Introduction
Myocardial ischemia has become a common
life-threatening disease causing many adverse effects on the heart,
including a decrease in myocardial cell aerobic metabolism and a
reduction in the capacity to maintain the heart activity during
periods of low oxygen supply (1).
Myocardial ischemia can initially present no symptoms but
subsequently lead to serious clinical consequences. Although
reperfusion therapy can rescue myocardial ischemia and myocardial
stunning, damaged myocardia and newly forming myocardial injuries
caused by reoxygenation still induce heart failure and ventricular
remodeling even in patients who received reperfusion therapy
(2,3). Ventricular remodeling following
myocardial ischemia is an important factor contributing to chronic
heart failure. Myocardial fibrosis and connexin43 (Cx43) remodeling
are important forms of ventricular remodeling which affect
myocardial systolic and diastolic function and lead to cardiac
arrhythmia, increasing the incidence of sudden death (4,5).
Myocardial fibrosis is characterized by an excessive
deposition of extracellular matrix proteins by cardiac fibroblasts
(CFs) in the myocardial tissue and imbalanced abundance of
different types of collagen, which reduces tissue compliance and
limits the diastolic filling pressure (6,7).
Fibrosis of coronary arteries thickens the wall of the heart,
causes luminal narrowing and decreases myocardial elasticity,
reducing the blood supply for myocardial cells (8). The majority of causes of heart
disease involve pathological myocardial fibrosis. Myocardial
fibrosis is a complex pathological process in which transforming
growth factors-β1 (TGF-β1) is the most important growth factor.
TGF-β1 can promote differentiation of CFs, activation of the
renin-angiotensin-aldosterone system and cause increased abundance
of niacinamide adenine dinucleotide phosphate (NADPH). All of the
above are mechanisms of fibrosis (9,10).
Mothers against decapentaplegic homologue (Smad) proteins are the
main effector molecules in the TGF-β1 signaling pathway, which
regulates the expression of downstream genes and promotes fibrosis
(11). The Smad protein family can
be divided into three groups: i) Receptor regulated smads (R-Smads)
including Smad1, 2, 3, 5 and 8; ii) common partner smads (Co-Smads)
including Smad4; and iii) inhibitory smads (I-Smads) including
Smad6 and 7. I-Smads can compete with R-Smads for binding with
their receptor. Therefore, I-Smads can prevent phosphorylation of
R-Smads and inhibit TGF-β1 signaling pathway. Therefore, the
expression of TGF-β1, Smad2 and 7 serve a role in myocardial
fibrosis.
Connexins are transmembrane ion-channel proteins
that form gap junctions between adjacent cardiomyocytes and
transfer ions or molecules between cells, coupling them
electrically (12). Cx43 is the
predominant isoform in the heart, mainly present in ventricular
tissues. Changes in Cx43 expression and distribution are closely
associated with electrical remodeling of ventricular tissues
(13).
In the treatment of myocardial ischemia, reperfusion
is not the main goal and preventing harmful changes in the
myocardial structure is equally important. Calcium channel blockers
(CCBs) are common cardiovascular medicines that have been
effectively used in the treatment of arrhythmia, myocardial
ischemia and hypertension. Although CCBs have been reported to
alleviate myocardial remodeling, previous studies have only been
limited to studying myocardial cells, including a preliminary study
on the quality index of the heart (14–16).
The underlying molecular mechanisms of structural and electrical
remodeling, have yet to be elucidated. In the present study, the
effects of diltiazem on isoproterenol-induced myocardial fibrosis
and Cx43 expression have been investigated.
Materials and methods
Materials
Male 8-week-old Sprague-Dawley rats (body weight,
200–250 g; n=36) were obtained from Xi'an Jiaotong University
(Xi'an, China). Rats were reared at a constant-temperature of
22–25°C with a 12-h light/dark cycle, and given a standard diet and
unlimited access to drinking water. The present study was approved
by the Institutional Laboratory Animal Care and Use Committee of
the School of Xi'an Jiaotong University (approval no. 2016-160).
Isoproterenol was purchased from Sigma-Aldrich (Merck KGaA,
Damstadt, Germany). Diltiazem was purchased from Simcare Drug Store
(Nanjing, China). Other reagents were of commercial analytical
grade.
Experimental protocol
Following acclimatization for 1 week, experimental
rats (n=36) were randomly divided into three groups, 12 rats each:
i) Control (CTL); ii) isoproterenol (ISO); and iii) isoproterenol
with diltiazem (ISO_DIL). Myocardial ischemia in rats (ISO and
ISO_DIL) was induced by subcutaneous isoproterenol injection
(isoproterenol was dissolved in physiological saline and
administered at 5 mg/kg body weight/day) at 24-h intervals for 7
days, as previously described (17). The CTL group was injected with
equal volumes of physiological saline at the same time intervals.
Following the second isoproterenol injection, the ISO_DIL group was
given 25 mg/kg/day diltiazem for 4 weeks. The CTL and ISO group
were given 1 ml/day physiological saline for 4 weeks. The
treatments involved oral administration. At 12 h following
administration of the final dose of diltiazem, cervical dislocation
was performed and a piece of tissue from the apex of the left
ventricle was excised immediately form each rat, washed with
chilled isotonic saline and used for analysis.
Determination of heart weight index
(HWI)
Whole hearts were excised (excluding large blood
vessels and connective tissue) wiped with filter paper and weighed.
HWI was calculated as heart weight/body weight.
Histopathological analysis
Hearts were fixed in 10% formalin solution and
embedded in paraffin wax at 4°C for 24 h. Transverse sections, 4-mm
thick, were cut and stained as previously described (18). Hematoxylin and eosin (H&E) and
Masson stains were used to observe microstructural changes and
detect the quantities of collagen (magnification, ×400). Briefly,
hematoxylin dye was applied for 5 min, the acid dip for 5 sec and
ammonia water for another 5 sec. Subsequently, sections were washed
with water for 1 h followed by washes with distilled water for
another 5 min. 70 and 90% alcohol dehydration step followed for 10
min. Finally, sections were put into alcohol eosin staining for 2–3
min. For the Masson stain, sections were washes with tap water and
distilled water for 2 min, followed by Harris's wood dye for 1–2
min. rinses with water followed for 30 min. All incubations
occurred at room temperature. Additionally, Masson acid dye was
applied for 5–10 min, 1% phosphomolybdic acid aqueous solution for
another 3–5 min and 1% aniline blue dye for 5 min. Finally, 1%
glacial acetic acid aqueous solution was added for 5 sec. All
incubations occurred at room temperature and the visualization
occurred using a bright field Olympus+CX23 microscope (Olympus
Corporation, Tokyo, Japan).
