Introduction
Traumatic brain injury (TBI), a serious health
condition that is increasing in prevalence, is one of the leading
causes of mortality and long-term neurological disability worldwide
(1,2). Head injury, particularly in young
men, is frequently caused by external physical forces, including
falls, sport injuries, motor vehicle accidents and firearm
incidents (3,4). Impairment of cognitive, physical and
psychosocial functions induced by TBI largely depends on the
severity of the initial mechanical injury to the head and
subsequent pathophysiological processes (5). It has been well established that the
pathophysiology of TBI is a highly complex process that involves
primary and secondary injury mechanisms (6). Primary injury, defined as the direct
result of the mechanical effects of initial impact, can cause
contusion, laceration, ischemia, diffuse axonal injury, diffuse
swelling and intracranial hemorrhage at the moment of insult, which
may result in neurological impairment (7). These effects are irreversible and can
only be avoided via primary prevention strategies. Secondary injury
is initiated by primary injury following the initial traumatic
insult, or within h to days and even weeks post-injury (8,9). A
secondary insult can induce a progressive cascade of associated
events, including inflammation, oxidative stress, calcium-mediated
damage, neurotransmitter release, mitochondrial dysfunction (that
subsequently triggers neuronal cell death), astrocyte proliferation
and microglia activation (10–12).
Secondary brain injury is thought to be responsible for the
development of brain edema, neurological deficits and cognitive
dysfunction following TBI (12).
The delay in the induction of secondary injury provides a window
for therapeutic TBI intervention. A series of biophysiological and
pathological reactions following TBI contribute to subsequent
neuronal cell death, including apoptosis, necrosis and necroptosis
(13). Apoptosis, a programmed
form of cell death, is implicated in pathological responses
post-TBI, and contributes to secondary brain damage following TBI
(14). Furthermore, rapid onset of
cell death typically leads to tissue necrosis. Necrotic cell death
is a consequence of acute disruption of cellular metabolism. In
contrast to apoptosis, necrotic cell death rarely serves the
requirements of the organism (15). A number of genes including Bcl-2
and Bax are known to regulate the apoptotic pathway, and the
impairment of apoptosis is often caused by overexpression of the
pro-survival protein Bcl-2 (16).
Determining the underlying pathological mechanisms
and developing potential therapeutic strategies for TBI are major
research areas that have attracted global attention. Despite the
fact that numerous clinical and basic studies have focused on TBI
treatment, there are limited pharmacological therapies available
for the treatment of neurological injury and the prognosis for
patients with TBI (17–19). Thus, an improved mechanistic
understanding of TBI pathogenesis is required in order to develop
therapeutic strategies for the treatment of TBI.
Dexmedetomidine (Dex), a highly selective
α2-adrenoceptor agonist, previously provided
neuroprotection against ischemia reperfusion-induced cerebral
injury (20,21), transient spinal ischemia (22) and isoflurane-induced neuroapoptosis
(23). However, the underlying
mechanisms of these processes have not been fully determined. In
the present study, whether Dex possesses neuroprotective potential
in a rat model of TBI was investigated. Furthermore, whether the
TBI-associated neuroprotective effects of Dex are associated with
neuronal apoptosis, and 70 kDa heat shock protein (HSP70) protein
expression levels in the hippocampus was investigated.
Materials and methods
Animals
A total of 90 adult male Sprague-Dawley (SD) rats,
weighing 350–375 g (Vital River Laboratory Animal Technology Co.,
Ltd., Beijing, China) were used in the present study. The animals
were housed with a controlled temperature (20–25°C) and humidity
(50–65%), a standard 12-h light/dark cycle, and had free access to
food and water prior to and following the surgery/sham operation.
All rats were sacrificed via cervical dislocation. All experimental
procedures were carried out in accordance with the guidelines of
the Chinese Council on Animal Protection (24) and were approved by the Animal
Ethics Committee of Hebei Medical University (Hebei, China).
Experimental TBI model
The rat model of TBI was induced using a modified
weight-drop device, as described previously by Marmarou et
al (25). Briefly, rats were
anaesthetised with intraperitoneal chloral hydrate (40 mg/kg).
Following this, a midline incision was made in order to expose the
skull between the bregma and lambda suture lines. Subsequently a
steel disc (10 mm in diameter and 3 mm in thickness) was adhered to
the skull using dental acrylic. Animals were then relocated onto a
foam mattress placed underneath a weight-drop device and, following
relocation, a weight of 450 g was dropped through a vertical tube
from a height of 1.5 m onto the steel disc. Sham-operated animals
underwent the same surgical procedure, however, they were not
subjected to cortical impact. Following surgery, the wound was
closed, and rats were then housed in individual cages and placed on
heat pads (37°C) for 24 h in order to maintain normal body
temperature during the recovery period.
Group and drugs administration
The total 90 male SD rats were randomly divided into
3 groups (n=30/group: Sham-operated, TBI and TBI treated with Dex
groups. Animals in the TBI + Dex group received an intravenous
injection of Dex (15 µg/kg; Jiangsu Hengrui Medicine Co., Ltd.,
Lianyungang, China) at 1 h post-TBI. The particular dose of Dex
used in the present study was selected according to a previous
study (26). Each group was then
further divided into 3 subgroups (n=10/subgroup); each subgroup of
rats was sacrificed at either 12, 24 or 72 h post-TBI,
respectively.
Neurological function assessment
Neurobehavioral testing was performed using the
Neurological Severity Score (NSS) assessment (24), which assesses motor, sensory,
reflex and coordination abilities. One point was awarded for each
failure to perform a particular task, and a score of 10 reflected
maximal impairment. NSS was evaluated at 3 and 7 days post-TBI. An
observer who was blinded to the group assignment of each rat,
carried out the assessments. The difference between the initial NSS
and that at any later time was calculated for each rat, and this
value (∆NSS) was understood to reflect either the spontaneous
recovery or the treatment-induced recovery of motor function.
Morris water maze test
The spatial learning ability of the rats was
assessed using a Morris water maze as previously described
(24). The Morris water maze
consisted of a black circular pool (180 cm diameter, 45 cm high)
filled with water (30 cm depth) at a temperature of 26°C and was
virtually divided into 4 equivalent quadrants: North (N), west (W),
south (S) and east (E). A 2 cm submerged escape platform (diameter
12 cm, height 28 cm; made opaque with paint) was placed in the
middle of one of the quadrants equidistant from the sidewall and
the center of the pool. Rats were trained to find the platform
prior to either the TBI or sham operation. For each trial, the rat
was randomly placed into a quadrant start point (N, S, E or W)
facing the wall of the pool and allowed a maximum of 60 sec to find
the escape platform. If the rat failed to escape within 90 sec, it
was permitted to remain on the platform for a maximum of 20 sec
prior to returning to its holding cage for a new trial (intertrial
interval, 20 sec). Maze performance was recorded using a video
camera suspended above the maze and interfaced with a video
tracking system (Huaibei Zhenghua Biological Instrument Co., Ltd.,
China). A total of 6 rats from each group were tested at day 3 and
6 different rats from each group were tested at day 7 (5 times
each); rats tested on day 3 were not tested on day 7 and vice
versa.
Investigation of brain edema
Brain edema was investigated via analysis of brain
water content using the wet-dry weight method. Briefly, rats were
anesthetized intraperitoneally using 10% chloral hydrate (0.3
ml/100 g) and sacrificed at 24 h post-TBI. The brains from rats in
each group were rapidly separated and weighed, and then placed in
an oven for 72 h at 100°C. Following this, the brains were then
reweighed in order to determine dry weight content. The percentage
of water in the tissues was calculated according to the formula:
Percent brain water=[(wet weight-dry weight)/wet weight]x100
(27).
