Introduction
α-hemolytic Streptococcus is the foremost
cause of pneumonia in age groups with the exception of newborns,
and occasionally predisposes individuals to peritonitis, otitis
media, sinusitis, and meningitis (1,2).
Streptococcus mitis is a gram-positive a-hemolytic species
of Streptococcus. It is the closest relative of
Streptococcus pneumonia with high pathogenicity that is due
to a variety of virulence factors, including pneumolysin (Ply), a
hemolytic cytolysin, the autolysin, LytA, and various surface
proteins involved in host cell interaction, and shares >900 core
genes with S. pneumonia (3).
The majority of previous studies have generally
described S. mitis as a normal commensal that colonizes the
human oropharynx, and is characterized by low pathogenicity
(4,5). However, diverse infectious
complications, such as infective endocarditis, bacteraemia and
septicaemia, occur in immunocompromised patients as a result of the
transition of S. mitis from a commensal to pathogenic
microorganism when it escapes from the colonizing site (6–8). A
recent study using multilocus sequence analysis revealed that
severe clinical diseases are more likely to occur in cancer
patients with S. mitis than in patients with
Streptococcus oralis (9).
S. mitis resists certain antibiotics and induces infective
endocarditis in combined immunocompromised patients (10). Its infection often combines with
other pathogenetic factors and appears to cause various
complications in patients with variable syndromes and signs,
leading to difficulties in treatment. Furthermore, there is a lack
of effective therapeutic strategies targeting these complications
(11,12). Therefore, developing an effective
vaccination to reduce the incidence of S. mitis
pathogenicity-induced diseases in immunocompromised patients is
considered to be important.
Establishing the complete genome sequence of a
free-living organism enables the development of reverse vaccinology
(RV), a novel approach to vaccine design for treatment of bacterial
infections, reliant on deciphering the information contained in the
genome of the bacterium. Marked progress has been made in
understanding the biology of the pathogens and the vaccination
development as a result of advances in genomics and RV (13). RV has been applied to group B
Streptococcus (14), S.
pneumonia (15), as well as
human herpes simplex viruses (16). In addition, Rickettsia
prowazekii T-cell antigens have been identified by combining RV
technology and in vivo screening (17). RV also facilitates identification
of vaccine candidates in Rhipicephalus microplus (18). Consequently, numerous antigen
candidates for these pathogens have been acquired, which
demonstrates the significance and power of RV. In addition to
guiding vaccine design, RV promoted understanding of the
pathogenesis of meningococcus (13).
The aim of the present study was to identify
potential antigens suitable for use in an effective vaccine. The
candidate antigens of the pathogenic bacterium were screened using
RV based on whole genome sequencing of S. mitis321A. The
biological functions and signaling pathways of the predicted genes
in the genome were also analyzed.
Materials and methods
Sample collection
The clinical strain S. mitis321A was
collected from a 70-year-old male patient with chronic obstructive
pulmonary disease in stable state (moderate severity) using
pharyngeal swabs at the Institute of Respiratory Disease, Xinqiao
Hospital of Third Military Medical University in February, 2011.
The S. mitis321A strain was seeded onto blood agar plates
containing 5% sheep blood and grown overnight at 37°C. A single
clone was subsequently cultured and grown to mid-logarithmic phase
in Todd-Hewitt broth (THB) supplemented with 0.5% yeast extract at
37°C [5–6 h; optical density (OD)=0.5–0.6 at a wavelength of 600
nm]. Bacterial DNA was extracted from overnight broth cultures
using a QIAamp DNA mini kit (Qiagen AG, Basel, Switzerland)
according to the manufacturer's protocols. The patient provided
informed consent prior to the present study.
Preprocessing and DNA sequencing
Large DNA fragments were sheared into small
fragments (≤800 bp) using a high throughput sonication instrument
(Covaris or BioRuptor). The sticky end of the small DNA fragments
was converted into a blunt end using T4 DNA Polymerase, Klenow DNA
Polymerase and T4 polynuleotide kinase (Illumina, Inc., San Diego,
CA, USA), followed by adaptors ligating to the ends. Subsequently,
the blunt-ended DNA fragments were subjected to electrophoretic
separation (2% agarose gel in TAE buffer; 120V; 60 min) to recover
the target DNA products, followed by polymerase chain reaction
amplification according to the manufacturer's instructions.
Briefly, the PCR reaction mix included DNA (1 µg), Phusion DNA
polymerase (Finnzymes; Thermo Fisher Scientific, Inc., Waltham, MA,
USA), PCR primer 1.1 (1 µl; Illumina, Inc.), PCR primer 2.1 (1 µl;
Illumina, Inc.) and deionized water (22 µl). Amplify protocols were
as follows: 98°C for 30 sec, 10 cycles of 98°C for 10 sec, 65°C for
30 sec, and 72°C for 30 sec, with a final extension at 72°C for 5
min. When the DNA library was ready, DNA clusters were formed and
sequenced on an Illumina HiSeq 2000 instrument (Illumina,
Inc.).
Raw data purification
The genomic DNA was used for constructing 500- and
6,000-bp random sequencing libraries. For DNA filtering,
low-quality data was deleted from the raw data generated on the
sequencing platform to increase the accuracy and reliability of
subsequent analyses. Consequently, clean data was obtained. The
500- and 6,000-bp libraries were handled as follows: i) 1- to 90-bp
sequence was intercepted from read1 and read2; ii) the reads
containing >36 consecutive bases with quality score ≤20 were
deleted (default 40%, the cutoff was set as 36 bp); iii) the reads
with the number of the bases containing N up to a certain degree
were removed (default 10%, the cutoff was set as 9 bp); iv) Adapter
sequences were deleted (default: Adapter sequence has 15 bp overlap
with read sequences); and v) duplicated sequences were removed.
Subsequent to the above process, 10–20% of the data
(small fragment DNA data) was removed and clean data was
obtained.