The degree of cardiac fibrosis was determined based
on the area of fibrosis/total tissue area (%), as previously
described (18).
Concentration of Ca2+
Myocardial tissue was homogenized by heat-treatment
for 6 h at 85°C. Centrifugation (7,500 × g at 4°C for 15 min)
separated the supernatant containing Ca2+ from the
pellet. Methyl phenol blue thyme in alkaline solution was added to
the supernatant and formed blue-colored complexes with
Ca2+. The concentration of Ca2+ in the
supernatant was evaluated by color comparison with a standard
solution, as previously described (19).
Western blotting
Western blotting analyses were performed according
to a previously described method (20). Frozen heart tissue was homogenized
in lysis buffer. Following two centrifugation extractions at 4°C,
12,000 × g for 15 min and at 4°C, 12,000 × g for 10 min, the
resulting supernatant was used for analysis. Protein content was
determined using the Bradford protein assay. Diluted tissues (20 µg
protein/lane) were separated by electrophoresis in 10%
polyacrylamide gels and the separated proteins were transferred
onto polyvinylidene fluoride membranes for visualization.
Nonspecific binding sites were blocked with 5% skimmed milk at room
temperature for 30 min. Following washing with 0.05% Tris-buffered
saline (TBST) with 0.05% Tween), membranes were incubated overnight
at 4°C with primary monoclonal antibodies specific for TGF-β1
(1:800, cat. no. sc-146; Santa Cruz Biotechnology, Inc., Dallas,
TX, USA), Smads (1:800, Smad2; cat. no. SAB4300251 and Smad7, cat.
no. SAB4200345; both from Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany, Cx43 (1:800, cat. no. 3512; Cell Signaling Technology,
Inc., Dancers, MA, USA) and GAPDH (1:1,000, cat. no. SAB2100894;
Sigma-Aldrich; Merck KGaA) and then with Goat anti rabbit IgG
(1;5,000, cat. no. 1706515; Bio-Rad Laboratories, Inc., Hercules,
CA, USA) at room temperature for 2 h. Following washing with TBS
with Tween-20, proteins were visualized using enhanced
chemiluminescence substrate and a ChemiDoc XRS Imaging system
(Bio-Rad Laboratories, Inc.). GAPDH was used as a loading
control.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Following removal and washing with cold saline,
hearts were frozen in liquid nitrogen and preserved at −80°C.
RT-qPCR was performed as previously described (21). Total RNA was extracted from
myocardial tissue using TRIzol reagent (Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA). RNA was then reverse
transcribed by RevertAid First-Strand cDNA Synthesis kit (Thermo
Fisher Scientific, Inc.). The mRNA expression level of Cx43 was
measured by RT-qPCR. The expression of the transcript was
normalized to the mean GAPDH mRNA expression (22) and determined using a 7300 Real-Time
PCR System SDS software version 1.4 (Applied Biosystems; Thermo
Fisher Scientific, Inc.). The primer sequences are summarized in
Table I.
 | Table I.Primer sequences used for reverse
transcription-quantitative polymerase chain reaction. |
Table I.
Primer sequences used for reverse
transcription-quantitative polymerase chain reaction.
| Gene/site | Forward primer
(5′-3′) | Reverse primer
(5′-3′) | Product length
(bp) |
|---|
| Cx43 |
GCCGCAATTACAACAAGCAA |
TTGGCATTCTGGTTGTCGTC | 142 |
| TGF-β1 |
GAGAAGCGGTACCTGAACCC |
GGCGAAAGCCCTCAATTTCC | 234 |
| Smad2 |
CCATCTTGCCATYCACTCCG |
AAGCTCATCTAATCGTCCTG | 176 |
| Smad7 |
CCATCACCTTAGCCGACTCTG |
AAATCCATCGGGTATCTGGAGTA | 69 |
| GAPDH |
GGAAAGCTGTGGCGTGAT |
AAGGTGGAAGAATGGGAGTT | 308 |
Immunofluorescence assay
As previously described (23), fixed hearts were coated with
optimal cutting temperature compound (Sakura Finetek USA, Inc.,
Torrance, CA, USA) and cut into 6 µm-thick sections. Sections of
left ventricular tissue were incubated with the primary antibodies
for Cx43 (1:100; cat. no. 3512; Cell Signaling Technology, Inc.)
overnight at 4°C. Following three washes with 0.1 mol/l
phosphate-buffered saline (PBS), 5 min each, the sections were
incubated with Fluorescein-Conjugated AffiniPure Goat Anti-Rabbit
IgG(H+L) (1:50 dilution, cat. no. ZF-0311; ZSGB-BIO; OriGene
Technologies, Inc., Rockville, MD, USA) at 37°C for 1 h in the
dark. Then, following rinsing with PBS three times, the images of
the sections were captured using a laser confocal microscope
(Olympus Soft Imaging Solutions, Münster, Germany). The absorbance
was measured at a wavelength of 488 nm and analyzed using Image
Pro-Plus software (version 7.0; Media Cybernetics, Inc., Rockville,
MD, USA).
Statistical analysis
The experimental data were analyzed by one-way
analysis of variance, followed by the Fisher's least significant
difference test. using SPSS software (version 10.0; SPSS, Inc.,
Chicago, IL, USA) and are expressed as the mean ± standard
deviation. P<0.05 was considered to indicate a statistically
significant difference.
Results
HWI
HWIs were significantly greater in ISO rats compared
with the CTL group and ISO_DIL groups (P<0.05; Table II).
| Table II.Effect of diltiazem on HWI. |
Table II.