Immunohistochemical analysis
Immunohistochemical analysis was performed in
accordance with the instructions of the StreptAvidin-Biotin Complex
immunohistochemistry kit (Wuhan Boster Biological Engineering,
Ltd., Wuhan, China). The brains were fixed in 4% paraformaldehyde
for 24 h at room temperature, dehydrated by graded ethanol, and
then embedded in paraffin. Following this, the sections (5 µm) were
then prepared and incubated with 3% H2O2 for
10 min and 5% bovine serum albumin solution (Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) for 20 min at room temperature.
Subsequently, sections were incubated overnight at 4°C with rabbit
anti-B-cell lymphoma-2 (Bcl-2; cat. no. sc-24511; 1:100) and
anti-Bcl-2-associated X protein (Bax; cat. no. sc-4239; both Santa
Cruz Biotechnology, Inc., Dallas, TX, USA; 1:100), and then with
horseradish peroxidase (HRP)-conjugated anti-rabbit immunoglobulin
(Ig)-G antibodies (Wuhan Boster Biotechnology Ltd., Wuhan, China;
cat. no. SA1021; 1:3,000) for 40 min at room temperature. Following
this, the detection reagent 3,3′-diaminobenzidine was used to
visualize the immunohistochemical reaction under light microscope
(BX53; Olympus, Tokyo, Japan). PBS was used as a substitute for the
primary antibody in order to act as the negative control.
Immunofluorescence assay
After the experimental protocol, rats were
anesthetized and sacrificed and the brain tissues were removed for
immunofluorescence staining. The frozen sections (−20°C, 15 µm)
were treated with 0.4% Triton-100 (Wuhan Boster Biotechnology Ltd.)
for 30 min and blocked in normal donkey serum (Wuhan Boster
Biotechnology Ltd.) for 1.5 h at room temperature. The sections
were then incubated in a mixture of rabbit anti-HSP70 polyclonal
antibody (cat. no. sc-221731; 1:20), mouse anti-neuronal nuclei
(NeuN; cat. no. sc-161127; 1:50; both Santa Cruz Biotechnology,
Inc.) and mouse anti-GFAP (cat. no. sc-33673; 1:50; both Santa Cruz
Biotechnology, Inc.) overnight at 4°C. The following day, the
sections were incubated in a mixture of fluorescein-conjugated
anti-rabbit IgG (cat. no. sc-2357; 1:1,000) and anti-mouse IgG
(cat. no. sc-2962.; 1:1,000; both Santa Cruz Biotechnology, Inc.)
for 2 h at 37°C in the dark. The nuclei were then counterstained
with DAPI for 15 min at room temperature. All microphotographs were
taken under a laser scanning confocal microscope (Leica TCS SP8
STED 3X; serial no. 8100001247; Wetzlar, Germany, magnification,
×1,000).
Western blot analysis
Brain tissues from damaged hippocampal regions in
different groups at 12, 24 or 72 h post-TBI were extracted and
homogenized in RIPA lysis buffer (Wuhan Boster Biotechnology Ltd.).
The lysates were centrifuged for 10 min at 12,000 × g, 4°C, and the
supernatants were transferred into a fresh tube. Total protein
concentration was determined using the bicinchoninic acid reagent
method. Samples (30 mg per sample) were separated on a 10% SDS-PAGE
gel, and subsequently transferred to polyvinylidene fluoride
membranes (Roche Diagnostics GmbH, Mannheim, Germany). The
membranes were blocked with 10% non-fat milk (w/v) at room
temperature for 2 h and then incubated overnight at 4°C with rabbit
anti-Bax polyclonal antibodies (cat. no. sc-4239; 1:500), rabbit
anti-Bcl-2 polyclonal antibodies (cat. no. sc-24511; 1:500), rabbit
anti-HSP70 polyclonal antibodies (cat. no. sc-221731; 1:500) or
rabbit anti-β-actin monoclonal antibodies (cat. no. sc-47778; all
Santa Cruz Biotechnology, Inc.; dilution, 1:500). Membranes were
then incubated with HRP-conjugated anti-rabbit IgG secondary
antibody (cat. no. sc-2376; Santa Cruz Biotechnology, Inc.;
dilution, 1:5,000) at room temperature for 1 h. Immunoreactivity
was detected using an enhanced chemiluminescence detection system
(ChemiDoc XRS; Bio-Rad Laboratories, Inc., Hercules, CA, USA) and
the densitometric signals were quantified using an Image Quant 5.2
software (GE Healthcare Life Sciences, Little Chalfont, UK).
Protein expression was normalized to the intensity of corresponding
bands for β-actin.
Statistical analysis
Data were expressed as the mean ± standard error
from three independent experiments. SPSS version 16.0 software
(SPSS, Inc., Chicago, IL, USA) was used for statistical analysis.
Statistical analysis was performed using one-way analysis of
variance followed by the Student-Newman-Keuls post-hoc test, or the
Student's t-test (for two means comparison). P<0.05 was
considered to indicate a statistically significant difference.
Results
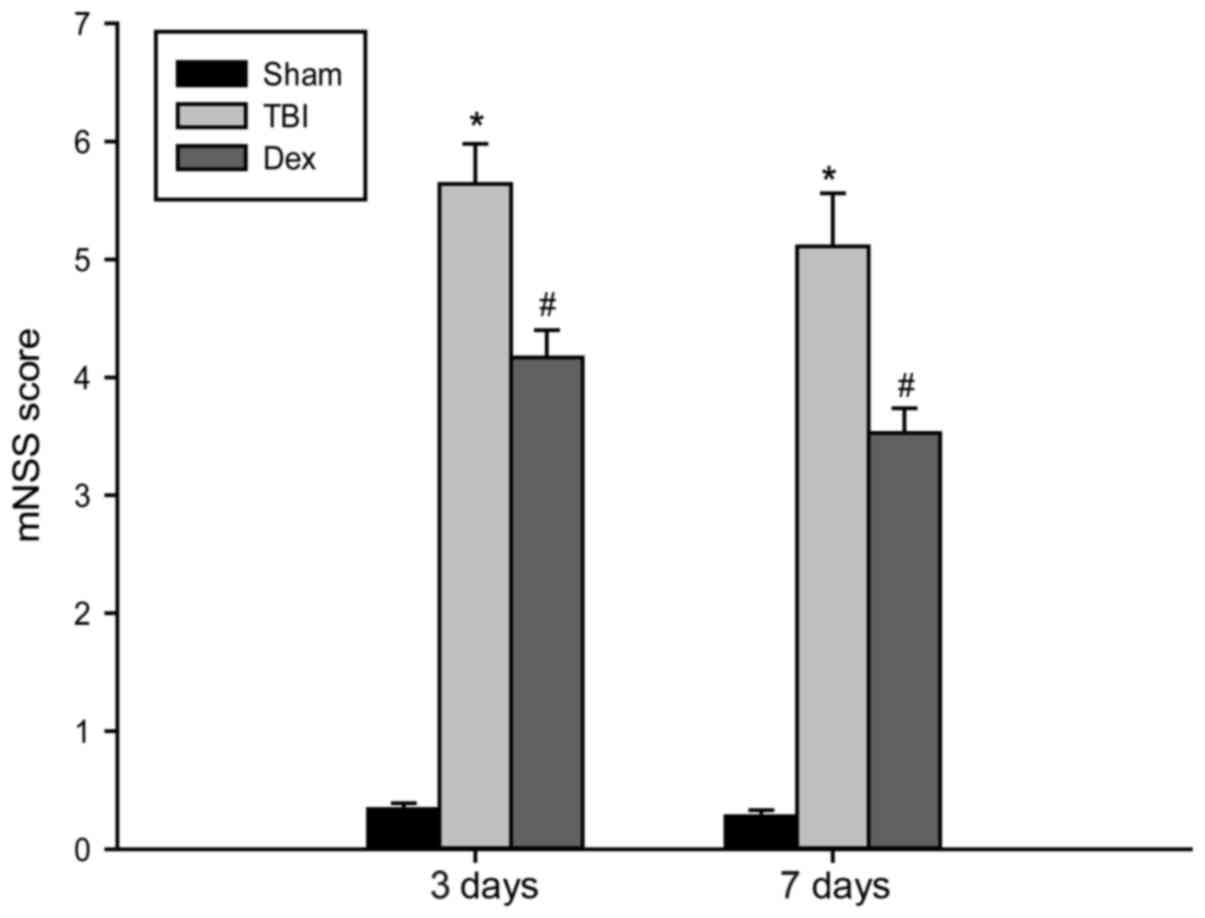
Dex alleviates neurological deficits
post-TBI
The NSS test was performed in order to investigate
the long-term neurological function of rats post-TBI. The temporal
alterations in functional recovery of the rat were expressed as
∆NSS. As revealed by the results of the NSS test presented in
Fig. 1, TBI elicited a significant
decline in neurological function, apparent according to the
increase in ∆NSS at 3 and 7 days post-TBI. However, Dex
administration significantly decreased the neurological score in
rats that underwent TBI, thereby suggesting that treatment with Dex
post-TBI may improve recovery of neurological function.
