxyl radical formation in washed human platelets.
(A) The reaction mixture contained human platelet suspensions
(4.5×108 platelets/ml; 150 µl) with (a) DMPO (100 mM)
and (b) GalN (600 µM). The inhibitors of (c) indo (10 µM), (d)
AA861 (10 µM) and (e) ABT (30 µM) were added to the GalN (600 µM)
contained platelet suspensions. The instrument parameters were the
same as those in Fig. 1.
**P<0.01, vs. the reaction of GalN incubated platelets. (B)
Effects of indo on baicalein induced semiquinone and hydroxyl
radical formation in washed human platelets. (a) Human platelets
(4.5×108 platelets/ml, 150 µl) were preincubated with
DMPO (100 mM) followed by the addition of baicalein at (b) 300 µM,
and (c) baicalein 300 µM + indomethacin (10 µM). The ESR spectra
are labeled to demonstrate their components: DMPO-hydroxyl radical
adducts (▼, Panel A); DMPO-semiquinone radical adduct (#, Panel B).
Data are presented as the mean ± standard error of the mean (n=4).
***P<0.001, vs. the reaction of baicalein treated platelets.
**P<0.01, vs. the reaction of GaIN treated platelets. DMPO,
5,5-dimethyl-1-pyrroline N-oxide; GalN, D-galactosamine;
ABT, 1-aminobenzotriazole; Indo, indomethacin; ESR, electron spin
resonance. |
Spin trapping of the baicalein-induced
semiquinone radicals in human platelets
The result suggesting that the COX enzyme mediated
GalN-induced OH• in human platelets was further
investigated using the COX inhibitor indomethacin on
baicalein-induced semiquinone radicals in a platelet suspension.
Fig. 2A demonstrates that
indomethacin significantly (P<0.01) inhibited GalN induced
OH• formation. Application of baicalein significantly
induced semiquinone radicals as well as OH•; however,
the COX inhibitor indomethacin suppressed baicalein-induced
semiquinone radicals (Fig. 2B)
without affecting the induced OH• formation. Therefore,
it was confirmed that COX serves a role in the formation of
GalN-induced OH• in platelets and LOX may involve
baicalein induced OH• in this reaction system.
GalN potentiates AA-induced
carbon-centered free radicals in mouse primary hepatocytes
A previous study demonstrated that carbon-centered
lipid-derived radicals are an intermediate of enhanced oxidative
stress product of lipid peroxidation in the liver (21). In the present study, it was
demonstrated that a six-line ESR signal (aN=15.9 G,
aH=22.8 G) was produced when the DMPO/hepatocyte-derived
adduct was subjected to AA (Fig.
3A). In the control reactions, without AA, GalN produced no ESR
signal (Fig. 3Aa). This six-line
signal of carbon-centered radicals stimulated by AA was
concentration dependently potentiated by GalN (Fig. 3Ab-e). The identity of the radical
species was presumed to be a carbon-centered radical adduct that
could be proved based on the close resemblance of the hyperfine
coupling constants of the observed signal (22).