The k-mer frequency distribution analysis for DNA
sequencing reads (19,20) is the preliminary step for
evaluating the size of the genome prior to DNA sequence assembly
using the obtained clean data and SOAPdenovo (http://soap.genomics.org.cn/soapdenovo.html) (-k:73)
short read assembler (21).
Genome analysis
Glimmer software is specifically designed to mine
genes in microbial DNA, such as bacteria, viruses and other
microorganisms. Compared with the previous versions, Glimmer 3.02
(http://ccb.jhu.edu/software/glimmer/index.shtml) (-l
linear) is more powerful for predicting the initiation site and
coding region, improving the accuracy of predicting GC-rich
sequences and effectively reducing the false positive rate
(22). In the present study,
Glimmer 3.02 was used to predict genes in reconstructed sequences
following DNA sequence assembly.
Tandem Repeat Finder (TRF)4.04 (2778010502000-d-h)
was applied to predict tandem repeats from which mini- and
micro-satellite sequences were screened according to the length and
number of repeats.
Functional annotation
Functional annotation for the obtained DNA sequences
was conducted using the Basic Local Alignment Search Tool (BLAST)
analysis of DNA sequences in the Kyoto Encyclopedia of Genes and
Genomes (KEGG, http://www.kegg.jp/) (23), Cluster of Orthologous Groups of
proteins (COG, http://www.ncbi.nlm.nih.gov/COG) (24), SwissProt (http://www.expasy.org/sprot/) (25), non-redundant protein database (NR,
http://www.ncbi.nlm.nih.gov/RefSeq/)
(26), Gene Ontology (GO,
http://www.geneontology.org/) (27), InterProScan (http://www.ebi.ac.uk/interpro/scan.html) and TrEMBL
(http://www.expasy.org/sprot/).
Specifically, the query amino acid sequence corresponding to the
obtained DNA was mapped to the known amino acid sequence in these
databases; identifying the known amino acid sequence that resembles
the query sequence above a certain threshold identified the
function of the query protein.
The COG database contains 2,091 COGs and covers
56–83% of the gene products extracted from the complete bacterial
and archaea genomes and facilitates protein classification. The
SWISS-PROT protein knowledgebase aims to provide detailed
annotation information for amino acid sequences, including the
function, domains structure, variants and modifications at a
post-translational level. TrEMBL is a supplement to the SWISS-PROT
database. InterProScan acts as a tool to predict the functions of a
given protein sequence based on the known information concerning
the protein domains and functional sites (28).
Screening vaccine antigen
RV integrated with bioinformatics approaches was
utilized to screen genes encoding antigens of S. mitis321A,
which elicited protective immune response in the human body.
There is a common consensus that the cell surface
antigens, secreted proteins and pathogenic protein of pathogenic
microorganisms may serve as antigens for vaccine development
(29,30); thus, genome sequences encoding the
secreted proteins, cell surface anchoring proteins and virulence
factor were selected in the study as follows:
Firstly, all information associated with secreted
proteins was downloaded from Cell PLoc (http://www.csbio.sjtu.edu.cn/bioinf/Cell-PLoc-2/), an
online package of multiple web servers, which comprises rich
knowledge on the subcellular locations of proteins involved in
diverse organisms (31). The
downloaded secreted proteins sequence was compared with the
retained target protein sequence to determine the protein sequence
sharing significant homology with the secreted protein (E-value,
<e-10) using BLAST, which is an algorithm for protein sequence
comparison to determine a library sequence that most resembles the
query sequence above a certain threshold (32). Specifically, the query sequence
corresponding to the obtained DNA was mapped to the known sequence.
Thus, the query sequence that resembled the known sequence above a
certain threshold was identified.
Subsequently, the pfam database (https://pfam.xfam.org/) provides detailed information
associated with protein multiple sequences alignments and families
(33). Through searching for cell
surface-expressed anchoring protein family from the pfam database,
the Ecm33 (glycosyl phosphatidyl inositol-anchored cell wall
organization protein) family and its protein sequence was obtained
and then compared with the target protein sequence of S.
mitis321A to determine significant protein sequences (E-value,
<e−10) using BLAST.
The Virulence Searcher (http://www.hpa-bioinfotools.org.uk/help/virfactfind_help.html)
online tool allows for convenient searches for putative genes
encoding virulence factors (34).
Using this tool, motif information associated with virulence
factors was acquired and integrated with functional annotation
result of the S. mitis321A to identify the possible gene
encoding virulence factor.
Finally, the obtained secreted proteins, anchoring
proteins and virulence factors were compared with the known
protective antigens of S. mitis321A that had been reported
in previous studies using BLAST to screen significant vaccine
candidate genes, which resembled the known protective antigens
above the threshold value (E-value, <e-10).
Essential gene screening for
developing antibacterial drugs
As essential genes are crucial for bacteria survival
and may serve as target genes for developing potent antimicrobial
drugs, the essential genes of S. mitis321A were screened to
identify potential target genes. Initially, BLAST was used with
target gene sequences against the essential gene sequences in the
Database of Essential Genes (DEG) database (E-value,
<e−10) (35) to
determine potential essential genes that may serve as vaccine
candidates.
A multiple sequence alignment of all the candidate
essential gene sequences was produced using the ClustalW2 program
(http://www.ebi.ac.uk/Tools/msa/clustalw2/) (36). From the alignment, a phylogenetic
tree was generated and visualized using the PHYLogeny Inference
Package (version 3.695; http://atgc.lirmm.fr/phyml) (37). A bootstrap analysis was conducted
with 1,000 replications to evaluate the robustness of the method.
All gaps and regions of the alignment with low confidence were
deleted from the phylogenetic analysis.
Results
Raw data purification
A total of 332 Mb of DNA sequence data was retained
following purification, and the details of the purification are
presented in Table I.
 | Table I.Genome sequencing data of
Streptococcus mitis321A. |
Table I.