Effect of diltiazem on HWI.
| Group | HWI (mg/g) |
|---|
| Control |
3.106±0.809 |
| Model |
3.916±0.235a |
| Diltiazem |
3.341±0.575b |
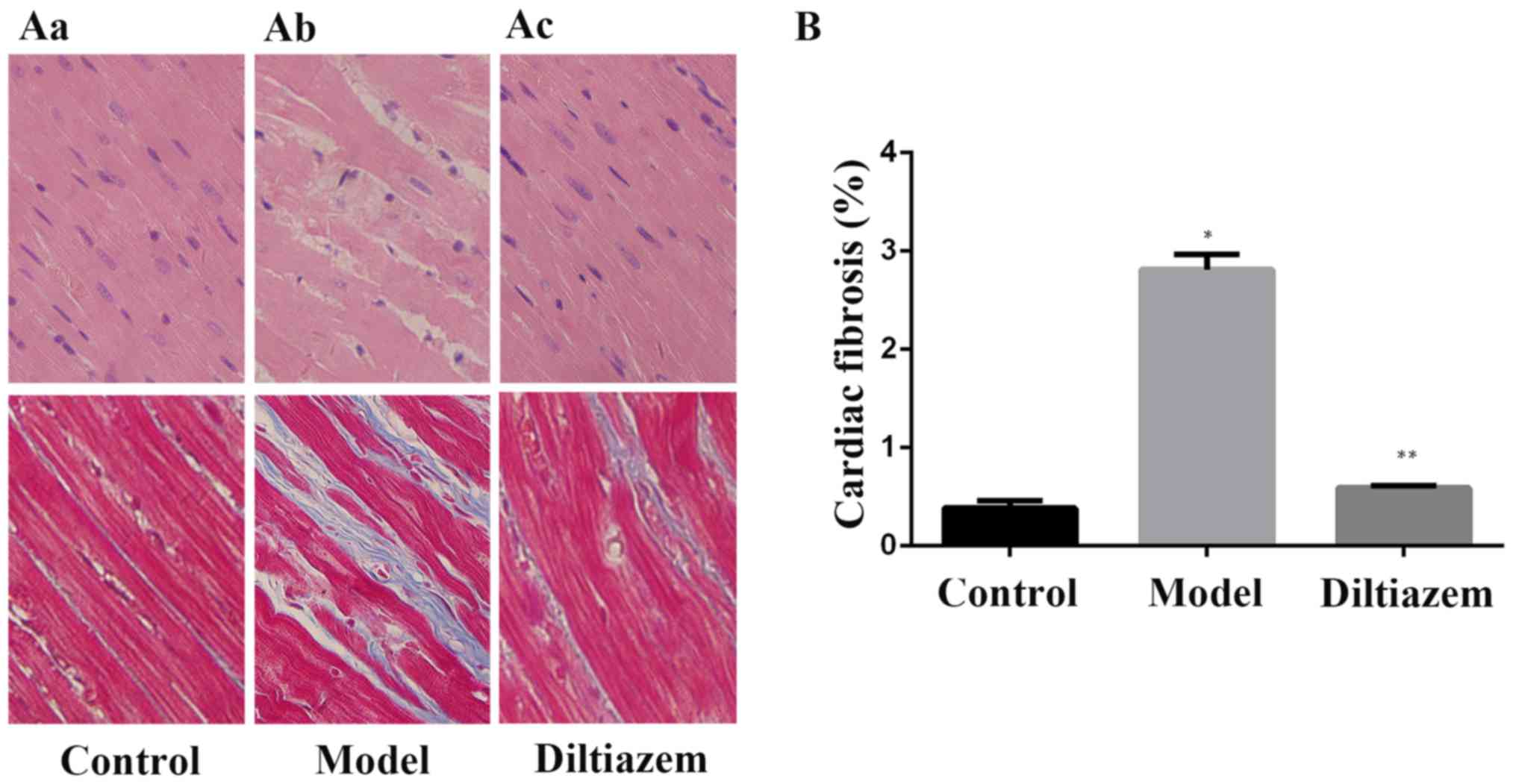
Ventricular histological
observation
To assess the differences in structural remodeling
between subgroups, ventricular histology was analyzed. Light
microscopy of H&E- and Masson-stained tissue sections from CTL
and ISO_DIL rats demonstrated myocardium with normal cellular
structure and continuous myofibril structure with adjacent
myofibrils. However, tissue sections in ISO rats exhibited obvious
myocardial cell swelling, degeneration and hyperplasia of large
connective tissues. The percentage of fibrotic area increased
significantly in the ISO group compared with the CTL group, and
decreased significantly in the ISO_DIL group compared with the ISO
model group (both P<0.001; Fig.
1).
Concentration of Ca2+
Compared with CTL rats, concentration of
Ca2+ in myocardium increased significantly in ISO rats.
It has been demonstrated that high calcium concentrations
contribute to the pathogenesis of myocardial ischemia injury
(24). Concentration of
Ca2+ significantly decreased the ISO_DIL group
(P<0.05; Table III).
| Table III.Concentration of Ca2+. |
Table III.
Concentration of Ca2+.
