Introduction
Hypoxia results in pathophysiological damage to
various cells and tissues. Cells possess a powerful regulatory
system involving hypoxia-inducible factors (HIFs) that respond to
hypoxic stress (1–3). HIF-1α regulates metabolic adaptation
to hypoxia and activates multiple target genes, including vascular
endothelial growth factor, erythropoietin and nuclear factor-κB
under hypoxic conditions (4).
Furthermore, HIF-1α has been implicated as a co-regulator of
autophagy activation and also participates in apoptosis (5).
Autophagy protects cells from various damaging
factors, including hypoxia or starvation and maintains
intracellular stability (6). It
has been demonstrated that autophagy is closely associated with
apoptosis under hypoxic condition (5). Apoptosis is a type of programmed cell
death which is triggered by intrinsic or extrinsic signals
(7). Hypoxia and cobalt (II)
chloride (CoCl2) treatment activate autophagy through the target
genes induced by HIF, such as mechanistic target of rapamycin
(2) and correlate with the
expression of certain pro-apoptotic factors, including caspase-9
and caspase-3 (8,9). These results suggest that autophagy
and apoptosis often occur in parallel following CoCl2 treatment,
with autophagy typically occurring prior to apoptosis. The
important role of autophagy may be associated with the regulation
of apoptosis. It has been reported that autophagy attenuates
cellular injury by inhibiting the induction of apoptosis (10).
Autophagy and apoptosis may serve pivotal roles in
neurodegenerative disease (11). A
previous report revealed that autophagy serves a dual role, by
exhibiting a protective role for cell survival, but autophagy may
also promote apoptosis associated with various lung diseases,
including chronic obstructive pulmonary disease, idiopathic
pulmonary fibrosis and sepsis (12). However, the molecular mechanism of
crosstalk between autophagy and apoptosis remains unclear. In the
present study, the association between autophagy and apoptosis
induced by long-term CoCl2 treatment in RLE-6TN cells was
investigated. The results suggested that autophagy provided a
survival strategy for RLE-6TN cells via apoptosis inhibition. The
present study may aid in elucidating the underlying molecular
mechanism of autophagy and apoptosis interaction in a hypoxic
environment, which may contribute to the development of novel
treatments for the therapy of lung diseases.
Materials and methods
Reagents and antibodies
CoCl2, 2,7-dichlorodihyfluorescein diacetate
(DCFH-DA), Tris, glycine, Tween-20, SDS, acridine orange (AO),
propidium iodide (PI), anti-microtubule associated proteins 1A/1B
light chain 3B (LC3I/II; cat. no. L7543) were purchased from
Sigma-Aldrich (Merck KGaA, Darmstadt, Germany). Trypsin-EDTA was
obtained from Thermo Fisher Scientific, Inc. (Waltham, MA, USA).
Trypsin solution without EDTA was purchased from Beyotime Institute
of Biotechnology (Hangzhou, China). The Annexin V-fluorescein
isothiocyanate (FITC)/PI apoptosis detection kit was purchased from
BD Biosciences (San Jose, CA, USA). Primary antibodies were
purchased from CST Biological Reagents, Co., Ltd. (Shanghai, China)
or Abcam (Cambridge, UK). Secondary antibodies were purchased from
OriGene Technologies, Inc. (Beijing, China). All other chemicals
were obtained from Sigma-Aldrich (Merck KGaA).
Radioimmunoprecipitation assay (RIPA) lysis buffer and
phenylmethylsulfonyl fluoride (PMSF) were purchased from Beijing
Solarbio Science & Technology Co., Ltd. (Beijing, China).
Cell culture and treatments
RLE-6TN cell line was provided by Binzhou Medical
University (Yantai, China). Cells were cultured in RPIM-1640 medium
(Hyclone; GE Healthcare Life Sciences, Logan, UT, USA) supplemented
with 10% fetal bovine serum (Hyclone; GE Healthcare Life Sciences)
and maintained at 37°C in a humidified atmosphere with 5% CO2.
Hypoxia was established with CoCl2 (100 µM) as previously described
(13,14). The concentration of the autophagy
inhibitor 3-methyladenine (3-MA; 1.5 mM) was determined by creating
a dose-response curve in a preliminary experiment (data not shown).
RLE-6TN cells were plated in 10-cm diameter plastic dish until
70–80% confluence was attained. Cells were treated with or without
CoCl2 and/or 3-MA for 1, 3, 5 and 7 days at 37°C.