Genome sequencing data of
Streptococcus mitis321A.
| Sample name | Insert size
(bp) | Reads length
(bp) | Raw data (Mb) | Adapter (%) | Duplication
(%) | Total reads | Filtered reads
(%) | Low quality
filtered reads (%) | Clean data
(Mb) |
|---|
| 321A | 464 | (90:90) | 227 | 0.07 | 0.53 | 2,527,772 |
2.86 | 1.14 | 221 |
| 321A | 600 | (90:90) | 125 | 2.07 | 0.71 | 1,378,728 | 11.13 | 1.94 | 111 |
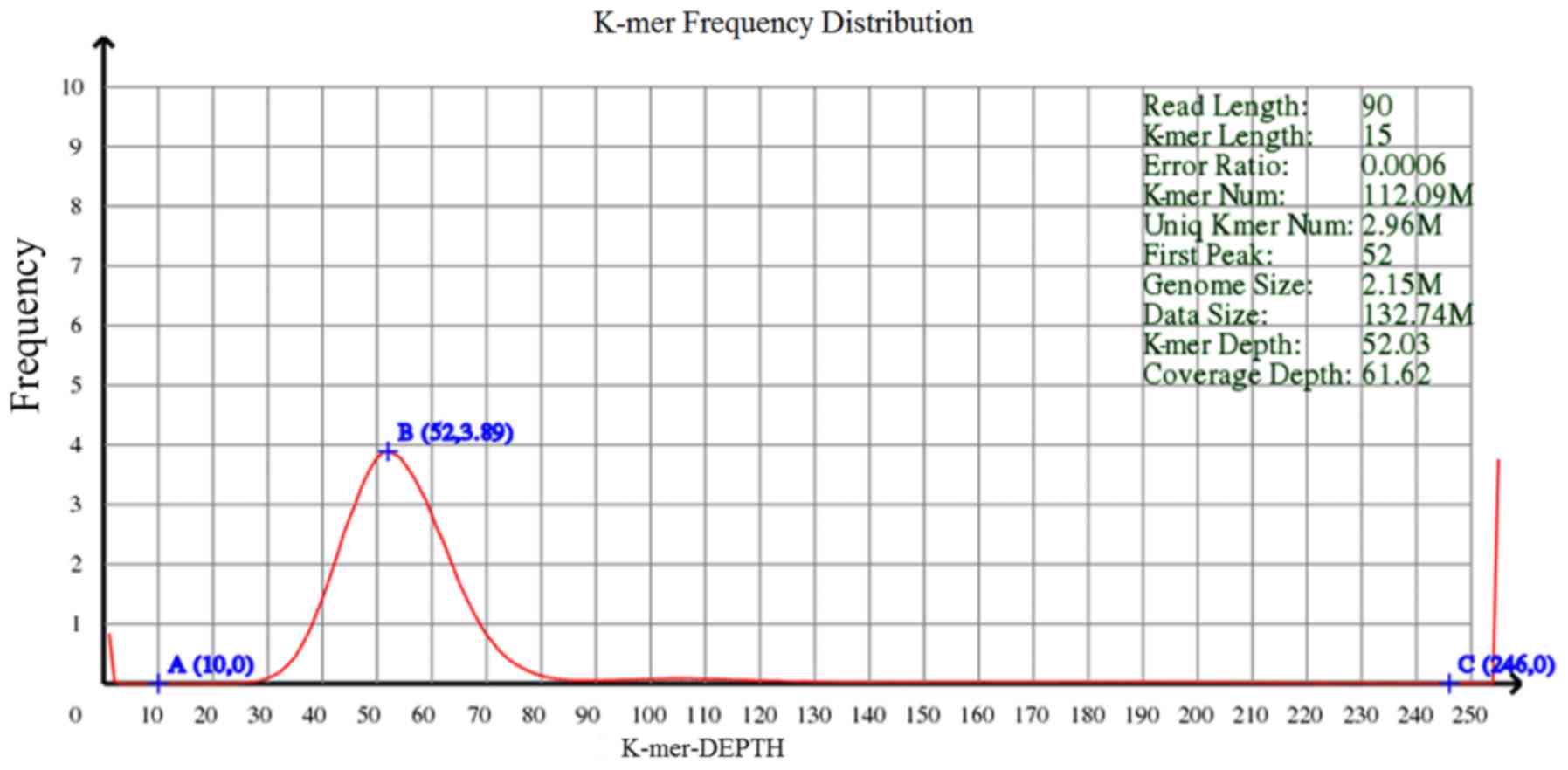
K-mer frequency distribution was analyzed to
calculate the genome size. As shown in Fig. 1, no apparent heterozygosity peak
and repeat peak was observed, indicating small degree of
heterozygosity and repeat in the DNA sequences. The result of DNA
sequence assembly is presented in Table II. The assembled genome size was
2,110,680 bp, with 40.12% GC, 6 scaffolds and 9 contig.
| Table II.DNA sequence assembly. |
Table II.
DNA sequence assembly.
| Index | Scaffold | Contig |
|---|
| Total number
(>500 bp) | 6 | 9 |
| Total length
(bp) | 2,110,680 | 2,109,125 |
| N50 length
(bp) | 2,100,529 | 1,460,616 |
| N90 length
(bp) | 2,100,529 | 636,033 |
| Max. length
(bp) | 2,100,529 | 1,460,616 |
| Min. length
(bp) | 515 | 515 |
| Sequence GC
(%) | 40.12 | 40.12 |
Genome analysis
A total of 1,944 protein-encoding genes were
predicted from the genome DNA, with mean gene length of 946 bp and
GC content of 40.9%. The total length of predicted genes and gene
interval occupied 87.1 and 34.86% of the whole genome,
respectively. The GC content in the gene interval was 34.86%.
The findings from the tandem repeat evaluation are
presented in Table III. In
total, 119 TRFs, 56 microsatellite DNAs, 10 minisatellite DNAs and
154 transposons were determined. The percentages of transposons and
TRFs in the whole genome were 0.6914 and 2.6943%, respectively.
Although the number of transposons was larger than the number of
tandem repeats, the percentage of the total length of the tandem
repeats was larger than that of transposons in the whole genome
length.
| Table III.Tandem repeats analysis. |
Table III.