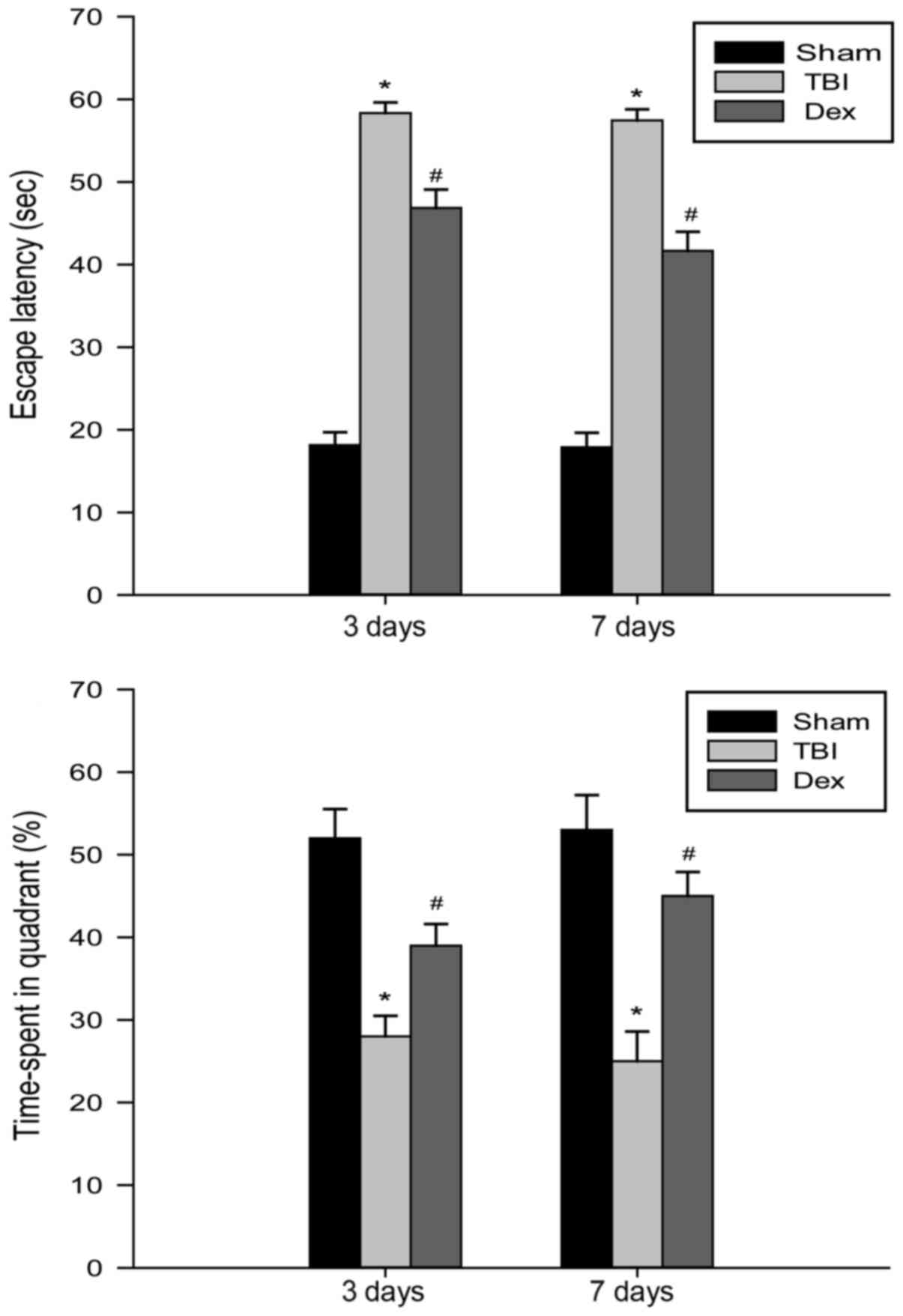
Administration of Dex improves spatial
learning and memory deficits post-TBI
The spatial learning and memory of rats was assessed
using the Morris water maze at 3 and 7 days following TBI. As
revealed by Fig. 2, all rats in
the TBI group exhibited increased escape latency time periods as a
result of their impaired ability to find the hidden platform
compared with the sham group at 3 and 7 days. The rats subjected to
Dex administration demonstrated a significant decrease in their
escape latency time period compared with the rats belonging to the
TBI group, thereby indicating that Dex administration may result in
spatial learning and memory functional recovery post-TBI.
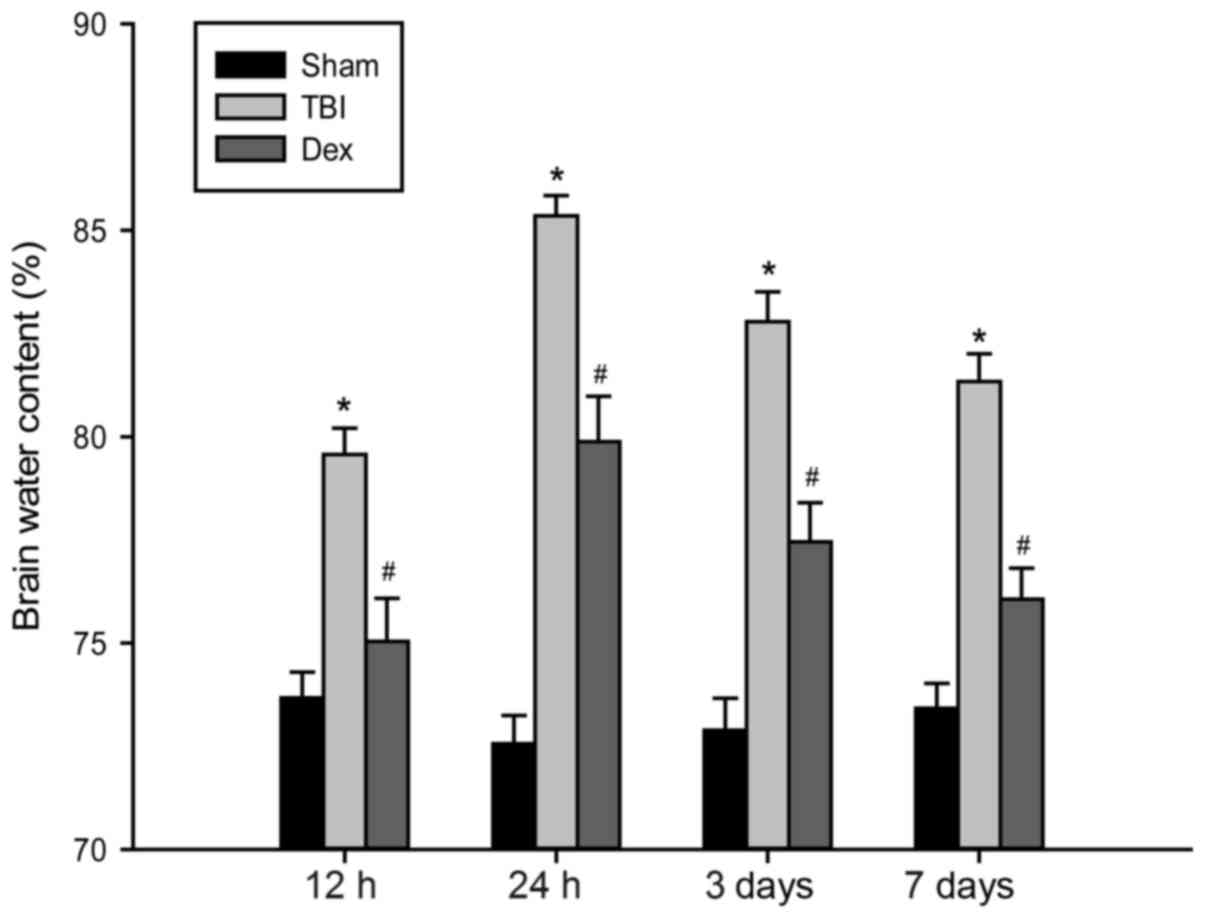
Administration of Dex attenuates brain
edema post-TBI
The severity of brain edema was investigated by
measuring the brain water content at set time points between 12 h
and 7 days post-TBI. In the present study, the wet-dry weight
method was used to determine brain water content. As demonstrated
by Fig. 3, the water content of
brain tissues post-TBI was significantly increased compared with
the sham group. The administration of Dex markedly decreased brain
water content compared with the TBI group; however, the brain water
content still remained higher than that of the sham group.