Transmission electron microscopy
(TEM)
For ultrastructural observation, cells were fixed
with 2.5% paraformaldehyde and 2% glutaraldehyde in 0.1 M phosphate
buffer (pH 7.4) for 2 h at 4°C. Cells were subsequently post-fixed
at 4°C with 1% osmium tetroxide for 1 h and dehydration was
performed using 50, 70, 80, 95 and 100 ethanol, and 100% acetone
sequentially every 15 min and embedded in Epon 812 resin at 37°C
for 12 h, at 45°C for 12 h and 60°C for 36 h. Following this, cells
were cut into ultrathin sections, imposed on a copper grid and
stained with uranyl acetate for 30 min and led citrate for 10 min
at room temperature. Observations and photographs were obtained
using a JEOL JEM 1,011 microscope with a MORADA camera (JEOL Ltd.,
Tokyo, Japan).
Reactive oxygen species (ROS)
assay
DCFH-DA was used to detect ROS levels. DCFH-DA
converts into the highly fluorescent DCF upon oxidation by ROS.
Cultured RLE-6TN cells were plated at a density of 1×105 cells/ml
in 6-well plates and incubated with RPIM-1640 medium supplemented
with 10% fetal bovine serum overnight at 37°C. DCFH-DA working
solution was added directly to the medium to reach 20 µM and
incubated at 37°C for 30 min. Cells were subsequently washed with
PBS three times and visualized with a fluorescence microscope
(excitation wavelength, 488 nm; magnification, ×200).
AO/PI double staining
RLE-6TN cells (80–90% confluence) were plated in 24
well-plates on a glass slide at 37°C for 24 h. Following
incubation, the cells in control group and treated RLE-6TN cells
were harvested and washed twice with PBS. Glass slides were
subsequently stained with 10 µg/ml of AO/PI for 5 min in the dark
at 37°C, followed by washing with PBS three times. Stained cells
were observed under a fluorescence microscope (excitation
wavelength, 488 nm; magnification, ×200).
Annexin V/PI apoptosis assay
To analyze apoptosis, the FITC Annexin V Apoptosis
Detection kit (BD Biosciences) was used according to the
manufacturer's protocols. Treated cells (1×106) were washed with
binding buffer of the aforementioned kit and resuspended in 100 µl
binding buffer containing 5 µl FITC and 5 µl PI. After 15 min of
incubation at room temperature in the dark, 400 µl binding buffer
was added and the apoptotic rate of cells was analyzed with a flow
cytometer (FACSDiva Software 6.0, BD Biosciences).
Western blot analysis
RLE-6TN cells were lysed in ice-cold RIPA lysis
buffer (1 ml RIPA; 10 µl PMSF) for 30 min. Protein concentration
was determined with a bicinchoninic acid protein assay kit and
proteins (50 µg/lane) were separated by 12 and 15% SDS-PAGE and
transferred to polyvinylidene fluoride membranes. Membranes were
blocked at room temperature for 2 h with 5% skimmed milk in
Tris-buffered saline with 0.1% Tween-20 (cat. no. ST825; Beyotime
Institute of Biotechnology) and subsequently probed with the
following antibodies at 4°C overnight: Anti-β-actin antibody
(1:2,000; cat. no. bs-0061R; BIOSS; Beijing, China), Anti-HIF-1α
(1:1,000; cat. no. 14179; CST Biological Reagents Co., Ltd.),
anti-LC3I/II (1:1,000; cat. no. L7543; Sigma-Aldrich; Merck KGaA),
anti-cleaved caspase-9 (1:300; cat. no. ab2325; Abcam),
anti-caspase-8 (1:1,000; cat. no. ab25901; Abcam), anti-cleaved
caspase-3 (1:1,000; cat. no. 9664; CST Biological Reagents Co.,
Ltd.). Following this, membranes were incubated with horseradish
peroxidase-conjugated goat anti-rabbit IgG (1:5,000; ZB2301;
OriGene Technologies, Inc.) antibody for 1 h at room temperature.
Proteins were visualized with an enhanced chemiluminescence
substrate kit (Clinx Science Instruments Co., Ltd., Shanghai,
China) and standard X-ray film development (ChemiScope 3,300 mini;
Clinx Science Instruments Co., Ltd.). Results were quantified with
ImageJ software v1.48 (National Institutes of Health, Bethesda, MD,
USA) and processed using Adobe Photoshop CS5 (Adobe Systems, Inc.,
San Jose, CA, USA).
Statistical analysis
All experiments were performed at least three times.
The data were expressed as the mean ± standard deviation.
Statistical analyses were performed using SPSS 17.0 (SPSS, Inc.,
Chicago, IL, USA). Data were analyzed by one-way analysis of
variance followed by Fisher's least significant difference.
P<0.05 was considered to indicate a statistically significant
difference.