Tandem repeats analysis.
| Category | Number | Repeat size
(bp) | Total length
(bp) | In genome (%) |
|---|
| Transposon | 154 | 13–674 | 14,651 | 0.6941 |
| Tandem repeat
finder | 119 | 6–1,353 | 56,867 | 2.6943 |
| Minisatellite
DNA | 56 | 15–60 | 19,574 | 0.9274 |
| Microsatellite
DNA | 10 | 6–10 | 451 | 0.0214 |
Function analysis
KEGG pathways involving the predicted DNAs were
identified and classified (Fig.
2). Of all the identified pathways, membrane transport
(environmental information processing) was the most significant
pathway containing the largest number of matched genes, and other
important pathways with a large numbers of genes comprised
xenobiotic biodegradation and metabolism, carbohydrate metabolism,
amino acid metabolism, translation, transcription, replication and
repair, and infectious disease pathways.
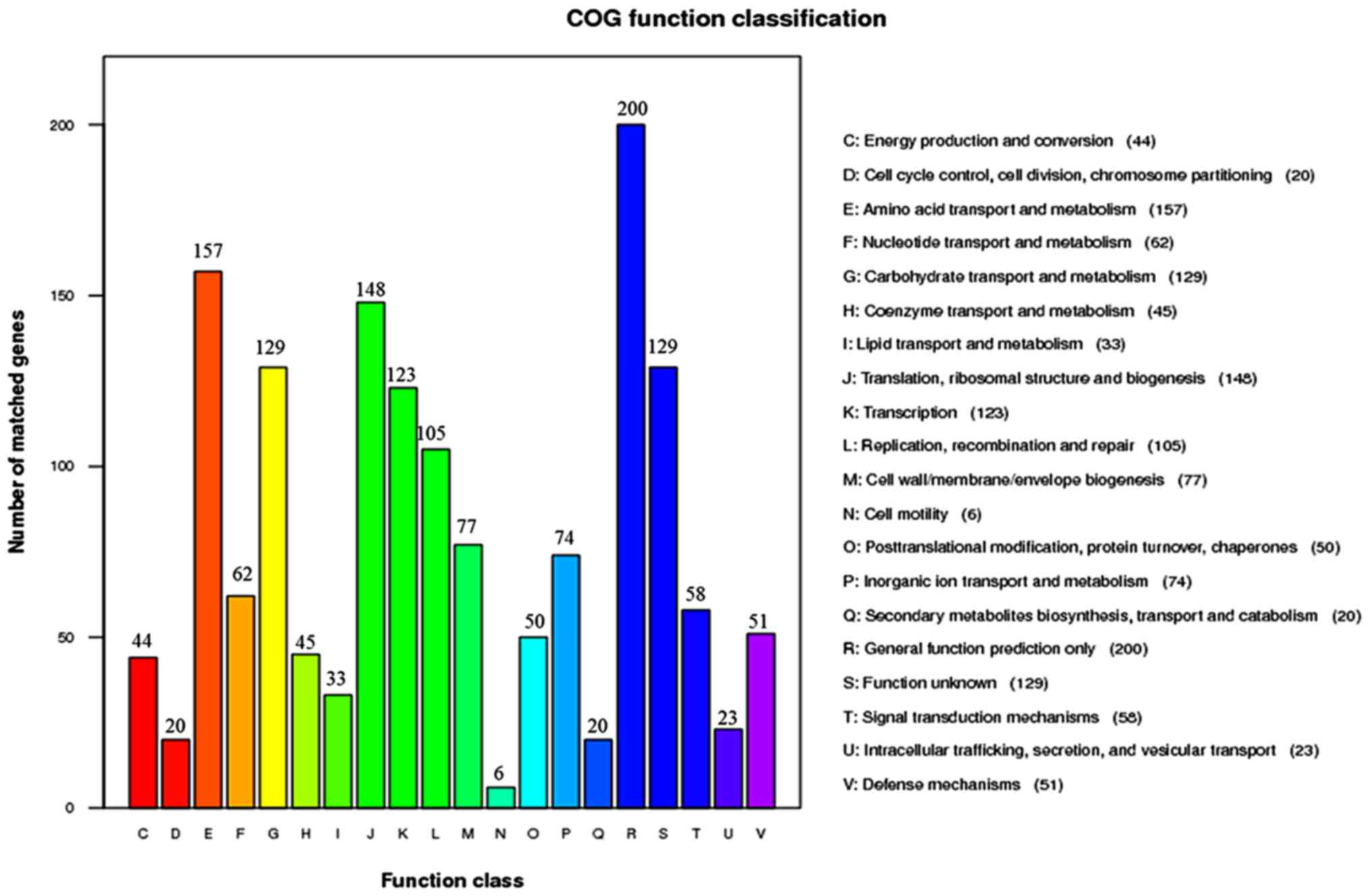
Regarding the result of COG database analysis
(Fig. 3), more genes were
clustered in cell wall/membrane/envelope biogenesis (M), signal
transduction mechanisms (T) and defense mechanisms (V) when
compared with other function classes, and the exact function of a
proportion of identified genes remained undefined (function
unknown; S).
With the BLAST analysis that aligned the obtained
genes encoding putative surface proteins, secreted proteins and
virulence factors with the previous studies, protective antigen
candidates were identified to be 321AGL000253, 321AGL000282,
321AGL000444, 321AGL000958 and 321AGL001626. Detailed functional
information of the five identified sequences is displayed in
Table IV: 321AGL000253 was
closely associated with Xaa-Pro aminopeptidase and hydrolase
activity; 321AGL000282 was associated with sensor histidine kinase
and signal transduction; 321AGL000444 was linked to competence
damage-inducible protein A (CinA); 321AGL000958 was associated with
manganese ABC transporter substrate-binding lipoprotein and metal
ion transport system; and 321AGL 001626 was linked to glutathione
reductase (NADPH) and glutathione-disulfide reductase activity.
| Table IV.Functional annotation information of
the five sequences based on NR, KEGG, COG, GO, InterProScan and
TrEMBL databases. |
Table IV.