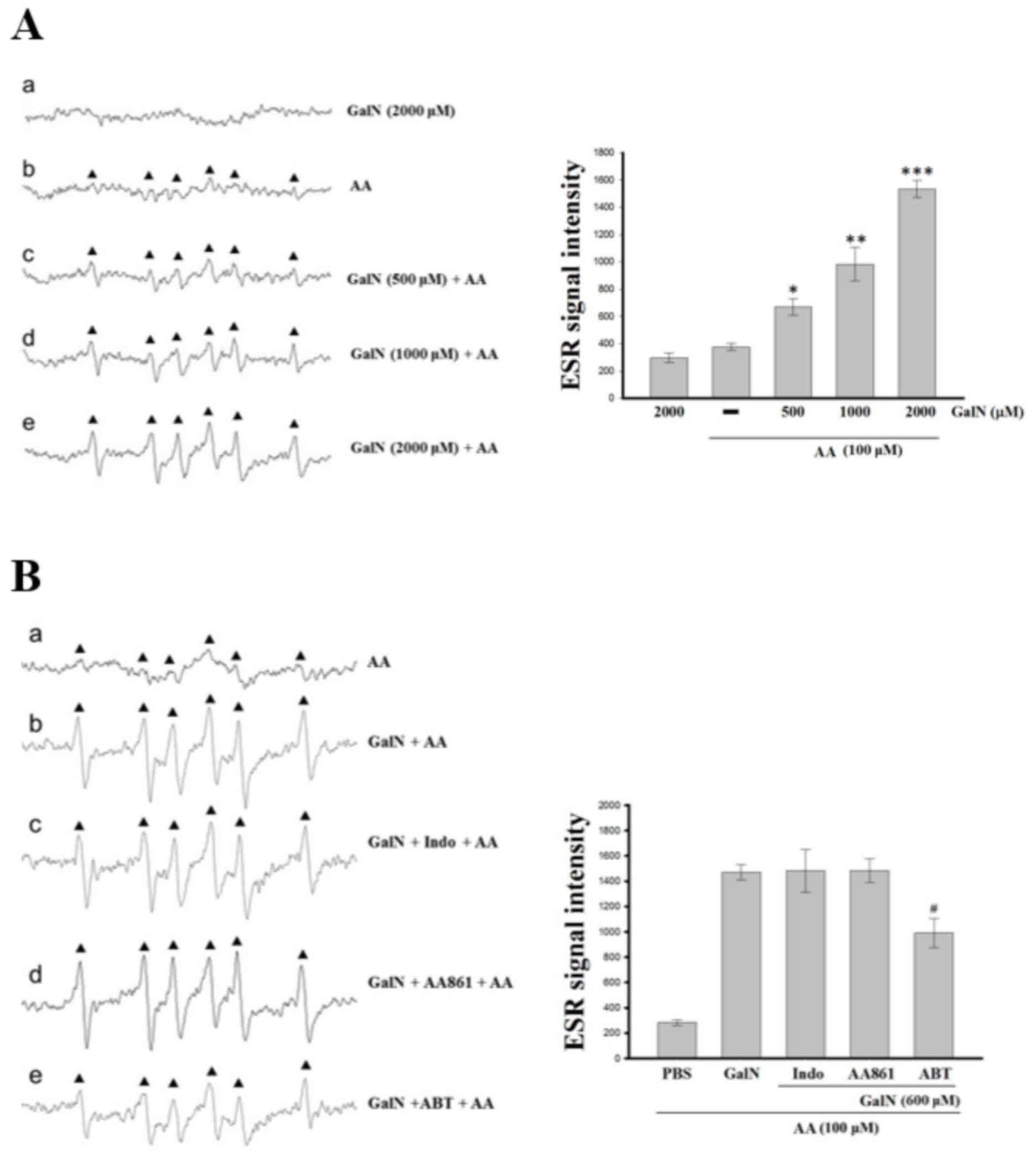 | Figure 3.ESR spectra analysis for GalN and
different inhibitor treated hepatocytes. (A) GalN potentiated the
AA induced carbon-centered radical formation in primary mouse
hepatocytes. Mouse hepatocytes (3×105 cells/ml, 150 µl)
were incubated with DMPO (100 mM) followed by the addition of GalN
at (a) 2,000 µM and (b) AA 100 µM. GalN (c) 500, (d) 1,000 and (e)
2,000 µM was added to the AA pretreated hepatocytes. (B) Effects of
(a) AA induced carbon-centered radical formation (b) with GalN
potentiation and (c) indo, (d) AA861 and (e) ABT. Instrument
parameters were as follows: Modulation amplitude, 1 G; time
constant, 164 msec; scanning for 42 sec with six scans accumulated.
The ESR spectra are labeled to show their components:
DMPO-carbon-centered radical adduct (▲). Data are presented as the
mean ± standard error of the mean (n=4). *P<0.05; **P<0.01
and ***P<0.001, vs. the reaction of AA treated hepatocytes;
#P<0.05, vs. the reaction of AA pretreated GalN +
inhibitor treated platelets. ESR, electron spin resonance; GalN,
D-galactosamine; AA, arachiodonic acid; DMPO,
5,5-dimethyl-1-pyrroline N-oxide; indo, indomethacin; ABT,
1-aminobenzotriazole. |
Role of cytochrome P450 on AA-induced
carbon-centered radicals in hepatocytes
GalN induced cytochrome P450 generate ROS that are
known to have greater chemical toxicity (23). A significant increase in hepatic
cytochrome P450 mRNA and protein expression was previously
established in the GalN-intoxicated rats (23). Supplementation with carvacrol, a
predominant monoterpenic phenol, suppressed cytochrome P450 mRNA
and protein expression in GalN induced rats (23). Therefore, evaluation of whether
cytochrome P450 serves role in AA-induced carbon-centered radicals
in hepatocytes was performed. As expected, the results demonstrated
that GalN potentiated the AA-induced six-line signals of
carbon-centered radical, which was inhibited by a non-isoform
specific cytochrome P450 inhibitor, though not by COX or LOX
inhibitors (Fig. 3Ba-e),
indicating that cytochrome P450 may serve a role in GalN
potentiation of AA-induced carbon-centered radicals in
hepatocytes.