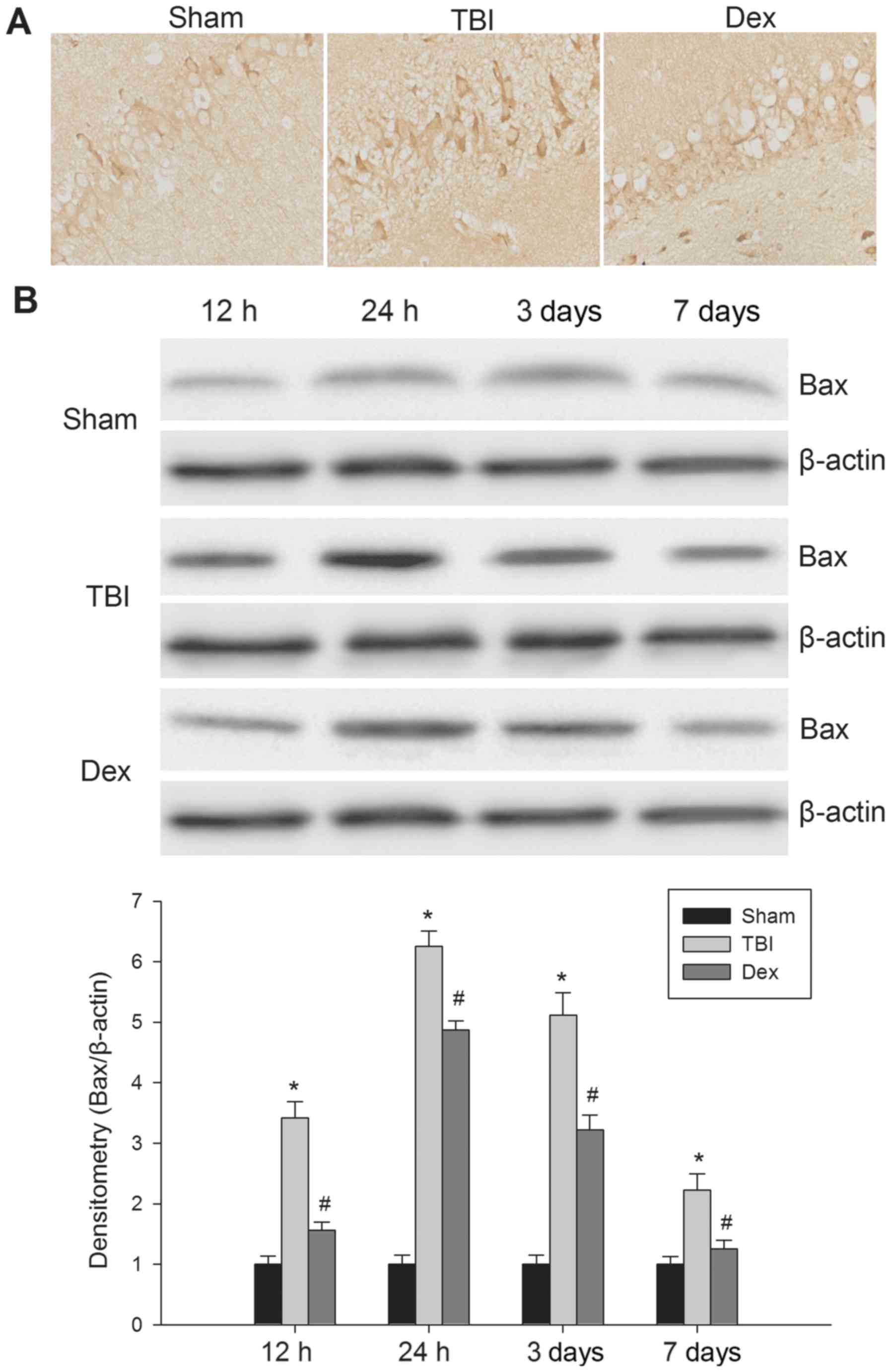
Administration of Dex downregulates
Bax expression post-TBI
At the 12 h post-TBI, Bax protein expression was
measured using immunohistochemistry in the paraffin-embedded brain
tissue sections. Bax protein expression was revealed to be
localized in the hippocampal neurons in all of rats. As
demonstrated by Fig. 4A, Bax
positive cells were observed within the hippocampus post-TBI;
however, the administration of Dex post-TBI markedly reduced the
number of Bax positive cells visualized. Following this, the
expression of Bax protein was detected using western blot analysis.
As revealed by Fig. 4B, basal Bax
protein expression was low in the brains of the rats in the sham
group. Compared with sham group rats, the level of Bax expression
in the hippocampal fraction was significantly increased at 12 h
post-TBI, reaching a maximum at 24 h post-TBI, which then steadily
decreased until 7 days post-TBI. When compared with the TBI group,
Bax protein expression levels were significantly reduced following
Dex administration post-TBI. Taken together, these results
suggested that Dex treatment may suppress the expression of Bax in
the hippocampus following TBI.