Functional annotation information of
the five sequences based on NR, KEGG, COG, GO, InterProScan and
TrEMBL databases.
| Gene_Id | 321AGL000253 | 321AGL000282 | 321AGL000444 | 321AGL000958 | 321AGL001626 |
|---|
| NR | [X-Pro
aminopeptidase (Streptococcus mitis)] | [Sensor histidine
kinase (Streptococcus mitis)] | [Damage-inducible
protein, CinA (Streptococcus mitis)] | [Manganese ABC
transporter substrate-binding lipoprotein (Streptococcus
mitis SK564)] |
[Glutathione-disulfide reductase
(Streptococcus mitis SK564)] |
| KEGG | [K01262 pepPXaa-Pro
aminopeptidase 3.4.11.9 metabolism; enzyme families; peptidases
(BR:ko01002)] | [K07718 yes M
two-component system, sensor histidine kinase Yes M 2.7.13.3
metabolism; enzyme families; protein kinases (BR:ko01001)
environmental information processing; signal transduction;
two-component system (PATH:ko02020) environmental information
processing; signal transduction; two-component system
(BR:ko02022)] | (NA) | [K09818 ABC.MN.S
manganese/iron transport system substrate-binding
protein-environmental information processing; membrane transport;
transporters (BR:ko02000)] | [K00383 E1.8.1.7,
GSR, glutathione oxidoreductase, glutathione reductase 1.8.1.7
metabolism; metabolism of other amino acids; glutathione metabolism
(PATH:ko00480)] |
| COG | (COG0006 Xaa-Pro
aminopeptidase E amino acid transport and metabolism) | (COG2972 Predicted
signal transduction protein with a C-terminal ATPase domain T
signal transduction mechanisms) | (NA) | (COG0803 ABC-type
metal ion transport system, periplasmic component/surface adhesin P
inorganic ion transport and metabolism) | [COG1249
pyruvate/2-oxoglutarate dehydrogenase complex, dihydrolipoamide
dehydrogenase (E3) component, and associated enzymes C energy
production and conversion] |
| SwissProt | [YQHT_BACSU
uncharacterized peptidase yqhT organism=Bacillus subtilis
(strain 168) GN=yqhT PE=3 SV=1] | (NA) | (NA) | (MTSA_STRAP
Manganese ABC transporter substrate-binding lipoprotein OS=
Streptococcus anginosus GN=psaA PE=3 SV=1) | (GSHR_STRTR
glutathione reductase; organism= Streptococcus thermophilus
gene Gene name=gor; protein inferred from homology=3; sequence
version=1) |
| TrEMBL | (G6NMA3_STRPN
XAA-pro aminopeptidase organism= Streptococcus pneumoniae
GA07643 GN=pepP PE=3 SV=1) | (I0T163_STRMT
histidine kinase OS=Streptococcus mitis) SK575 GN=
HMPREF1048_1531 PE=4 SV=1 | (F9MKF5_STRMT
putative uncharacterized protein organism=Streptococcus
mitis SK569 GN= HMPREF9959_0223 PE=4 SV=1) | (E1LLV4_STRMT
manganese ABC transporter substrate-binding lipoprotein OS=
Streptococcus mitis SK564 GN=SMSK564_0925 PE=3 SV=1) | (E1LK57_STRMT
glutathione-disulfide reductase organism= Streptococcus
mitis SK564 GN=gor PE=3 SV=1) |
| Interprocan | (IPR000587;
creatinase IPR000994; peptidase M24, structural domain IPR001131;
peptidase M24B, X-Pro dipeptidase/aminopeptidase P, conserved
site) | (IPR003594;
ATPase-like, ATP-binding domain IPR003660; HAMP linker domain
IPR010559; signal transduction histidine kinase, internal
region) | (NA) | (IPR006127; ABC
transporter, metal-binding lipoprotein IPR006128; Adhesion
lipoprotein IPR006129; Adhesin B) | [IPR004099;
pyridine nucleotide-disulphide oxidoreductase, dimerization
IPR006322; glutathione reductase, eukaryote/bacterial IPR012999;
pyridine nucleotide-disulphide oxidoreductase, class I, active site
IPR013027; Flavin adenine dinucleotide (FDA)-dependent pyridine
nucleotide-disulphide oxidoreductase IPR016156; FAD/NAD-linked
reductase, dimerization IPR023753; pyridine nucleotide-disulphide
oxidoreductase, FAD/NAD (P)-binding domain] |
| (GO) | (GO:0009987;
cellular process; biological process GO:0016787; hydrolase
activity; molecular function) | [GO:0000155;
two-component sensor activity; molecular function GO:0000160;
two-component signal transduction system (phosphorelay); biological
process GO:0004871; signal transducer activity; molecular function
GO:0005524; ATP binding; molecular function GO:0007165; signal
transduction; biological process GO:0016021; integral to membrane;
cellular component] | (NA) | (GO:0007155; cell
adhesion; biological process GO:0030001; metal; ion transport
biological) process GO:0046872; metal ion binding; molecular
function | (GO:0004362;
glutathione-disulfide reductase activity; molecular function
GO:0005737; cytoplasm; cellular component GO:0006749; glutathione
meta bolic process; biological process GO:0016491; oxidoreductase
activity; molecular function GO:0016668; oxidoreductase activity,
acting on a sulfur group of donors, NAD or NADP as accep tor;
molecular function GO:0045454; cell redox homeo stasis; biological
process GO:0050660; flavin adenine dinucleotide binding; molecular
function GO:0050661; NADP binding; molecular function GO:0055114;
oxidation-reduction process; Biological Process) |
Phylogenetic tree analysis
Following alignment using CLUSTALW2, 27 essential
genes were identified. The identified genes were subjected to
phylogenetic tree analysis showing that essential genes
(321AGL000129 and 321AGL000299) on the same branch belonged to the
same phylogenetic lineage, and may act as the same type of
antibacterial drug target genes (Fig.