Involvement of COX and cytochrome P450
in GalN-stimulated radicals was confirmed using a cell free Fenton
reaction system
A cell free Fenton reaction containing iron and
H2O2 was studied in the absence or presence
of GalN and thioacetamide to elucidate the role of endogenous
enzymes COX and cytochrome P450 in GalN-induced free radicals in
cell systems (platelets and hepatocytes). As presented in Fig. 4, GalN and thioacetamide did not
produce OH• in the cell free Fenton reaction system, as
they demonstrated very similar signal spectra to that of the PBS
only treated reaction. These results suggested that the endogenous
enzymes of COX, LOX and cytochrome p450 may require GalN inducing
free radicals in the cell systems.
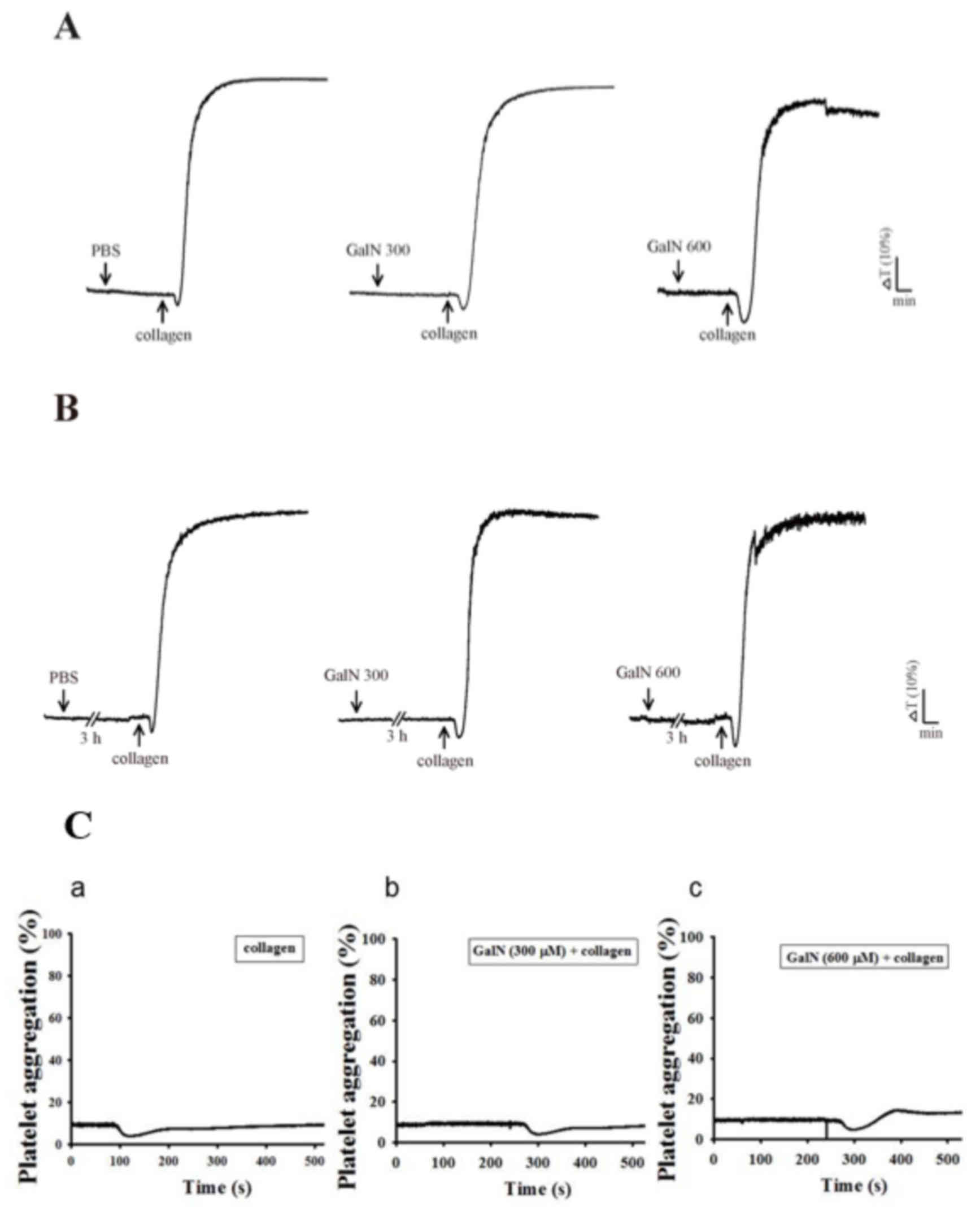
GalN on platelet aggregation
Khandoga et al (24) hypothesized that activated platelets
may be able to generate ROS and possess the potential to induce
liver injury. Despite the apparent involvement of platelets in
liver injury, Woolbright and Jaeschke (25) questioned whether the available
reports clearly corroborate a mediatory role of platelets in
post-ischemic liver injury or platelet activation. Therefore, the
present study evaluated whether GalN induces platelet aggregation
and whether GalN induced OH•production is dependent on
its agrregatory effects. GalN (300 and 600 µM) did not induce
platelet aggregation when stimulated with short (3 min) or long
(180 min) periods of incubation (Fig.
5A and B) in the human platelet suspensions. In addition, GalN
for 120 min incubation with the same concentrations (300 and 600
µM) did not affect collagen-induced aggregation (Fig. 5C). This result indicated that GalN
induced free radical formation in platelets is not a result of the
induction of platelet aggregation.
Discussion
The present study utilized ESR spin trapping