| Group | Ca2+
(nmol/l) |
|---|
| Control |
161.976±7.476 |
| Model |
429.973±23.592a |
| Diltiazem |
253.636±25.250b |
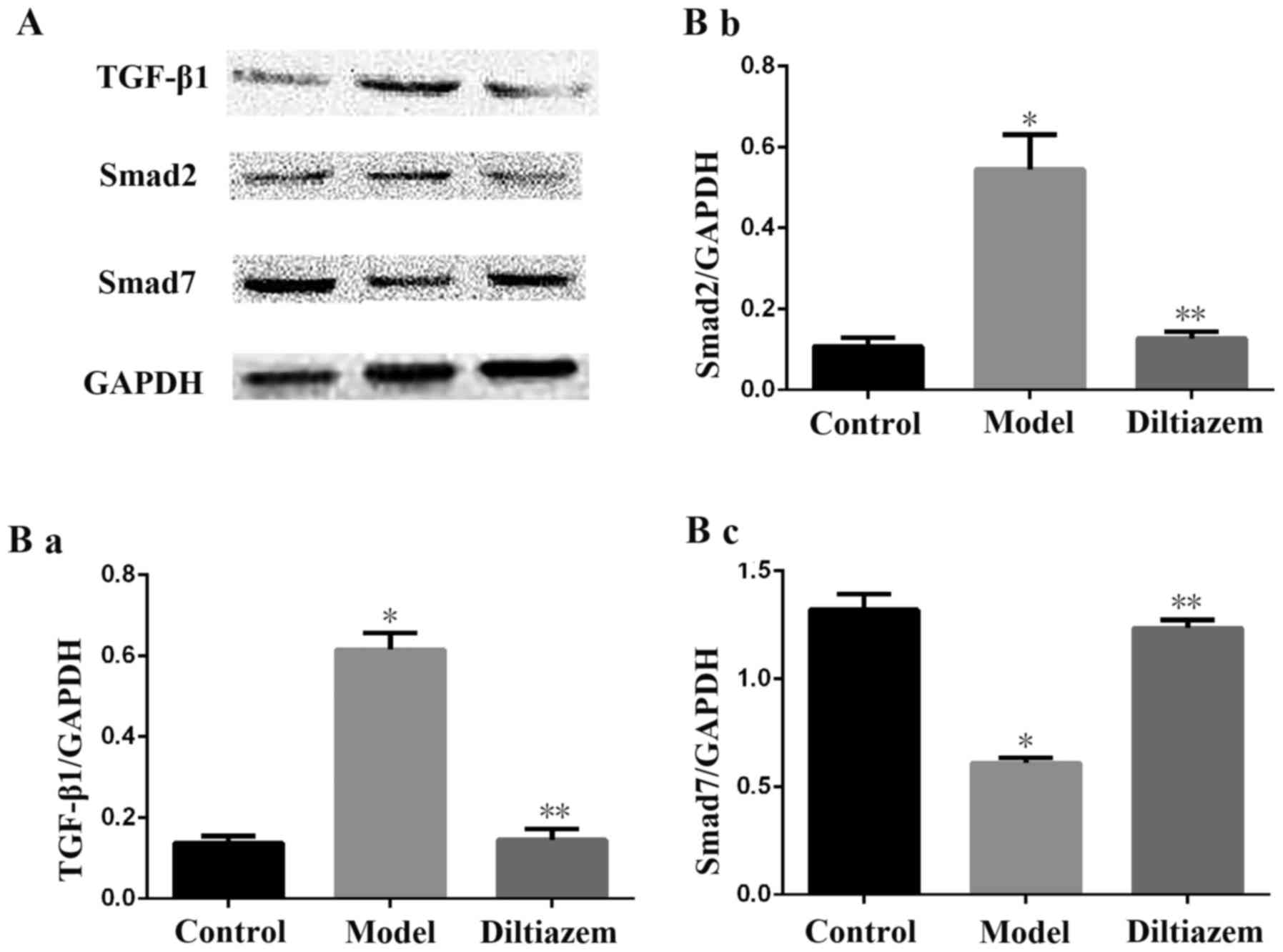
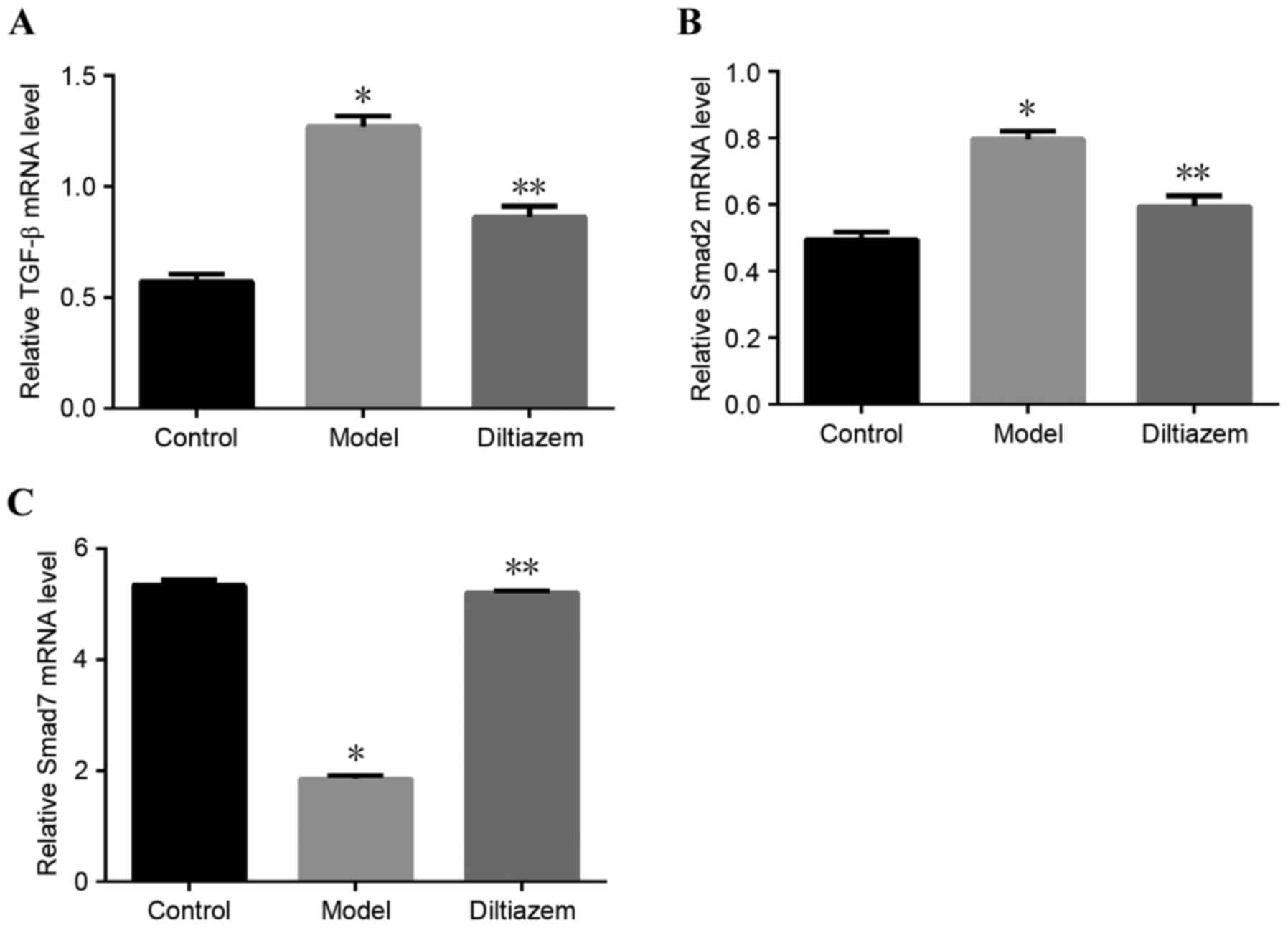
Changes in the TGF-β1/Smads signaling
pathway
Both protein (Fig.