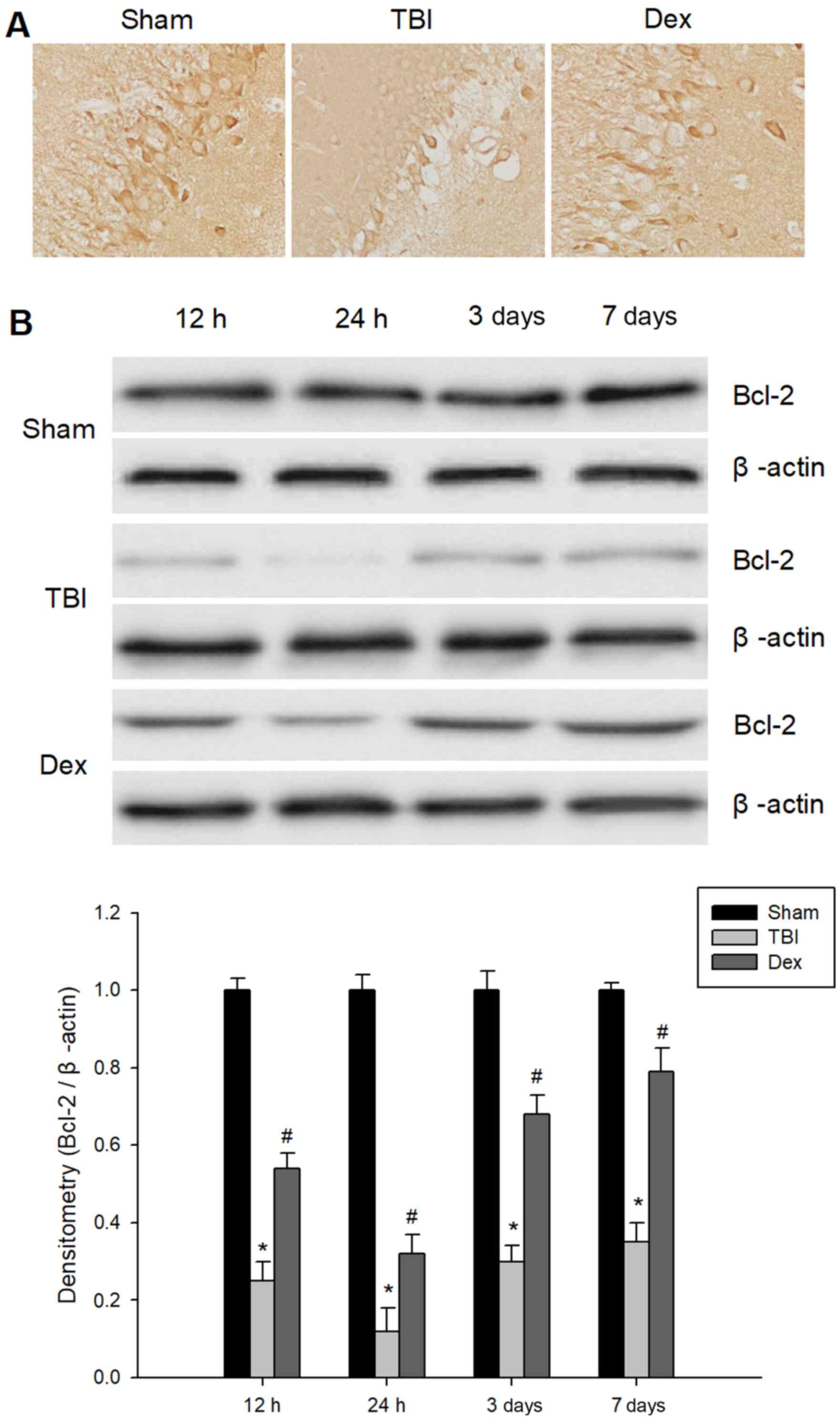
Administration of Dex upregulates
Bcl-2 expression post-TBI
Using immunohistological staining, the expression
levels of Bcl-2 in samples of hippocampal brain tissue at different
time intervals following TBI was investigated. As revealed by
Fig. 5A, less Bcl-2 positive
staining was visible in rats subjected to TBI when compared with
the sham group. Furthermore, Fig.
5A revealed that rats treated with Dex demonstrated an increase
in the number of Bcl-2 positive cells in the injured hippocampus
compared with the TBI group. Following this, western blot analysis
was performed in order to determine Bcl-2 protein expression
levels. As demonstrated in Fig.
5B, basal expression of Bcl-2 was detected in the hippocampus
samples from the sham group. Compared with the sham group, the
expression of Bcl-2 was significantly reduced in the TBI group 12 h
post-injury and reached a minimum concentration at 24 h post-TBI.
However, Bcl-2 protein expression level in the hippocampal tissue
was significantly increased in the Dex group when compared with the
TBI group. Taken together, these results indicated that the