techniques to detect the free radical formation stimulated induced
by GalN in platelets and primary mouse hepatocytes, and also
established the potential role of endogenous enzymes in this
phenomenon. The results demonstrated that GalN stimulates
OH• and baicalein induces semiquinone radicals in
platelets, these radicals were suppressed by a COX inhibitor. GalN
also potentiated AA-induced carbon-centered radicals in mouse
hepatocytes and was inhibited by cytochrome P450 inhibitor.
However, a cell free Fenton reaction system demonstrated that GalN
did not induce radical formation and also did not respond to
platelet aggregation when it was incubated alone or in collagen
pretreated platelets.
Cardiac cirrhosis or congestive hepatopathy includes
a spectrum of hepatic imbalances arising from right-sided heart
failure (26). GalN is a
hepatotoxic agent metabolized exclusively in hepatocytes. The
administration of GalN causes the development of lethal liver
injury, which resembles the biochemical and metabolic alterations
observed in severe hepatic failure (27). Oxygen free radicals have been
implicated in the pathogenesis of toxic liver injury (28), and oxidative stress during liver
cirrhosis is responsible for cardiac dysfunction (29). As free radicals in biological
systems are characterized by their high reactivity, short lifetimes
and low concentrations, the type of spin trap used is a key factor
in determining how informative and sensitive the spin trapping
technique may be for a given radical species. In the present study,
the frequently used spin trap DMPO was used. DMPO possesses
significant advantages over other nitrone spin traps, as it is the
most redox inactive and has a comparatively higher dependence on
the structure of the trapped radical in ESR spectra than the common
nitrone spin traps, including α-phenyl-N-tert-butylnitrone
and α-(4-pyridyl-1-oxide)-N-tert-butylnitrone.