2) and mRNA (Fig. 3)
expression of TGF-β1 and Smad2 decreased in ISO_DIL rats compared
with the ISO group, while the expression of Smad7 increased in
ISO_DIL group compared with the ISO group (all P<0.001).
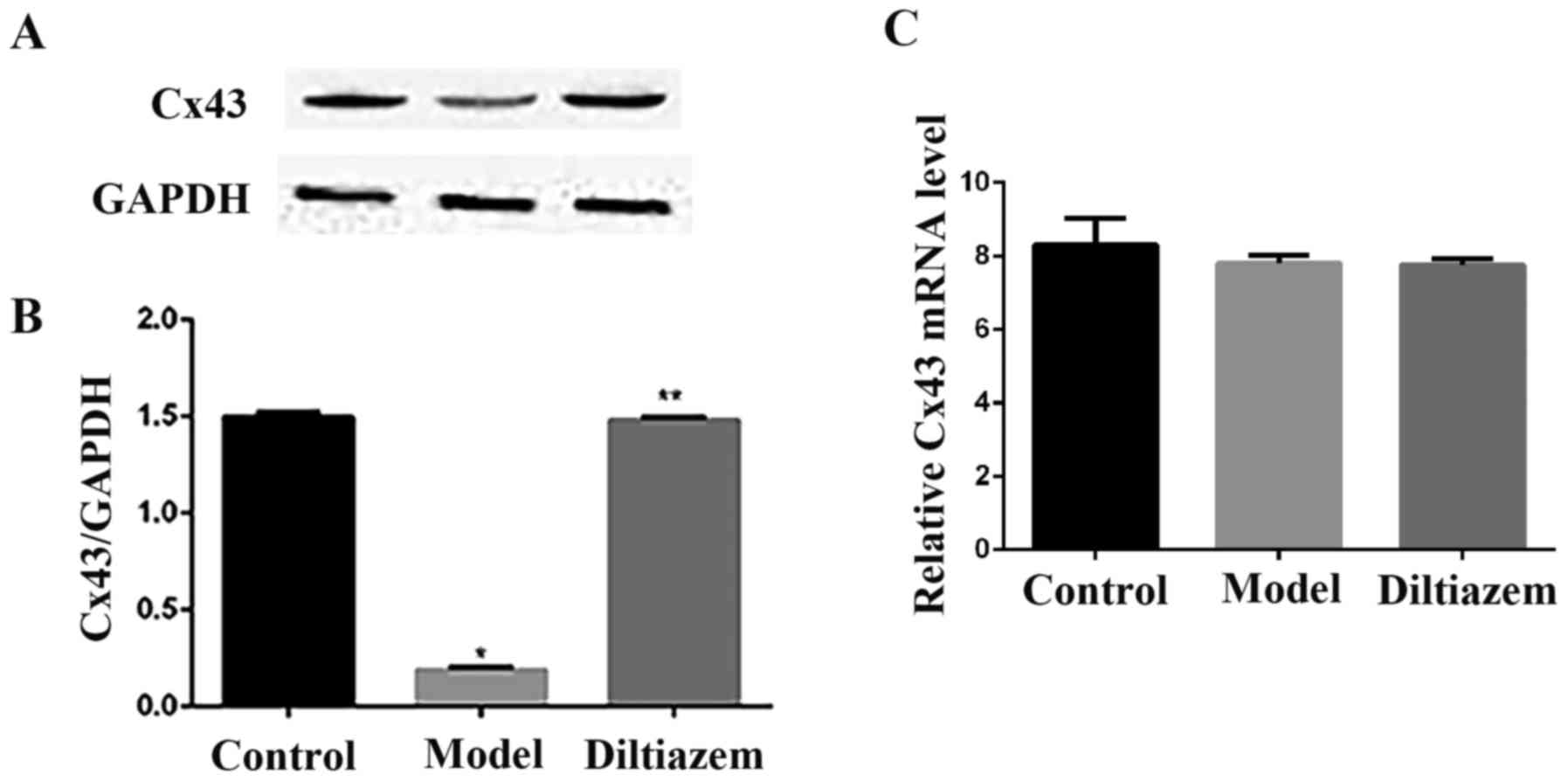
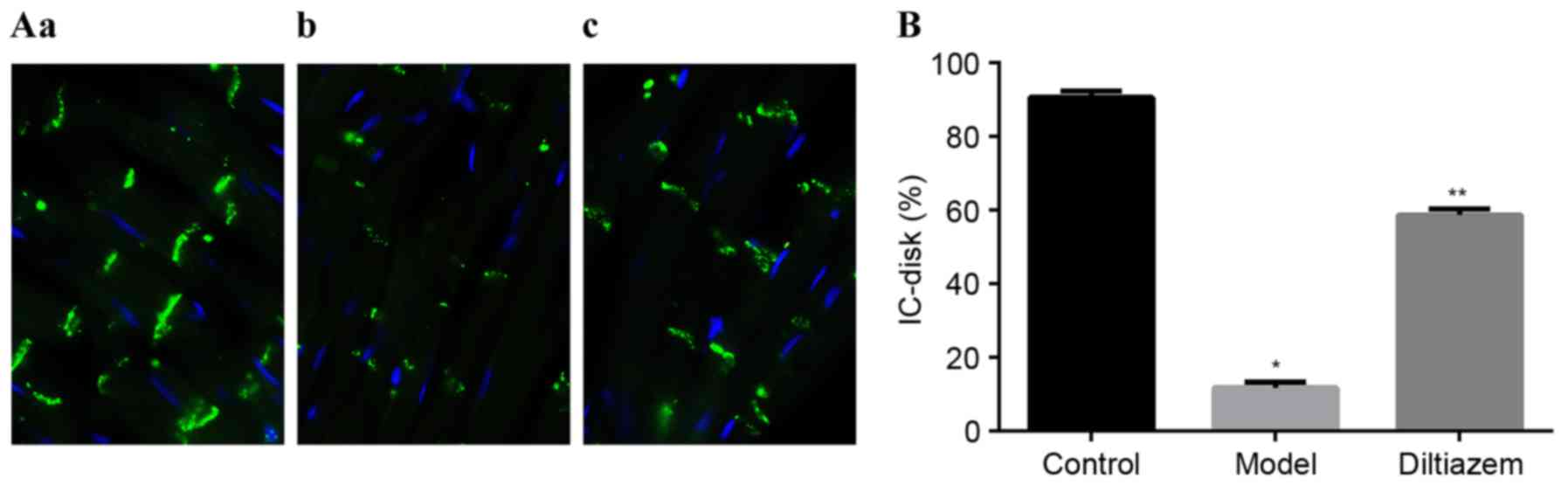
Remodeling of Cx43
As presented in Fig. 4A
and B, the protein expression of Cx43 was decreased in the
model group compared with the ISO_DIL and CTL groupa. However, the
expression of Cx43 mRNA did not differ significantly between all
three groups (Fig. 4C). This
observation suggests that diltiazem can improve the remodeling of
connexins in a quantitative manner following myocardial ischemia,
but the mechanism maybe not involve control at the mRNA level.