administration of Dex may upregulate the expression of Bcl-2 in the
hippocampus following TBI.
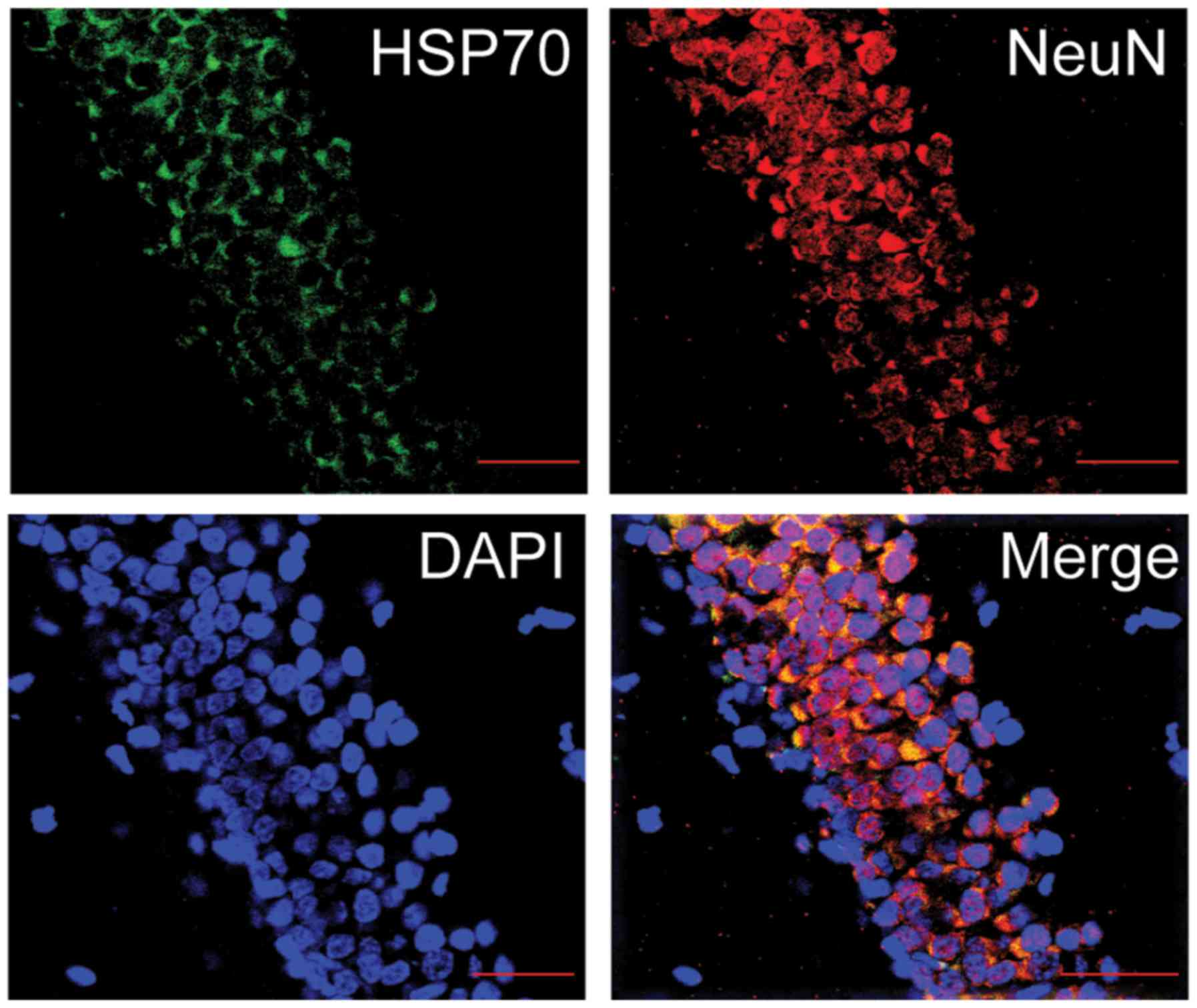
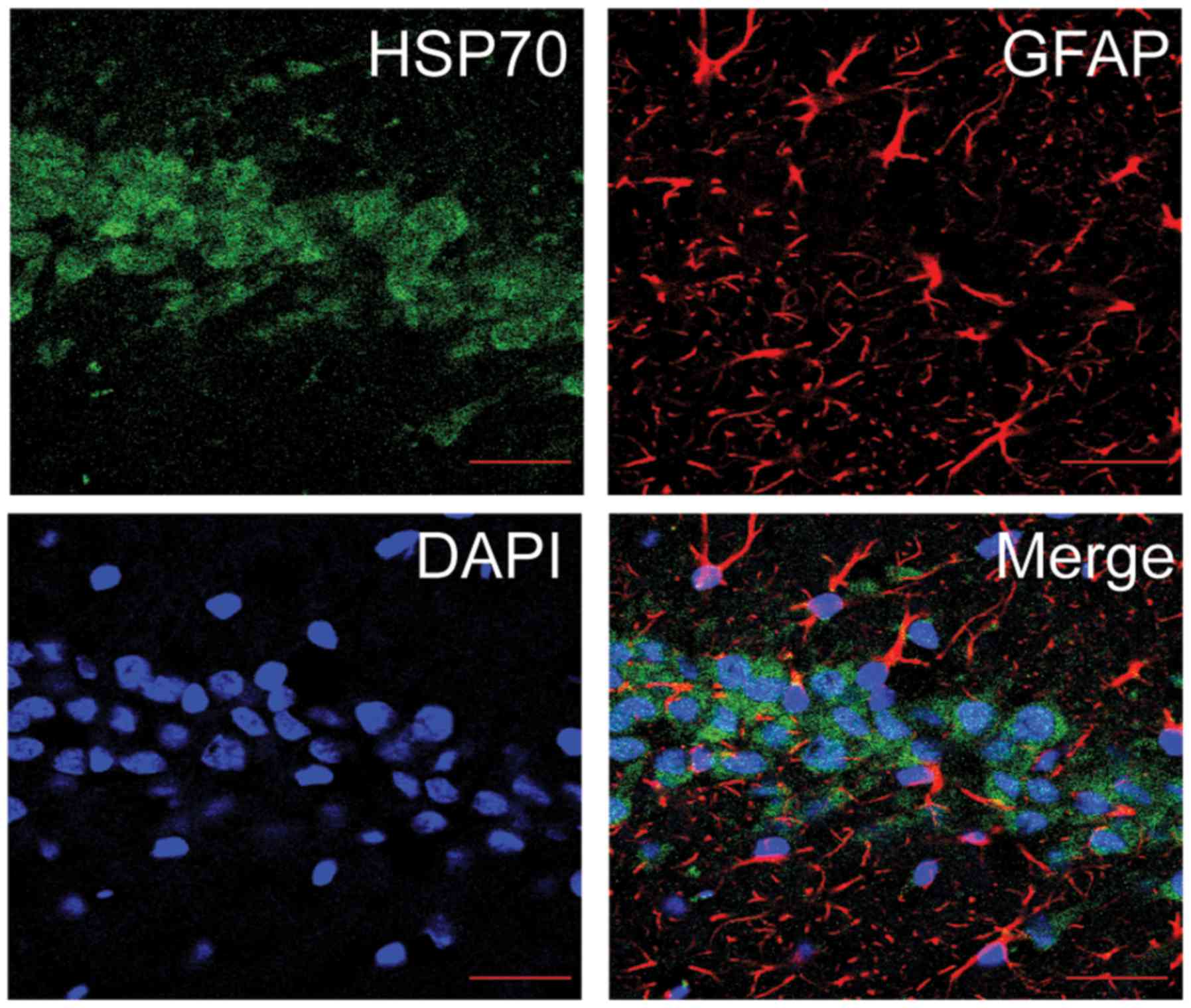
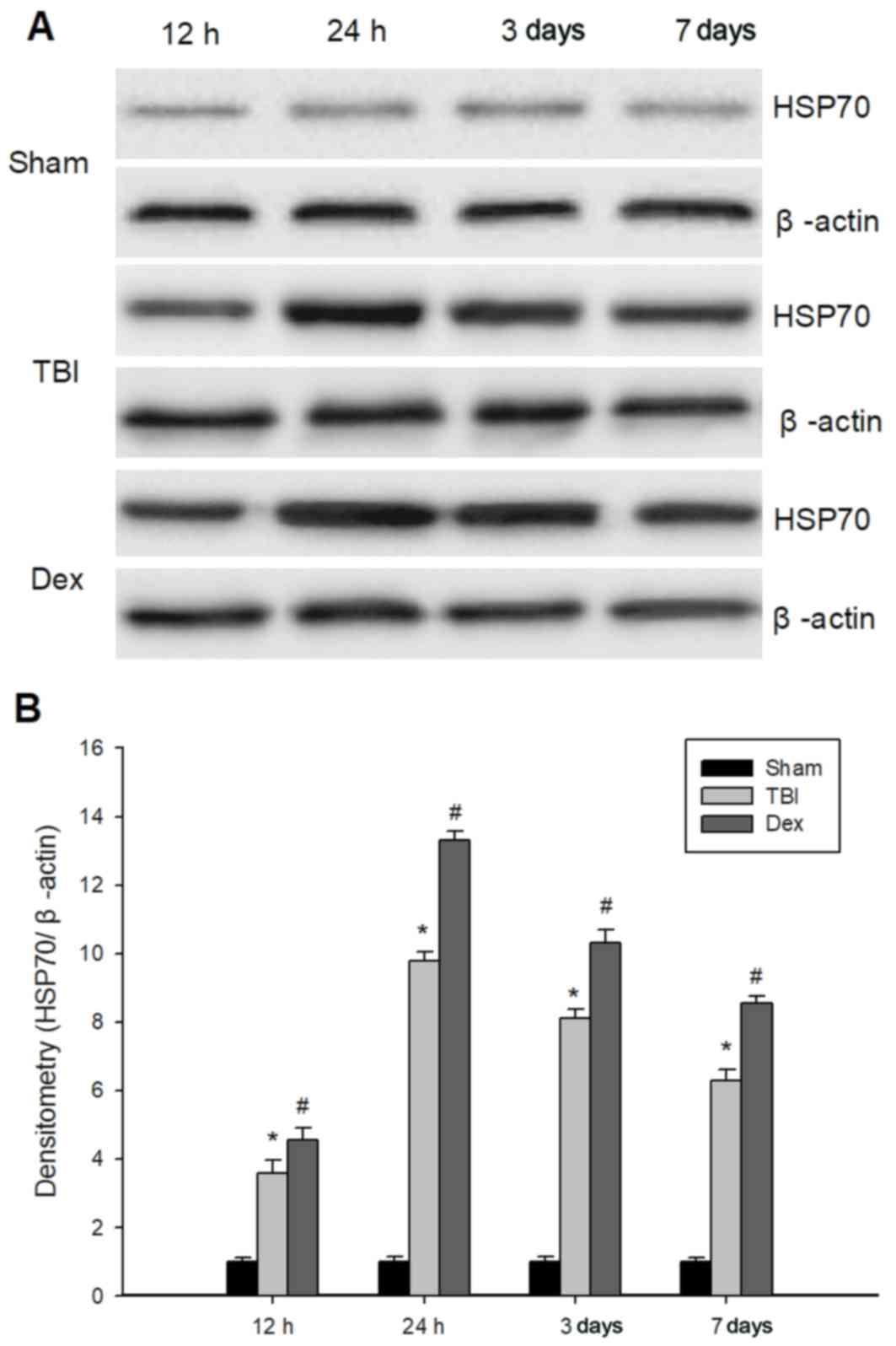
Administration of Dex enhances HSP70
expression post-TBI
In order to determine whether Dex protects against
TBI via the modulation of HSP70 expression, the expression levels
of HSP70 in the hippocampus of the different experimental rat
groups were investigated. Firstly, the co-expression of HSP70 and
either NeuN (Fig. 6) or GFAP
(Fig. 7) in the hippocampus of
rats was observed using a laser scanning confocal microscope. It
was revealed that HSP70 primarily presented itself in the neurons
of the hippocampal CA1 region. Additionally, the protein expression
levels of HSP70 in the hippocampus at 12, 24 h, 3 and 7 days
post-TBI were investigated using western blot. As demonstrated in
Fig. 8, HSP70 expression levels
were significantly higher in the TBI group compared with the sham
group and peaked at 24 h post-TBI. Furthermore, Dex treatment
significantly enhanced the protein expression of HSP70 post-TBI
(Fig. 8).