Results
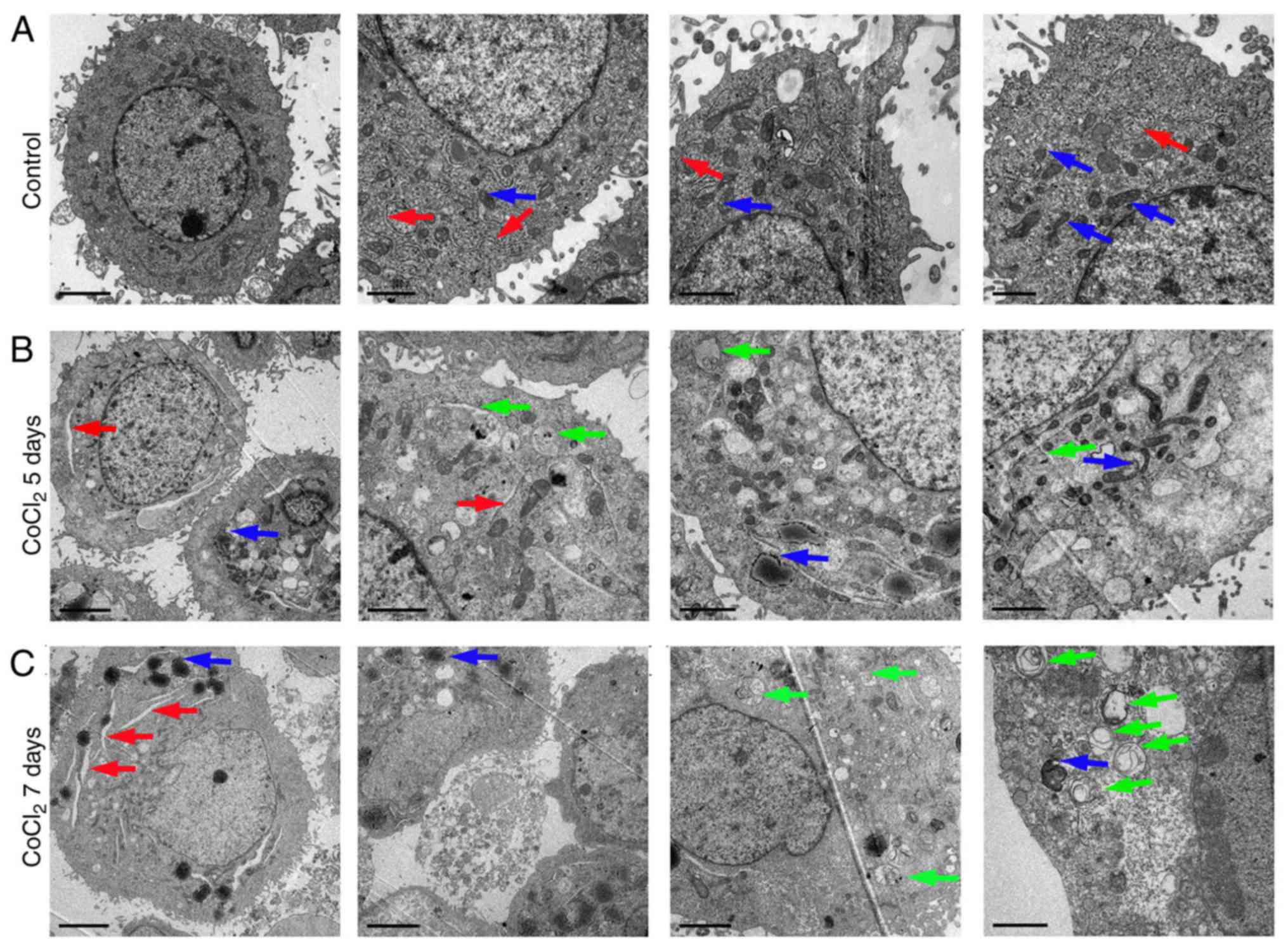
Hypoxia induces organelle impairment
and autophagosome generation
Previous studies have demonstrated that cancer cells
adapt to hypoxia or nutrient deprivation through autophagy
activation (15,16). In the present study, TEM was used
to observe the ultrastructure following CoCl2 treatment in RLE-6TN
cells (Fig. 1). Compared with
untreated cells (Fig. 1A),
numerous autophagosomes consisting of double membranes were
observed in the CoCl2-treated RLE-6TN cells on days 5 and 7
(Fig. 1B and C, respectively). No
notable autophagosomes were observed after 1 and 3 days following
treatment with CoCl2 (data not shown). Cytoplasmic material and/or
membrane vesicles were encapsulated in the autophagosomes.
Furthermore, swollen endoplasmic reticuli and damaged mitochondria
were also observed following CoCl2 treatment (Fig. 1B and C). These results indicated
that 100 µm CoCl2 treatment resulted in severe organelle impairment
and induced autophagy generation.