Localization of Cx43 was determined using immunohistochemistry and
confocal imaging. Only longitudinal sections of myocytes were
evaluated. Intercalated disks of all myocytes were identified in
high-power field, and image quantification software was used to
quantify the signal intensity and to calculate the ratio of
intercalated disk connexins to the total intensity
(IC-disk%=IC-disk intensity/total intensity ×100%). In the ISO_DIL
group, the percentage of Cx43 at intercalated disks did not change
compared with the CTL group. However, in the ISO group, the
relative percentage of Cx43 at intercalated disks decreased
significantly compared with the CTL and ISO_DIL groups (both
P<0.001; Fig. 5).
Discussion
In the present study, the cardioprotective effects
of diltiazem in myocardial ischemic rats induced by isoproterenol
were evaluated. By studying the histopathological changes,
translation and expression of the associated proteins, an attempt
was made to determine possible mechanisms underlying the
therapeutic efficiency of diltiazem. The results of the present
study demonstrated that diltiazem served a protective role during
myocardial ischemia, especially in fibrosis and Cx43
remodeling.
Isoproterenol, a known b-adrenoceptor agonist,
induces myocardial ischemia by increasing the force and frequency
of myocardial contractions (18,25–27)
and the concentration of intracellular Ca2+ (28). Ca2+ serves an important
role in maintaining myocardial systolic and diastolic functions,
and normal myocardial rhythmicity and excitability. As a secondary
intracellular messenger, Ca2+ participates in a variety
of pathophysiological processes and signal transduction pathways
(29).
During ischemia, anaerobic myocardial metabolism
predominates, reducing the amount of ATP produced. As a result, the
activity of sodium-potassium (Na+−K+) pumps
on cell membranes, which requires energy from ATP, gradually
declines. Therefore, the material exchange dependent on the
Na+−K+ electrochemical potential energy is
distorted, leading to an increase in the intracellular
Na+ concentration. The aforementioned changes lead to an
increase in anaerobic glycolysis and accumulation of lactic acid,
which in turn causes intracellular acidification, increased
Na+−H+ exchange and increased intracellular
Na+ concentration. Accumulation of intracellular
Na+ activates Na+−Ca2+ exchanging
proteins located on the plasma membrane and accelerates
Ca2+ transport into the cytoplasm, increasing
intracellular Ca2+ concentration. In the present study,
Ca2+ concentration in myocardium increased significantly
in ISO rats compared with the CTL group. Diltiazem effectively
decreased the Ca2+ concentration.
Accumulation of intracellular Ca2+, which
promotes mitosis, can induce cell proliferation, especially of
myocardial fibroblasts. The results of the present study were
consistent with those of previous studies (30–32).
Compared with the CTL and ISO_DIL groups, HWI in the ISO rats was
greater and the morphology of ventricular tissue was more abnormal,
demonstrating cardiomyocyte hypertrophy, disordered tissue
arrangement and connective tissue hyperplasia. These results
confirmed the possibility of myocardial reconstruction following
ischemia. At present, mechanisms underlying Ca2+−induced
proliferation of fibroblasts remain to be elucidated. According to
one hypothesis, secondary intracellular messenger Ca2+
affects the formation of fibroblasts and promotes their
proliferation (33). By
participating in signal transduction pathways of some cell growth
factors, Ca2+ can also promote myocardial fibrosis. The
present study demonstrated that the expression of TGF-β1 and Smad2
was decreased in ISO rats, while Smad7 was upregulated. These
results demonstrated similar patterns in protein and mRNA levels
with previous studies. Smad7 is an endogenous inhibitory factor of
TGF-β1. Following myocardial ischemia, expression of Smad7 is
reduced, which can lead to the excessive activation of TGF-β1
(34). Previous research
demonstrated that Ca2+ can interact with angiotensin II
and aldosterone to stimulate fibroblast proliferation and collagen
synthesis. Angiotensin II-aldosterone-stimulated myocardial
fibrosis in rats is reduced dramatically by CCBs (35). The present study demonstrated that
diltiazem serves a protective role in myocardial fibrosis but
further studies are required to investigate the underlying
mechanisms.
Remodeling of Cx43, the major connexin isoform in
the ventricle, is one of the most harmful consequences of
myocardial ischemia. Decreased Cx43 expression, its heterogeneity
and lateralization of connexin distribution are considered forms of
Cx43 remodeling. According to the present study, the expression of
Cx43 decreased markedly in the ISO group compared with CTL and
ISO_DIL groups. Percentage Cx43 expression localized to
intercalated disks was evaluated using immunohistochemistry and
confocal imaging. Relative abundance of Cx43 at intercalated disks
did not differ significantly between CTL and ISO_DIL groups.
However, there was a significant decrease in the ISO group compared
with both CTL and ISO_DIL groups. Cx43 distribution, particularly
its redistribution from cell-cell gap junctions to lateral margins,
was investigated in the present study. Given the implication of
connexin in cell-to-cell coupling, changes in connexin expression
and distribution were expected to have an influence on cardiac
conduction (36). Decreased Cx43
expression and heterogeneity could alter the isotonicity of
electric conduction and lead to reduction of the rate of electrical
conduction and reentry. Lateralization of Cx43 can lead to delayed
afterdepolarization (37,38). The aforementioned remodeling of
Cx43 in pathological conditions can lead to arrhythmia.