Platelets are highly active, anucleate cells, that
are rich in two representative peroxidase systems, prostaglandin H
synthase (PHS) and 12-LOX, a subtype of LOX (30). These enzymes serve an active role
in platelet function through the production of bioactive mediators
from AA including thromboxane A2, 12-hydroxyperoxyeicosatetraenoic
acid and 12-hydroxyeicosatetraenoic acid (31). Furthermore, platelets have been
frequently reported as a vulnerable target of xenobiotic-induced
cytotoxicity and peroxidase action on xenobiotic compounds may
contribute to platelet cytotoxicity. The authors' previous study
(32) demonstrated that 12-LOX and
PHS were involved in OH• formation induced by natural
flavonoids in platelets and suggested a role for these peroxidase
systems in the xenobiotic-compound activation. COX-2 has been
recognized as a potent pro-inflammatory cytokine and has been
implicated in platelet aggregation (33). COX-2 inhibitory activity has
previously been reported to be associated with
1,1-diphenyl-2-picrylhydrazyl radical scavenging activity (34). In the present study, OH•
production by GalN in platelets was significantly suppressed by the
COX inhibitor, however, not by the LOX or cytochrome P450
inhibitor, suggesting that COX may serve a vital role in GalN
stimulated OH• formation in platelets.
It has been suggested that the autoxidation of
flavonoids generates semiquinone radicals and superoxide radicals
(35). Semiquinone free radicals
were also generated when polyphenols were incubated with
H2O2 (36).
The authors' earlier study demonstrated that semiquinone free
radicals were generated when baicalein was incubated with
H2O2 or platelets/AA, and also demonstrated
that baicalein-induced OH• formation in human platelets
is independent of autoxidation reactions (32). A previous study has also
demonstrated that baicalein inhibited 12-LOX activity without
affecting COX in human platelets (37). In the present study, in platelets
the baicalein-stimulated semiquinone free radicals were inhibited
by the COX inhibitor without affecting the formation of
OH•. Therefore, it was hypothesized that baicalein may
produce OH• via LOX in human platelets.
Greenley and Davies (38) used ESR spin trapping to demonstrate
the production of free radicals in rat liver microsomal
preparations. In the present study, the free radicals reacted with
a nitrone or nitroso compound to form a relatively stable ESR
detectable radical adduct. In addition, the peroxyl and
carbon-centered radical adducts were detected in rat liver
microsomes (38). Cytochrome P450
belongs to a superfamily of haem proteins responsible for the
metabolic activation or inactivation of the majority of clinically
used drugs and a number of toxins. A previous study has
demonstrated that cytochrome P450 isoforms and their activities are
suppressed in animal models of endotoxemia as well as in cultured
hepatocytes stimulated by endotoxin (39). The inactivation of the P450 enzyme
can be attributed to the formation of a carbon-centered free
radical intermediate, which may attack the haem prosthetic group of
the enzyme (40). To the best of
our knowledge, the present study demonstrated, for the first time,
in vitro generation of carbon-centered free radicals in
mouse primary hepatocytes following exposure to AA, and revealed
that this production was further potentiated by GalN. The result
demonstrating that GalN-potentiated AA-induced carbon-centered
radicals were conceivably recovered by the cytochrome P450
inhibitor ABT, indicated that cytochrome P450 may act as an
intermediate of GalN enhanced carbon-centered radicals in
hepatocytes.
A previous study presented the role of platelets in
oxidative stress in the context of hepatic injury (41). In addition, it has also been
postulated that platelets are essential for the liver to regenerate
properly following a partial liver resection (42). Platelet aggregation has been
associated with microvascular perfusion defects, apoptotic cell
death, vascular oxidative stress and an inflamed endothelium
(43), all suggesting a
pathophysiological connection between platelets and hepatocytes. In
the present study, GalN induced OH• radicals in
platelets; however, it did not cause platelet aggregation when
analyzed alone or in collagen pretreated conditions. This result
demonstrated that platelet activation does not involve the GalN
induced production of OH• radicals.
In conclusion, the present study demonstrated that
GalN induces hydroxyl radical formation in platelets without GalN
activation or aggregation and potentiated AA-induced
carbon-centered free radical in the primary mouse hepatocytes. COX
and cytochrome P450 may serve a role in GalN induced free radical
production in platelets and hepatocytes, respectively. Thus, the
present study provides evidence for the pathophysiological
association between platelets and hepatocytes in which free
radicals are believed to be involved. This way, GalN holds an
important role in inducing free radicals in cardiohepatic diseases,
however, a further study is required to confirm this
hypothesis.
Acknowledgements
The present study was supported by grants from the
Ministry of Science and Technology of Taiwan (grant nos.