4).
Discussion
Generally, S. mitis is considered to be a
commensal oral Streptococcus posing little immunological
threat to the majority of individuals; however, elderly,
immunocompromised and cancer patients undergoing cytotoxic
chemotherapy are susceptible to it (38). In addition, it may occasionally
affect normal healthy infants and adults (8). Therefore, the aim of the present
study was to establish antigen candidates for developing potent
vaccines against the S. mitis pathogen. In the current
study, a 332-Mb sequence of the S. mitis321A genome was
predicted to encode a total of 1,944 genes with 40.9% GC content.
By contrast, S. mitis B5 genome sequencing determines
two15-Mb sequences with mean GC content of 39.98%, which is similar
to the genome of S. pneumonia (2.04–2.24 Mb and ~40% GC)
(3). Different strains of S.
mitis displayed varied genomes. The predicted genes of S.
mitis321A were closely associated with membrane transport
(environmental information processing), carbohydrate metabolism,
amino acid metabolism, translation, transcription, replication and
repair KEGG pathways. Of most importance was the membrane transport
pathway with 335 matched genes. Consistently, more genes were
involved in the wall/membrane/envelope biogenesis function class
when compared with the other function classes, as demonstrated in
the COG classification analysis, confirming that genes encoding
putative membrane proteins were critical for the pathogenicity of
S. mitis.
Another consideration of the present study was that
xenobiotic biodegradation and metabolism, carbohydrate metabolism
and amino acid metabolism, translation, transcription, replication
and repair pathways appeared to be associated with a large number
of genes, leading to the hypothesis that S. mitis may
deteriorate the condition of vulnerable patients by impairing
energy mechanisms and interrupting DNA synthesis, transcription and
translation processes in host cells, thus triggering severe
clinical consequences. Consistently, the COG classification
analysis indicated that amino acid transport and metabolism,
carbohydrate transport and metabolism, transcription, replication,
recombination and repair function classes were closely linked to
the identified genes.
Furthermore, through BLAST analysis, 321AGL000253,
321AGL000282, 321AGL000444, 321AGL000958 and 321AGL001626 were
identified to be candidate antigens of S. mitis321A. The
putative biological function of the five sequences appeared to be
varied. As suggested by the present study, 321AGL000253 was closely
associated with Xaa-Pro aminopeptidase and hydrolase activity.
Xaa-Pro aminopeptidase hydrolyzes Xaa-Pro bonds. A previous study
has shown that Xaa-Pro aminopeptidase is involved in aminolysis
reactions in Lactococcuslactis (39). However, to the best of our
knowledge, the Xaa-Pro aminopeptidase in S. mitis has not
previously been defined; thus, requires further investigation to
clarify its association with vaccine design and development.
Additionally, 321AGL000282 was associated with sensor histidine
kinase and signal transduction, while 321AGL001626 was linked to
NADPH and glutathione metabolism activity. Histidine kinase is a
multifunctional transferase family that is implicated in upstream
signal transduction pathways of various virulent pathways (40). It has been demonstrated as a
critical component of the virulence of certain fungal strains
(41). Furthermore, it has been
revealed that glutathione peroxidase may contribute to the
virulence of S. pyogenes (42). These findings indicated that
321AGL000282 and 321AGL001626 may be virulence factors of S.
mitis321A. Furthermore, the 321AGL000444 was linked to CinA,
which has been found to mediate the membrane association in
Helicobacter pylori and S. pneumoniae (1,43).
321AGL000958 was associated with manganese ABC transporter
substrate-binding lipoprotein, which is a transmembrane protein for
adenosine triphosphate (44,45).
These evidence indicate that 321AGL000444 and 321AGL000958 may
encode the membrane anchoring protein of the bacteria.
Essential genes were defined as pivotal genes for
organism survival, which are often involved in metabolism, DNA
replication and translation into proteins (46). Notably, they are increasingly
recognized as potential target genes for developing novel agents
against various pathogenic microorganisms (47,48).
There are numerous studies based on genome analysis that have
provided a selection of essential genes, which is promising for
selecting and validating antimicrobial agents (49,50).
Thus, essential genes of S. mitis321A were screened based on
the DEG database, not including genes encoding surface proteins,
secreted proteins or virulence factors. As a result, 27 essential
genes were obtained. Phylogenetic tree analysis was used to analyze
the homologue of the essential genes. The majority of essential
genes appeared to belong to the same phylogenetic lineage, with the
exception of 321AGL000176, 321AGL001082 and 321AGL001586. Essential
genes on the same branch, such as 321AGL000129 and 321AGL000299 may
be the target genes for the same type of antibacterial agents.
The findings of this preliminary study require
validation with experimental data. Subsequent trials will evaluate
the efficacy of the vaccines that targeted the putative antigen
targets provided by the present study, and provide insight into the
biological function of the antigen targets and differences in
genomes between S. mitis321A and other strains of S.
mitis.
In conclusion, the genome sequencing of S.
mitis321A predicted 1,944 genes with 40.9% GC content. The
predicted genes were associated with a variety of signaling
pathways and biological functions regarding membrane transport and
energy metabolism. Five gene sequences encoding putative surface
proteins, secreted proteins and virulence factors, and several
essential genes were determined to be antigen candidates for
developing potent vaccines to prevent the diseases driven by the
S. mitis321A pathogen.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the
National Natural Science Foundation of China (grant nos. 81100007
and 81000004).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
QZ conceived and designed the research and drafted
the manuscript. KL, CW, ZX and LY acquired data, analyzed and
interpreted data and statistical analysis. QM conceived and
designed the research, and revised the manuscript for important
intellectual content. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of the Xinqiao Hospital of Third Military Medical
University, Chongqing, China.