Discussion
Apoptosis, the morphological manifestation of
programmed cell death occurring under physiological and
pathological conditions, is characterized by DNA fragmentation,
chromatin compaction, nuclear shrinkage and cytoplasmic
condensation (28). TBI triggers a
complex cascade of apoptotic events, which contribute to neuronal
damage and neurological dysfunction (29). The hippocampus, a key brain
structure associated with functional impairments, is particularly
vulnerable to brain injury. These early changes of TBI occur in the
hippocampal region (30).
The aim of the present study was to investigate
brain edema, behavioral impairment and neuronal apoptosis in the
hippocampus following experimental TBI. A rat model of TBI
utilizing a simple weight-drop device was established, and this
simple model was capable of producing a graded brain injury in the
rodent without initiating hypertensive surge or excessive
brain-stem damage. The results of the present study demonstrated
that the brain water content and neurological deficit scores of
rats subjected to TBI were significantly lower at 24 h post-TBI
than those demonstrated by sham group.
Members of the Bcl-2 family are important regulators
of cellular apoptosis. The Bax protein promotes neuronal apoptosis
via direct activation of proapoptotic factors and inactivation of
antiapoptotic Bcl-2 family members (31). By contrast, the Bcl-2 protein
inhibits apoptosis and is implicated in neuroprotective events
post-TBI. The ratio of Bcl-2 and Bax expression determines the
extent of cellular apoptosis. In addition, HSP70, a highly
conserved molecular chaperone, confers cellular protection against
insults as it possesses potent anti-apoptotic properties (32). In the present study, the expression
levels of Bax, Bcl-2 and HSP70 in the hippocampus post-TBI were
determined by immunohistochemistry and western blot analysis. It
was revealed that Bc1-2 expression decreased post-TBI, while the
expression of Bax and HSP70 increased. These results suggested that
neuronal apoptosis may be implicated in the pathophysiological
process of TBI. Taken together, these results suggested that TBI
may initiate a neuronal apoptosis cascade that contributes to
substantial neuronal damage, brain edema and behavioral
impairment.
The results of the present study suggested that
anti-apoptotic proteins may serve as potential therapeutic targets
for the protection of the damaged brain in cases of TBI. Previous
clinical and experimental studies have demonstrated that the
administration of Dex is an effective intervention strategy for
several neurological diseases (33–35).
Furthermore, it has recently been reported that Dex has the ability
to pass freely across the blood brain barrier, and its metabolism
predominantly occurs in the liver (33). In addition, a further recent study
demonstrated that patients with liver disease have a significantly
decreased clearance rate of Dex (36). A large proportion of experimental
data has suggested that Dex is critically implicated in
ischemic/reperfusion models in different organs, including the
brain (21), heart (37), kidney (38) and lung (39). Furthermore, Dex has been
extensively studied due to its wide range of modulatory activities,
including modulation of inflammatory processes, gene expression,
oxidative stress reaction, transmitter release and channel
activation (40). In the present
study, the determination of the function of Dex in the
pathophysiological process of brain damage, cerebral edema and
neurological function impairment was investigated post-TBI in
comparison with the sham group at 24 h post-TBI. The results
revealed that Dex administration markedly attenuated the brain
water content post-TBI. In addition, following Dex treatment for
TBI, the ∆NSS was significantly lower than that of non-treated rats
at 24 h post-TBI. A previous study demonstrated that Dex has been
confirmed to confer neuroprotection for rats subjected to
weight-drop contusion injury (41). However, the exact mechanisms
underlying this neuroprotective process remain undetermined.
The present study then investigated the effects of
Dex on neuronal apoptosis and HSP70 protein expression at 12, 24
and 72 h post-TBI. The results indicated that the administration of
Dex had the potential to attenuate the otherwise increased
expression of Bax and reduced expression of Bcl-2 levels, and to
enhance the expression of HSP70 in the injured hippocampus
following TBI. These results suggested that Dex may possess a
neuroprotective function and thus in turn, may be a potent
therapeutic agent for TBI treatment. The results also indicated
that the underlying mechanisms of this process may be associated
with the modulation of neuronal apoptosis. Therefore, Dex may serve
as a novel therapeutic target for the treatment of TBI.
In conclusion, the results of the present study
suggested that the administration of Dex post-TBI may improve
clinical outcomes by reducing brain edema and neurological
functional impairment via anti-apoptotic mechanisms involving the
modulation of apoptosis-associated proteins Bax, Bcl-2 and HSP70
expression levels. This may be an important mechanism through which
Dex provides an important neuroprotective effect following TBI.
Thus, administration of Dex may be considered as a candidate for
clinical trials investigating alternative treatments for patients
with TBI. For future study, potential cellular and molecular
mechanisms implicated in the neuroprotective effect of Dex
administration, as well as signal transduction pathways involved in
this process, should be investigated.
Acknowledgements
The present study was supported by a grant of
Natural Science Foundation of Hebei Province. The authors would
like to thank Junling Gao who provided technical help and writing
assistance, as well as Yanxia Tian who contributed certain reagents
and provided technical support.
Funding
The present study was supported by a grant from the
Natural Science Foundation of Hebei Province (grant nos.
H2012401071 and 2014105079).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
MZ made substantial contributions to conception,
design, acquisition of data, analysis and interpretation of data,
was responsible for writing and revision of the manuscript. JC
contributed to the conception, design, writing, and revision of the
manuscript. YF and HZ were responsible for acquisition of data, or
analysis and interpretation of data. XZ and KW carried out the
experiments. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
All experimental procedures were approved by the
Animal Ethics Committee of Hebei Medical University (Hebei,
China).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
TBI
|
traumatic brain injury
|
|
Dex
|
dexmedetomidine
|
|
HSP70
|
70 kDa heat shock proteins
|
|
SD
|
Sprague-Dawley
|
References
|
1
|
Cheng G, Fu L, Zhang HY, Wang YM, Zhang LM
and Zhang JN: The role of mitochondrial calcium uniporter in
neuroprotection in traumatic brain injury. Med Hypotheses.
80:115–117. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kumar A and Loane DJ: Neuroinflammation
after traumatic brain injury: Opportunities for therapeutic
intervention. Brain Behav Immun. 26:1191–1201. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Summers CR, Ivins B and Schwab KA:
Traumatic brain injury in the United States: An epidemiologic
overview. Mt Sinai J Med. 76:105–110. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Stein SC, Georgoff P, Meghan S, Mizra K
and Sonnad SS: 150 years of treating severe traumatic brain injury:
A systematic review of progress in mortality. J Neurotraum.
27:1343–1353. 2010. View Article : Google Scholar
|
|
5
|
Smania N, Avesani R, Roncari L, Ianes P,
Girardi P, Varalta V, Gambini MG, Fiaschi A and Gandolfi M: Factors
predicting functional and cognitive recovery following severe
traumatic, anoxic and cerebrovascular brain damage. J Head Trauma
Rehab. 28:131–140. 2013. View Article : Google Scholar
|
|
6
|
Weber JT: Altered calcium signaling
following traumatic brain injury. Front Pharmacol. 3:602012.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Harvey LA and Close JC: Traumatic brain
injury in older adults: Characteristics, causes and consequences.
Injury. 43:1821–1826. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mangat HS: Severe traumatic brain injury.