Previous studies confirmed that intracellular
Ca2+ concentration regulates the permeability of gap
junctions composed of Cx43 (39,40).
Increased abundance of intracellular Ca2+ can activate
calcium-dependent proteolytic enzymes (calpains), which can degrade
a variety of proteins, and change cell structure and function
(41). Therefore, activation of
calpains caused by the accumulation of intracellular
Ca2+ may be one of the most important mechanisms in
connexin remodeling following myocardial ischemia. In the present
study, the expression of Cx43 mRNA did not change between the three
groups of interest, whereas the expression of Cx43 proteins
decreased markedly in the ISO group compared with the CTL and
ISO_DIL groups. These results are in line with previous research
(42). Calpains degrade Cx43 at
the protein level, but do not affect the transcription of Cx43. The
mechanism underlying lateralization of connexin following
hydrolysis remain to be elucidated, but may be due to myocardial
fibrosis, which changes myocardial normal cell morphology and cell
spacing, leading to abnormal localization of connexin and
ventricular remodeling following ischemia.
Diltiazem, a CCB inhibiting inward Ca2+
flow and reducing intracellular Ca2+ concentration, can
effectively expand cardiac blood vessels, increase coronary blood
flow and improve myocardial energy metabolism; therefore, it can be
widely used in the treatment of cardiovascular diseases (43). Previous research demonstrated that
diltiazem can effectively reduce the myocardial cell damage
following myocardial hypoxia/ischemia (44), but the majority of evidence is
limited to changes in serum markers (45–47).
In conclusion, in the present study, chronic administration of
isoproterenol induced myocardial fibrosis and was associated with
the upregulation of Ca2+. Furthermore, diltiazem
improved myocardial remodeling of myocardial ischemia, especially
in fibrosis and Cx43. Taken together, the results of the present
study suggest that diltiazem serves a protective role in myocardial
ischemia.
References
|
1
|
Pimple P, Shah AJ, Rooks C, Bremner
Douglas J, Nye J, Ibeanu I, Raggi P and Vaccarino V: Angina and
mental stress-induced myocardial ischemia. J Psychosom Res.
78:433–437. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wu H, Ye M, Yang J and Ding J: Endoplasmic
reticulum stress-induced apoptosis: A possible role in myocardial
ischemia-reperfusion injury. Int J Cardiol. 208:65–66. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shintani-Ishida K and Yoshida K:
Mitochondrial m-calpain opens the mitochondrial permeability
transition pore in ischemia-reperfusion. Int J Cardiol. 197:26–32.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Eguchi A, Naito Y, Iwasaku T, Okuhara Y,
Morisawa D, Sawada H, Nishimura K, Oboshi M, Fujii K, Mano T, et
al: Association of dietary iron restriction with left ventricular
remodeling after myocardial infarction in mice. Heart Vessels.
31:222–229. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhang X, Ambale-Venkatesh B, Bluemke DA,
Cowan BR, Finn JP, Kadish AH, Lee DC, Lima JA, Hundley WG,
Suinesiaputra A, et al: Information maximizing component analysis
of left ventricular remodeling due to myocardial infarction. J
Transl Med. 13:3432015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Travers JG, Kamal FA, Robbins J, Yutzey KE
and Blaxall BC: Cardiac fibrosis: The fibroblast awakens. Circ Res.
118:1021–1040. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hayden MR, Chowdhury N, Govindarajan G,
Karuparthi PR, Habibi J and Sowers JR: Myocardial myocyte
remodeling and fibrosis in the cardiometabolic syndrome. J
Cardiometab Syndr. 1:326–333. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
McLenachan JM and Dargie HJ: Ventricular
arrhythmias in hypertensive left ventricular hypertrophy.
Relationship to coronary artery disease, left ventricular
dysfunction, and myocardial fibrosis. Am J Hypertens. 3:735–740.
1990. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jiang F, Liu GS, Dusting GJ and Chan EC:
NADPH oxidase-dependent redox signaling in TGF-β-mediated fibrotic
responses. Redox Biol. 2:267–272. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Samarakoon R, Overstreet JM and Higgins
PJ: TGF-β signaling in tissue fibrosis: Redox controls, target
genes and therapeutic opportunities. Cell Signal. 25:264–268. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tandon A, Tovey JC, Sharma A, Gupta R and
Mohan RR: Role of transforming growth factor Beta in corneal
function, biology and pathology. Curr Mol Med. 10:565–578. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kato T, Iwasaki YK and Nattel S: Connexins
and atrial fibrillation: Filling in the gaps. Circulation.
125:203–206. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lambiase PD and Tinker A: Connexins in the
heart. Cell Tissue Res. 360:675–684. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Borger MA and Weisel RD: Calcium channel
blockers in myocardial and cerebral ischemia: A clinician's review
from bench to bedside. Can J Cardiol. 15:333–340. 1999.PubMed/NCBI
|
|
15
|
Ohyama Y, Funao K, Kawabe E, Hayashi D,
Yamazaki T, Iga T, Koide D, Ohe K and Kubota K: Calcium channel
blockers and myocardial infarction: A case-control study in a
Japanese hospital. Pharmacoepidemiol Drug Saf. 11:487–492. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Williams D, Kim KS and Adams-Campbell LL:
Survival benefit from calcium channel blockers in elderly blacks
following acute myocardial infarction. Ethn Dis. 12:229–233.
2002.PubMed/NCBI
|
|
17
|
Zhang YG, Li YG, Liu BG, Wei RH, Wang DM,
Tan XR, Bu DF, Pang YZ and Tang CS: Urotensin II accelerates
cardiac fibrosis and hypertrophy of rats induced by isoproterenol.
Acta Pharmacol Sin. 28:36–43. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hori Y, Kunihiro S, Sato S, Yoshioka K,
Hara Y, Kanai K, Hoshi F, Itoh N and Higuchi S: Doxycycline
attenuates isoproterenol-induced myocardial fibrosis and matrix
metalloproteinase activity in rats. Biol Pharm Bull. 32:1678–1682.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ferreira AJ, Santos RA and Almeida AP:
Angiotensin-(1–7): Cardioprotective effect in myocardial
ischemia/reperfusion. Hypertension. 38:665–668. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Axelsen LN, Calloe K, Braunstein TH,
Riemann M, Hofgaard JP, Liang B, Jensen CF, Olsen KB, Bartels ED,
Baandrup U, et al: Diet-induced pre-diabetes slows cardiac
conductance and promotes arrhythmogenesis. Cardiovasc Diabetol.