Consent for publication
The patients provided informed consent prior to the
present study.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
BLAST
|
Basic Local Alignment Search Tool
|
|
KEGG
|
Kyoto Encyclopedia of Genes and
Genomes
|
|
COG
|
Cluster of Orthologous Groups of
proteins
|
|
Ply
|
pneumolysin
|
|
RV
|
reverse vaccinology
|
|
THB
|
Todd-Hewitt broth
|
|
PNK
|
polynuleotide kinase
|
|
GO
|
Gene Ontology
|
|
CinA
|
competence damage-inducible protein
A
|
|
NADPH
|
glutathione reductase
|
References
|
1
|
Tettelin H, Nelson KE, Paulsen IT, Eisen
JA, Read TD, Peterson S, Heidelberg J, DeBoy RT, Haft DH, Dodson
RJ, et al: Complete genome sequence of a virulent isolate of
Streptococcus pneumoniae. Science. 293:498–506. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Alvarez EF, Olarte KE and Ramesh MS:
Purpura fulminans secondary to Streptococcus pneumoniae
meningitis. Case Rep Infect Dis. 2012:5085032014.
|
|
3
|
Denapaite D, Brückner R, Nuhn M, Reichmann
P, Henrich B, Maurer P, Schähle Y, Selbmann P, Zimmermann W and
Wambutt R: The genome of Streptococcus mitis B6-what is a
commensal? PloS One. 5:e94262010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kilian M, Poulsen K, Blomqvist T,
Håvarstein LS, Bek-Thomsen M, Tettelin H and Sørensen UB: Evolution
of Streptococcus pneumoniae and its close commensal
relatives. PloS One. 3:e26832008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hakenbeck R, Madhour A, Denapaite D and
Brückner R: Versatility of choline metabolism and choline-binding
proteins in Streptococcus pneumoniae and commensal
streptococci. FEMS Microbiol Rev. 33:572–586. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kohno K, Nagafuji K, Tsukamoto H, Horiuchi
T, Takase K, Aoki K, Henzan H, Kamezaki K, Takenaka K, Miyamoto T,
et al: Infectious complications in patients receiving autologous
CD34-selected hematopoietic stem cell transplantation for severe
autoimmune diseases. Transpl Infect Dis. 11:318–323. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Han XY, Kamana M and Rolston KV: Viridans
streptococci isolated by culture from blood of cancer patients:
Clinical and microbiologic analysis of 50 cases. J Clin Microbiol.
44:160–165. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mitchell J: Streptococcus mitis:
Walking the line between commensalism and pathogenesis. Mol Oral
Microbiol. 26:89–98. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shelburne SA, Sahasrabhojane P, Saldana M,
Yao H, Su X, Horstmann N, Thompson E and Flores AR:
Streptococcus mitis strains causing severe clinical disease
in cancer patients. Emerg Infect Dis. 20:762–771. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Matsui N, Ito M, Kuramae H, Inukai T,
Sakai A and Okugawa M: Infective endocarditis caused by
multidrug-resistant Streptococcus mitis in a combined
immunocompromised patient: An autopsy case report. J Infect
Chemother. 19:321–325. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bochud PY, Eggiman P, Calandra T, Van
Melle G, Saghafi L and Francioli P: Bacteremia due to viridans
Streptococcus in neutropenic patients with cancer: Clinical
spectrum and risk factors. Clin Infect Dis. 18:25–31. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Husain E, Whitehead S, Castell A, Thomas
EE and Speert DP: Viridans streptococci bacteremia in children with
malignancy: Relevance of species identification and penicillin
susceptibility. Pediatr Infect Dis J. 24:563–566. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Delany I, Rappuoli R and Seib KL:
Vaccines, reverse vaccinology, and bacterial pathogenesis. Cold
Spring Harb Perspect Med. 3:a0124762013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen VL, Avci FY and Kasper DL: A maternal
vaccine against group B Streptococcus: Past, present, and
future. Vaccine. 31 Suppl 4:D13–D19. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Talukdar S, Zutshi S, Prashanth KS, Saikia
KK and Kumar P: Identification of potential vaccine candidates
against Streptococcus pneumoniae by reverse vaccinology
approach. Appl Biochem Biotechnol. 172:3026–3041. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Xiang Z and He Y: Genome-wide prediction
of vaccine targets for human herpes simplex viruses using Vaxign
reverse vaccinology. BMC Bioinformatics. 14 Suppl 4:S22013.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Caro-Gomez E, Gazi M, Goez Y and Valbuena
G: Discovery of novel cross-protective Rickettsia prowazekii
T-cell antigens using a combined reverse vaccinology and in vivo
screening approach. Vaccine. 32:4968–4976. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Maritz-Olivier C, Van Zyl W and Stutzer C:
A systematic, functional genomics, and reverse vaccinology approach
to the identification of vaccine candidates in the cattle tick,
Rhipicephalus. Ticks Tick Borne Dis. 3:179–187. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Melsted P and Pritchard JK: Efficient
counting of k-mers in DNA sequences using a bloom filter. BMC
Bioinformatics. 12:3332011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li R, Zhu H, Ruan J, Qian W, Fang X, Shi
Z, Li Y, Li S, Shan G and Kristiansen K: De novo assembly of human
genomes with massively parallel short read sequencing. Genome Res.
20:265–272. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Luo R, Liu B, Xie Y, Li Z, Huang W, Yuan
J, He G, Chen Y, Pan Q, Liu Y, et al: SOAPdenovo2: An empirically
improved memory-efficient short-read de novo assembler.
Gigascience. 1:182012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Craig JW, Chang FY, Kim JH, Obiajulu SC
and Brady SF: Expanding small-molecule functional metagenomics
through parallel screening of broad-host-range cosmid environmental
DNA libraries in diverse proteobacteria. Appl Environ Microbiol.
76:1633–1641. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kanehisa M, Goto S, Kawashima S, Okuno Y
and Hattori M: The KEGG resource for deciphering the genome.