Continuum (Minneap Minn). 18:532–546. 2012.PubMed/NCBI
|
|
9
|
Zink BJ, Szmydynger-Chodobska J and
Chodobski A: Emerging concepts in the pathophysiology of traumatic
brain injury. Psychiatr Clin North Am. 33:741–756. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hart T, Brenner L, Clark AN, Bogner JA,
Novack TA, Chervoneva I, Nakase-Richardson R and Arango-Lasprilla
JC: Major and minor depression after traumatic brain injury. Arch
Phys Med Rehabi. 92:1211–1219. 2011. View Article : Google Scholar
|
|
11
|
Bye N, Carron S, Han X, Agyapomaa D, Ng
SY, Yan E, Rosenfeld JV and Morganti-Kossmann MC: Neurogenesis and
glial proliferation are stimulated following diffuse traumatic
brain injury in adult rats. J Neurosci Res. 89:986–1000. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Loane DJ and Faden AI: Neuroprotection for
traumatic brain injury: Translational challenges and emerging
therapeutic strategies. Trends Pharmacol Sci. 31:596–604. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Blennow K, Hardy J and Zetterberg H: The
neuropathology and neurobiology of traumatic brain injury. Neuron.
76:886–899. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xu X, Jiang R, Gong P, Liu Q, Chen Y, Hou
S, Yuan D, Shi J and Lan Q: Up-regulation of FOS-like antigen 1
contributes to neuronal apoptosis in the cortex of rat following
traumatic brain injury. Metab Brain Dis. 33:115–125. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sato A: Novel anticancer strategy
targeting switch mechanisms in two types of cell death: Necrosis
and apoptosis. Yakugaku Zasshi. 1315–1321. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Anderson MA, Huang D and Roberts A:
Targeting BCL2 for the treatment of lymphoid malignancies. Semin
Hematol. 51:219–227. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hartings JA, Bullock MR, Okonkwo DO,
Murray LS, Murray GD, Fabricius M, Maas AI, Woitzik J, Sakowitz O,
Mathern B, et al: Spreading depolarisations and outcome after
traumatic brain injury: A prospective observational study. Lancet
Neurol. 10:1058–1064. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Andriessen TM, Horn J, Franschman G, van
der Naalt J, Haitsma I, Jacobs B, Steyerberg EW and Vos PE:
Epidemiology, severity classification and outcome of moderate and
severe traumatic brain injury: A prospective multicenter study. J
Neurotraum. 28:2019–2031. 2011. View Article : Google Scholar
|
|
19
|
Zhang J, Jiang R, Liu L, Watkins T, Zhang
F and Dong JF: Traumatic brain injury-associated coagulopathy. J
Neurotraum. 29:2597–2605. 2012. View Article : Google Scholar
|
|
20
|
Wang Z, Kou D, Li Z, He Y, Yu W and Du H:
Effects of propofol-dexmedetomidine combination on ischemia
reperfusion-induced cerebral injury. NeuroRehabilitation.
35:825–834. 2014.PubMed/NCBI
|
|
21
|
Zhu YM, Wang CC, Chen L, Qian LB, Ma LL,
Yu J, Zhu MH, Wen CY, Yu LN and Yan M: Both PI3K/Akt and ERK1/2
pathways participate in the protection by dexmedetomidine against
transient focal cerebral ischemia/reperfusion injury in rats. Brain
Res. 1494:1–8. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Goyagi T and Tobe Y: Dexmedetomidine
improves the histological and neurological outcomes 48 h after
transient spinal ischemia in rats. Brain Res. 1566:24–30. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liao Z, Cao D, Han X, Liu C, Peng J, Zuo
Z, Wang F and Li Y: Both JNK and P38 MAPK pathways participate in
the protection by dexmedetomidine against isoflurane-induced
neuroapoptosis in the hippocampus of neonatal rats. Brain Res Bull.
107:69–78. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cui C, Cui Y, Gao J, Sun L, Wang Y, Wang
K, Li R, Tian Y, Song S and Cui J: Neuroprotective effect of
ceftriaxone in a rat model of traumatic brain injury. Neurol Sci.
35:695–700. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Marmarou A, Foda MA, van den Brink W,
Campbell J, Kita H and Demetriadou K: A new model of diffuse brain
injury in rats. Part I: Pathophysiology and biomechanics. J
Neurosurg. 80:291–300. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yang NL, Xu DM and Ming GF: Intervention
effect of dexmedetomidine on inflammatory response following
traumatic brain injury in rats. Pract Prev Med. 17:243–245.
2010.
|
|
27
|
Roof RL, Duvdevani R, Heyburn JW and Stein
DG: Progesterone rapidly decreases brain edema: Treatment delayed
up to 24 h is still effective. Exp Neurol. 138:246–251. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang X, Chen Y, Jenkins LW, Kochanek PM
and Clark RS: Bench-to-bedside review: Apoptosis/programmed cell
death triggered by traumatic brain injury. Crit Care. 9:66–75.
2005. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Stoica BA and Faden AI: Cell death
mechanisms and modulation in traumatic brain injury.
Neurotherapeutics. 7:3–12. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Fanselow MS and Dong HW: Are the dorsal
and ventral hippocampus functionally distinct structures? Neuron.
65:7–19. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Shamas-Din A, Brahmbhatt H, Leber B and
Andrews DW: BH3-only proteins: Orchestrators of apoptosis. Biochim
Biophys Acta. 1813:508–520. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Feder ME and Hofmann GE: Heat-shock
proteins, molecular chaperones and the stress response:
Evolutionary and ecological physiology. Annu Rev Physiol.
61:243–282. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Xiong B, Shi QQ and Miao CH:
Dexmedetomidine renders a brain protection on hippocampal formation
through inhibition of nNOS-NO signalling in endotoxin-induced shock
rats. Brain Inj. 28:1003–1008. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wang X, Ji J, Fen L and Wang A: Effects of
dexmedetomidine on cerebral blood flow in critically ill patients
with or without traumatic brain injury: A prospective controlled
trial. Brain Inj. 27:1617–1622. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
James ML, Olson DM and Graffagnino C: A
pilot study of cerebral and haemodynamic physiological changes
during sedation with dexmedetomidine or propofol in patients with
acute brain injury. Anaesth Intens Care. 40:949–957. 2012.
|
|
36
|
Nasrallah N A, Thomas M, Kuehl S and Diab
K: The use of dexmedetomidine for longer than 48 h with evaluation
of its secondary outcome in patients with liver disease. Chest.
150:228A2016. View Article : Google Scholar
|
|
37
|
Guler L, Bozkirli F, Bedirli N, Unal Y,
Guler A, Oztas Y, Balta S, Cakar M, Demirkol S, Arslan Z and Unlu
M: Comparison of the effects of dexmedetomidine vs. ketamine in
cardiac ischemia/reperfusion injury in rats-preliminary study. Adv
Clin Exp Med. 23:683–689. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Si YN, Bao HG, Xu L, Wang XL, Shen Y, Wang
JS and Yang XB: Dexmedetomidine protects against
ischemia/reperfusion injury in rat kidney. Eur Rev Med Pharmaco
Sci. 18:1843–1851. 2014.
|
|
39
|
Jiang L, Li L, Shen J, Qi Z and Guo L:
Effect of dexmedetomidine on lung ischemiareperfusion injury. Mol
Med Rep. 9:419–426. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Cai Y, Xu H, Yan J, Zhang L and Lu Y:
Molecular targets and mechanism of action of dexmedetomidine in
treatment of ischemia/reperfusion injury. Mol Med Rep. 9:1542–1550.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Shen M, Wang S, Wen X, Han XR, Wang YJ,
Zhou XM, Zhang MH, Wu DM, Lu J and Zheng YL: Dexmedetomidine exerts
neuroprotective effect via the activation of the PI3K/Akt/mTOR
signaling pathway in rats with traumatic brain injury. Biomed
Pharmacother. 95:885–893. 2017. View Article : Google Scholar : PubMed/NCBI
|