MOST103-2320-B-038-017, MOST104-2622-B-038-003 and
MOST104-2320-B-038-045-MY2), Cathay General Hospital-Taipei Medical
University (grant no. 104CGH-TMU-01-3) and Taipei Medical
University-National Taiwan University of Science and Technology
(grant no. TMU-NTUST-103-02).
References
|
1
|
Boelsterli UA and Lim PL: Mitochondrial
abnormalities-a link to idiosyncratic drug hepatotoxicity? Toxicol
Appl Pharmacol. 220:92–107. 2007. View Article : Google Scholar
|
|
2
|
Gómez-Lechón MJ, Tolosa L, Castell JV and
Donato MT: Mechanism-based selection of compounds for the
development of innovative in vitro approaches tohepatotoxicity
studies in the LIINTOP project. Toxicol In Vitro. 24:1879–1889.
2010. View Article : Google Scholar
|
|
3
|
Keppler D, Lesch R, Reutter W and Decker
K: Experimental hepatitis induced by D-galactosamine. Exp Mol
Pathol. 9:279–290. 1968. View Article : Google Scholar
|
|
4
|
Quintero A, Pedraza CA, Siendones E,
ElSaid Kamal AM, Colell A, García-Ruiz C, Montero JL, De la Mata M,
Fernández-Checa JC, Miño G and Muntané J: PGE1 protection against
apoptosis induced by D-galactosamine is not related to the
modulation of intracellular free radical production in primary
culture of rat hepatocytes. Free Radic Res. 36:345–355. 2002.
View Article : Google Scholar
|
|
5
|
Lekić N, Cerný D, Hořínek A, Provazník Z,
Martínek J and Farghali H: Differential oxidative stress responses
to D-galactosamine-lipopolysaccharide hepatotoxicity based on real
time PCR analysis of selected oxidant/antioxidant andapoptotic gene
expressions in rat. Physiol Res. 60:549–558. 2011.
|
|
6
|
Choi JH, Kang JW, Kim DW, Sung YK and Lee
SM: Protective effects of Mg-CUD against D-galactosamine-induced
hepatotoxicity in rats. Eur J Pharmacol. 657:138–143. 2011.
View Article : Google Scholar
|
|
7
|
Bruck R, Aeed H, Avni Y, Shirin H, Matas
Z, Shahmurov M, Avinoach I, Zozulya G, Weizman N and Hochman A:
Melatonin inhibits nuclear factor kappa B activation and oxidative
stress and protects against thioacetamide induced liver damage in
rats. J Hepatol. 40:86–93. 2004. View Article : Google Scholar
|
|
8
|
von Sonntag C: Free-radical-induced DNA
damage and its repair. Springer-Verlag; Berlin: 2006, View Article : Google Scholar
|
|
9
|
Black M: Acetaminophen hepatotoxicity.
Annu Rev Med. 35:577–593. 1984. View Article : Google Scholar
|
|
10
|
Lam FW, Vijayan KV and Rumbaut RE:
Platelets and their interactions with other immune cells. Compr
Physiol. 5:1265–1280. 2015. View Article : Google Scholar :
|
|
11
|
van Golen RF, van Gulik TM and Heger M:
Mechanistic overview of reactive species-induced degradation of the
endothelial glycocalyx during hepatic ischemia/reperfusion injury.
Free Radic Biol Med. 52:1382–1402. 2012. View Article : Google Scholar
|
|
12
|
Khandoga A, Biberthaler P, Enders G,
Axmann S, Hutter J, Messmer K and Krombach F: Platelet adhesion
mediated by fibrinogen-intercelllular adhesion molecule-1 binding
induces tissue injury in the postischemic liver in vivo.
Transplantation. 74:681–688. 2002. View Article : Google Scholar
|
|
13
|
Cook JA: Eicosanoids. Crit Care Med. 33 12
Suppl:S488–S491. 2005. View Article : Google Scholar
|
|
14
|
Ito S, Ito Y, Katagiri H, Suzuki T, Hoka
S, Yokomizo T, Shimizu T and Majima M: Leukotriene B4/leukotriene
B4 receptor pathway is involved in hepatic microcirculatory
dysfunction elicited by endotoxin. Shock. 30:87–91. 2008.