Nucleic Acids Res. 32:(Database Issue). D277–D780. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tatusov RL, Galperin MY, Natale DA and
Koonin EV: The COG database: A tool for genome-scale analysis of
protein functions and evolution. Nucleic Acids Res. 28:33–36. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Boeckmann B, Bairoch A, Apweiler R,
Blatter MC, Estreicher A, Gasteiger E, Martin MJ, Michoud K,
O'Donovan C, Phan I, et al: The SWISS-PROT protein knowledgebase
and its supplement TrEMBL in 2003. Nucleic Acids Res. 31:365–370.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Pruitt KD, Tatusova T and Maglott DR: NCBI
reference sequence (RefSeq): A curated non-redundant sequence
database of genomes, transcripts and proteins. Nucleic Acids Res.
33:(Database Issue). D501–D5D4. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Consortium GO: The Gene Ontology (GO)
database and informatics resource. Nucleic Acids Res. 32:(Database
Issue). D258–D261. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zdobnov EM and Apweiler R: InterProScan-an
integration platform for the signature-recognition methods in
InterPro. Bioinformatics. 17:847–848. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sette A and Rappuoli R: Reverse
vaccinology: Developing vaccines in the era of genomics. Immunity.
33:530–541. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Maione D, Margarit I, Rinaudo CD,
Masignani V, Mora M, Scarselli M, Tettelin H, Brettoni C, Iacobini
ET, Rosini R, et al: Identification of a universal Group B
Streptococcus vaccine by multiple genome screen. Science.
309:148–150. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chou KC and Shen HB: Cell-PLoc: A package
of Web servers for predicting subcellular localization of proteins
in various organisms. Nat Protoc. 3:153–162. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lobo I: Basic local alignment search tool
(BLAST). Nat Educ. 1:22008.
|
|
33
|
Bateman A, Coin L, Durbin R, Finn RD,
Hollich V, Griffiths-Jones S, Khanna A, Marshall M, Moxon S,
Sonnhammer EL, et al: The Pfam protein families database. Nucleic
Acids Res. 32:(Database Issue). D138–D141. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Underwood A, Mulder A, Gharbia S and Green
J: Virulence Searcher: A tool for searching raw genome sequences
from bacterial genomes for putative virulence factors. Clin
Microbiol Infect. 11:770–772. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhang R and Lin Y: DEG 5.0, a database of
essential genes in both prokaryotes and eukaryotes. Nucleic Acids
Res. 37:D455–D458. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Larkin MA, Blackshields G, Brown N, Chenna
R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A and
Lopez R: Clustal W and Clustal X version 2.0. Bioinformatics.
23:2947–2948. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Guindon S, Lethiec F, Duroux P and Gascuel
O: PHYML online-a web server for fast maximum likelihood-based
phylogenetic inference. Nucleic Acids Res. 33:(Web Server Issue).
W557–W559. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Balkundi DR, Murray DL, Patterson MJ, Gera
R, Scott-Emuakpor A and Kulkarni R: Penicillin-resistant
Streptococcus mitis as a cause of septicemia with meningitis
in febrile neutropenic children. J Pediatr Hematol Oncol. 19:82–85.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yoshpe-Besancon I, Gripon JC and
Ribadeau-Dumas B: Xaa-Pro-dipeptidyl-aminopeptidase from
Lactococcus lactis catalyses kinetically controlled
synthesis of peptide bonds involving proline. Biotechnol Appl
Biochem. 20:131–140. 1994.PubMed/NCBI
|
|
40
|
Kowluru A: Identification and
characterization of a novel protein histidine kinase in the islet β
cell: Evidence for its regulation by mastoparan, an activator of
G-proteins and insulin secretion. Biochem Pharmacol. 63:2091–2100.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Torosantucci A, Chiani P, De Bernardis F,
Cassone A, Calera JA and Calderone R: Deletion of the two-component
histidine kinase gene (CHK1) of Candida albicans contributes to
enhanced growth inhibition and killing by human neutrophils in
vitro. Infect Immun. 70:985–987. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Brenot A, King KY, Janowiak B, Griffith O
and Caparon MG: Contribution of glutathione peroxidase to the
virulence of Streptococcus pyogenes. Infect Immun.
72:408–413. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Fischer W and Haas R: The RecA protein of
Helicobacter pylori requires a posttranslational
modification for full activity. J Bacteriol. 186:777–784. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kurokawa K, Lee H, Roh KB, Asanuma M, Kim
YS, Nakayama H, Shiratsuchi A, Choi Y, Takeuchi O, Kang HJ, et al:
The triacylated ATP binding cluster transporter substrate-binding
lipoprotein of Staphylococcus aureus functions as a native
ligand for Toll-like receptor 2. J Biol Chem. 284:8406–8411. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kolenbrander PE, Andersen RN, Baker RA and
Jenkinson HF: The adhesion-associated sca operon in
Streptococcus gordonii encodes an inducible high-affinity
ABC transporter for Mn2+ uptake. J Bacteriol.
180:290–295. 1998.PubMed/NCBI
|
|
46
|
Griffin JE, Gawronski JD, DeJesus MA,
Ioerger TR, Akerley BJ and Sassetti CM: High-resolution phenotypic
profiling defines genes essential for mycobacterial growth and
cholesterol catabolism. PLoS Pathog. 7:e10022512011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Akerley BJ, Rubin EJ, Novick VL, Amaya K,
Judson N and Mekalanos JJ: A genome-scale analysis for
identification of genes required for growth or survival of
Haemophilus influenzae. Proc Natl Acad Sci USA. 99:966–971. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Sassetti CM, Boyd DH and Rubin EJ:
Comprehensive identification of conditionally essential genes in
mycobacteria. Proc Natl Acad Sci USA. 98:12712–12779. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Weigel LM, Clewell DB, Gill SR, Clark NC,
McDougal LK, Flannagan SE, Kolonay JF, Shetty J, Killgore GE and
Tenover FC: Genetic analysis of a high-level vancomycin-resistant
isolate of Staphylococcus aureus. Science. 302:1569–1571.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Ramaswamy S and Musser JM: Molecular
genetic basis of antimicrobial agent resistance in Mycobacterium
tuberculosis: 1998 update. Tuber Lung Dis. 79:3–29. 1998.
View Article : Google Scholar : PubMed/NCBI
|