14:872015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kudo-Sakamoto Y, Akazawa H, Ito K, Takano
J, Yano M, Yabumoto C, Naito AT, Oka T, Lee JK, Sakata Y, et al:
Calpain-dependent cleavage of N-cadherin is involved in the
progression of post-myocardial infarction remodeling. J Biol Chem.
289:19408–19419. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nademanee K, Raju H, de Noronha SV,
Papadakis M, Robinson L, Rothery S, Makita N, Kowase S, Boonmee N,
Vitayakritsirikul V, et al: Fibrosis, connexin-43, and conduction
abnormalities in the brugada syndrome. J Am Coll Cardiol.
66:1976–1986. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dorado Garcia D, Ruiz-Meana M, Inserte J,
Rodriguez-Sinovas A and Piper HM: Calcium-mediated cell death
during myocardial reperfusion. Cardiovasc Res. 94:168–180. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bloom S and Davis DL: Calcium as mediator
of isoproterenol-induced myocardial necrosis. Am J Pathol.
69:459–470. 1972.PubMed/NCBI
|
|
26
|
Monasky MM, Varian KD and Janssen PM:
Gender comparison of contractile performance and beta-adrenergic
response in isolated rat cardiac trabeculae. J Comp Physiol B.
178:307–313. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hori Y, Uechi M, Ebisawa T, Yamano S,
Yoshioka K and Mutoh K: The influence of gender on cardiac fibrosis
induced by sympathetic stimulation. Chin J Physiol. 51:146–151.
2008.PubMed/NCBI
|
|
28
|
Singal PK, Beamish RE and Dhalla NS:
Potential oxidative pathways of catecholamines in the formation of
lipid peroxides and genesis of heart disease. Adv Exp Med Biol.
161:391–401. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Glukhov AV, Balycheva M, Sanchez-Alonso
JL, Ilkan Z, Alvarez-Laviada A, Bhogal N, Diakonov I, Schobesberger
S, Sikkel MB, Bhargava A, et al: Direct evidence for
microdomain-specific localization and remodeling of functional
L-type calcium channels in rat and human atrial myocytes.
Circulation. 132:2372–2384. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Olwin BB and Storm DR: Calcium binding to
complexes of calmodulin and calmodulin binding proteins.
Biochemistry. 24:8081–8086. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Pang X and Sun NL: Calcineurin-NFAT
signaling is involved in phenylephrine-induced vascular smooth
muscle cell proliferation. Acta Pharmacol Sin. 30:537–544. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kalyanasundaram A, Lacombe VA, Belevych
AE, Brunello L, Carnes CA, Janssen PM, Knollmann BC, Periasamy M
and Gyørke S: Up-regulation of sarcoplasmic reticulum Ca(2+) uptake
leads to cardiac hypertrophy, contractile dysfunction and early
mortality in mice deficient in CASQ2. Cardiovasc Res. 98:297–306.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wei HY, Guo ZQ and Cui SJ: Apocalmodulin
and Ca2+−independent calmodulin-binding proteins. Prog
Biochem Biophys. 34:124–131. 2007.(In Chinese).
|
|
34
|
Wei LH, Huang XR, Zhang Y, Li YQ, Chen HY,
Heuchel R, Yan BP, Yu CM and Lan HY: Deficiency of Smad7 enhances
cardiac remodeling induced by angiotensin II infusion in a mouse
model of hypertension. PLoS One. 8:e701952013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Icekson Kessler G, Schlesinger H, Freimann
S and Kessler E: Expression of procollagen C-proteinase enhancer-1
in the remodeling rat heart is stimulated by aldosterone. Int J
Biochem Cell Biol. 38:358–365. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Saffitz JE: Douglas P. Zipes Lecture.
Biology and pathobiology of cardiac connexins: From cell to
bedside. Heart Rhythm. 3:102–107. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Söhl G and Willecke K: Gap junctions and
the connexin protein family. Cardiovasc Res. 62:228–232. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Severs NJ, Bruce AF, Dupont E and Rothery
S: Remodelling of gap junctions and connexin expression in diseased
myocardium. Cardiovasc Res. 80:9–19. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Halidi N, Alonso F, Burt JM, Bény JL,
Haefliger JA and Meister JJ: Intercellular calcium waves in primary
cultured rat mesenteric smooth muscle cells are mediated by
connexin43. Cell Commun Adhes. 19:25–37. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lurtz MM and Louis CF: Intracellular
calcium regulation of connexin43. Am J Physiol Cell Physiol.
293:C1806–C1813. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Goll DE, Thompson VF, Li H, Wei W and Cong
J: The calpain system. Physiol Rev. 83:731–801. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhang W, Ma X, Zhong M, Zheng Z, Li L,
Wang Z and Zhang Y: Role of the Calpain system in pulmonary vein
connexin remodeling in dogs with atrial fibrillation. Cardiology.
112:22–30. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Anderson ME: Calmodulin kinase and L-type
calcium channels; a recipe for arrhythmias? Trends Cardiovasc Med.
14:152–161. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kröner A, Seitelberger R, Schirnhofer J,
Bernecker O, Mallinger R, Hallström S, Ploner M and Podesser BK:
Diltiazem during reperfusion preserves high energy phosphates by
protection of mitochondrial integrity. Eur J Cardiothorac Surg.
21:224–231. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Hegde A, Amaria T, Mandke A and Mandke NV:
Comparative study of perioperative infusion of diltiazem and
nicorandil on myocardial protection during OPCAB surgery. Ann Card
Anaesth. 8:49–54. 2005.PubMed/NCBI
|
|
46
|
Chouairi S, Carrie D and Puel J:
Myocardial protection with calcium-channel blockers during
ischaemia and reperfusion by PTCA. Eur Heart J. 16 Suppl H:S3–S8.
1995. View Article : Google Scholar
|
|
47
|
Rousseau G, Provost P and Latour JG:
Sustained myocardial protection by clentiazem (TA-3090) after a
90-minute coronary occlusion and 72 hours of reperfusion in dogs
with collateral flow. J Cardiovasc Pharmacol. 22:264–272. 1993.
View Article : Google Scholar : PubMed/NCBI
|