|
|
15
|
Reddy RC, Chen GH, Tateda K, Tsai WC,
Phare SM, Mancuso P, Peters-Golden M and Standiford TJ: Selective
inhibition of COX-2 improves early survival in murine endotoxemia
but not in bacterial peritonitis. Am J Physiol Lung Cell Mol
Physiol. 281:L537–L543. 2001.
|
|
16
|
Hsiao G, Lin KH, Chang Y, Chen TL, Tzu NH,
Chou DS and Sheu JR: Protective mechanisms of inosine in platelet
activation and cerebral ischemic damage. Arterioscler Thromb Vasc
Biol. 25:1998–2004. 2005. View Article : Google Scholar
|
|
17
|
Sun P, Zhang P, Wang PX, Zhu LH, Du Y,
Tian S, Zhu X and Li H: Mindin deficiency protects the liver
against ischemia/reperfusion injury. J Hepatol. 63:1198–1211. 2015.
View Article : Google Scholar
|
|
18
|
Hsu YP, Chen RJ, Fang JF, Lin BC, Huang
TL, Cheng ML, Chiu DT and Tsay PK: Increased susceptibility to
oxidant injury in hepatocytes from rats with intra-abdominal
hypertension. J Trauma. 57:569–575. 2004. View Article : Google Scholar
|
|
19
|
Chou DS, Hsiao G, Lai YA, Tsai YJ and Sheu
JR: Baicalein induces proliferation inhibition in B16F10 melanoma
cells by generating reactive oxygen species via 12-lipoxygenase.
Free Radic Biol Med. 46:1197–1203. 2009. View Article : Google Scholar
|
|
20
|
Chou DS, Hsiao G, Shen MY, Tsai YJ, Chen
TF and Sheu JR: ESR spin trapping of a carbon-centered free radical
from agonist-stimulated human platelets. Free Radic Biol Med.
39:237–248. 2005. View Article : Google Scholar
|
|
21
|
Shvedova AA, Kisin ER, Murray AR,
Mouithys-Mickalad A, Stadlerd K, Masond RP and Kadiisk M: ESR
evidence for in vivo formation of free radicals in tissue of mice
exposed to single-walled carbon nanotubes. Free Radic Biol Med.
73:154–165. 2014. View Article : Google Scholar
|
|
22
|
Qian SY, Wang HP, Schafer FQ and Buettner
GR: EPR detection of lipid-derived free radicals from PUFA, LDL,
and cell oxidations. Free Radic Biol Med. 29:568–579. 2000.
View Article : Google Scholar
|
|
23
|
Aristatile B, Al-Assaf AH and Pugalendi
KV: Carvacrol ameliorates the PPAR-A and cytochrome p450 expression
on D-galactosamine induced hepatotoxicity rats. Afr J Tradit
Complement Altern Med. 11:118–123. 2014. View Article : Google Scholar :
|
|
24
|
Khandoga A, Biberthaler P, Messmer K and
Krombach F: Platelet-endothelial cell interactions during hepatic
ischemia-reperfusion in vivo: A systematic analysis. Microvasc Res.
65:71–77. 2003. View Article : Google Scholar
|
|
25
|
Woolbright BL and Jaeschke H: Heme
oxygenase-1 and platelets in hepatic ischemia reperfusion injury. J
Gastroenterol Hepatol. 28:756–757. 2013. View Article : Google Scholar
|
|
26
|
Kubo SH, Walter BA, John DH, Clark M and
Cody RJ: Liver function abnormalities in chronic heart failure.
Influence of systemic hemodynamics. Arch Intern Med. 147:1227–1230.
1987. View Article : Google Scholar
|
|
27
|
Wu YH, Hu SQ, Liu J, Cao HC, Xu W, Li YJ
and Li LJ: Nature and mechanisms of hepatocyte apoptosis induced by
D-galactosamine/lipopolysaccharide challenge in mice. Int J Mol
Med. 33:1498–1506. 2014. View Article : Google Scholar :
|
|
28
|
Sumida Y, Niki E, Naito Y and Yoshikawa T:
Involvement of free radicals and oxidative stress in NAFLD/NASH.
Free Radic Res. 47:869–880. 2013. View Article : Google Scholar
|
|
29
|
Yang YY, Liu H, Nam SW, Kunos G and Lee
SS: Mechanisms of TNFalpha-induced cardiac dysfunction in
cholestatic bile duct-ligated mice: Interaction between TNFalpha
and endocannabinoids. J Hepatol. 53:298–306. 2010. View Article : Google Scholar :
|
|
30
|
Maskrey BH, Bermúdez-Fajardo A, Morgan AH,
Stewart-Jones E, Dioszeghy V, Taylor GW, Baker PR, Coles B, Coffey
MJ, Kühn H and O'Donnell VB: Activated platelets and monocytes
generate four hydroxyphosphatidylethanolamines via lipoxygenase. J
Biol Chem. 282:20151–20163. 2007. View Article : Google Scholar
|
|
31
|
Johnson EN, Brass LF and Funk CD:
Increased platelet sensitivity to ADP in mice lacking platelet-type
12-lipoxygenase. Proc Natl Acad Sci USA. 95:3100–3105. 1998.
View Article : Google Scholar :
|
|
32
|
Chou DS, Lee JJ, Hsiao G, Hsieh CY, Tsai
YJ, Chen TF and Sheu JR: Baicalein induction of hydroxyl radical
formation via 12-lipoxygenase in human platelets: An ESR study. J
Agric Food Chem. 55:649–655. 2007. View Article : Google Scholar
|
|
33
|
Morteau O: Prostaglandins and
inflammation: The cyclooxygenase controversy. Arch Immunol Ther Exp
(Warsz). 48:473–480. 2000.
|
|
34
|
Ko CH, Shen SC, Lin HY, Hou WC, Lee WR,
Yang LL and Chen YC: Flavanones structure-related inhibition on TPA
induced tumor promotion through suppression of extracellular
signal-regulated protein kinases: Involvement of prostaglandin E2
in anti-promotive process. J Cell Physiol. 193:93–102. 2002.
View Article : Google Scholar
|
|
35
|
Hodnick WF, Milosavljević EB, Nelson JH
and Pardini RS: Electrochemistry of flavonoids. Relationships
between redox potentials, inhibition of mitochondrial respiration,
and production of oxygen radicals by flavonoids. Biochem Pharmacol.
37:2607–2611. 1988. View Article : Google Scholar
|
|
36
|
Bors W, Michel C and Stettmaier K:
Electron paramagnetic resonance studies of radical species of
proanthocyanidins and gallate esters. Arch Biochem Biophys.
374:347–355. 2000. View Article : Google Scholar
|
|
37
|
You KM, Jong HG and Kim HP: Inhibition of
cyclooxygenase/lipoxygenase from human platelets by
polyhydroxylated/methoxylated flavonoids isolated from medicinal
plants. Arch Pharm Res. 22:18–24. 1999. View Article : Google Scholar
|
|
38
|
Greenley TL and Davies MJ: Radical
production from peroxide and peracid tumour promoters: EPR spin
trapping studies. Biochim Biophys Acta. 1157:23–31. 1993.
View Article : Google Scholar
|
|
39
|
Morgan ET: Regulation of cytochrome p450
by inflammatory mediators: Why and how? Drug Metab Dispos.
29:207–212. 2001.
|
|
40
|
Tolando R, Zanovello A, Ferrara R, Iley JN
and Manno M: Inactivation of rat liver cytochrome P450 (P450) by
N,N-dimethylformamide and N,N-dimethylacetamide. Toxicol Lett.
124:101–111. 2001. View Article : Google Scholar
|
|
41
|
van Golen RF, Stevens KM, Colarusso P,
Jaeschke H and Heger M: Platelet aggregation but not activation and
degranulation during the acute post-ischemic reperfusion phase in
livers with no underlying disease. J Clin Transl Res. 1:107–115.
2015.
|
|
42
|
Starlinger P, Assinger A, Haegele S, Wanek
D, Zikeli S, Schauer D, Birner P, Fleischmann E, Gruenberger B,
Brostjan C and Gruenberger T: Evidence for serotonin as a relevant
inducer of liver regeneration after liver resection in humans.
Hepatology. 60:257–266. 2014. View Article : Google Scholar
|
|
43
|
Mende K, Reifart J, Rosentreter D,
Manukyan D, Mayr D, Krombach F, Rentsch M and Khandoga A: Targeting
platelet migration in the postischemic liver by blocking
protease-activated receptor 4. Transplantation. 97:154–160. 2014.
View Article : Google Scholar
|