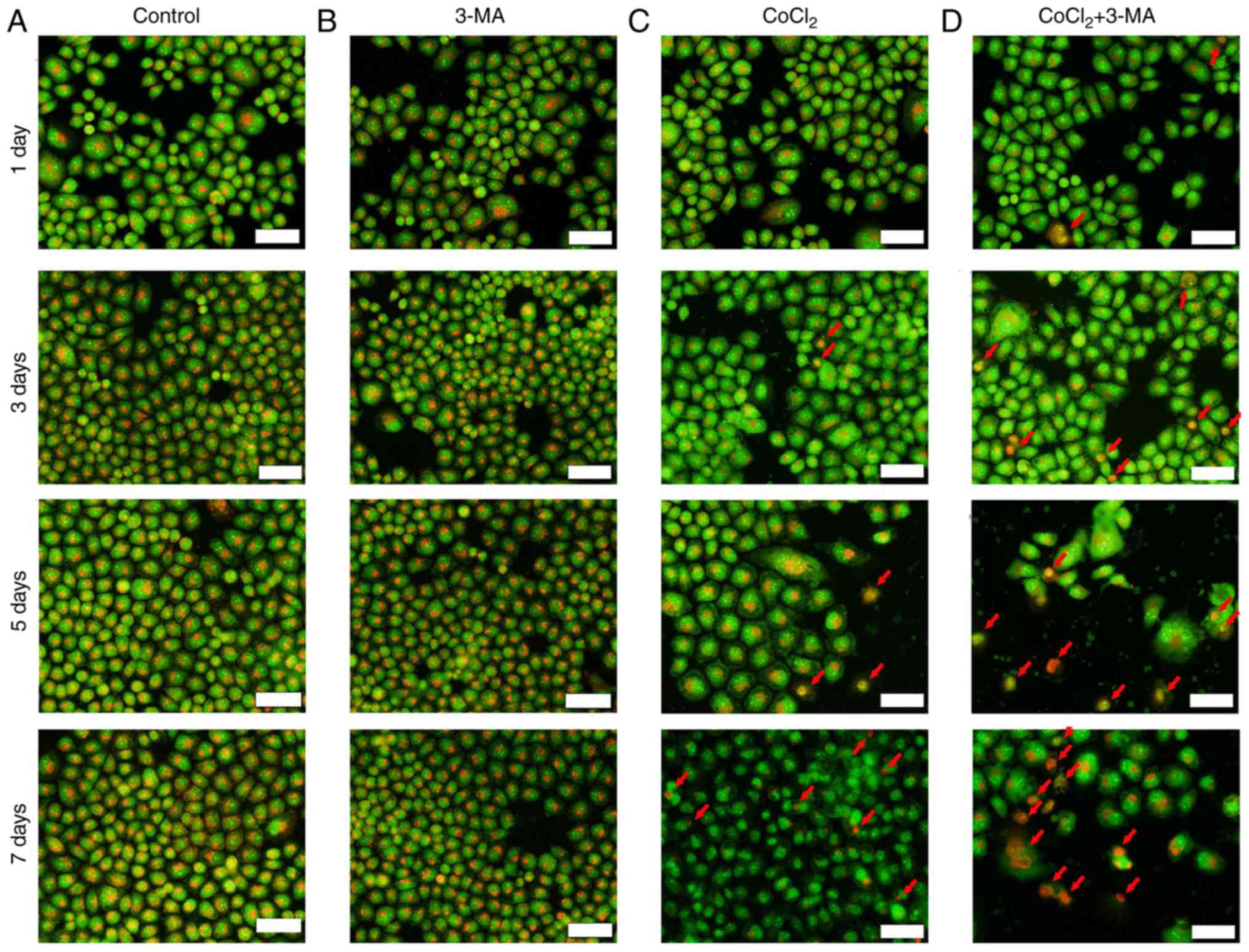
3-MA enhances apoptosis in hypoxic
conditions
Apoptosis is characterized by distinct morphological
features and energy-dependent biochemical mechanisms. Cell
pyknosis, cell shrinkage and chromatin condensation typically occur
in early apoptosis (17,18). Budding in late apoptosis is a
result of extensive blebbing of the plasma membrane to form
apoptotic bodies containing tightly packed organelles (19). In the present study, fluorescence
microscopy was used to detect the morphological changes in RLE-6TN
cells following CoCl2 and/or 3-MA treatment for 1, 3, 5 and 7 days
(Fig. 2A-D). CoCl2 treatment
resulted in a reddish-orange color in nucleus due to the binding of
PI to denatured DNA. Lysosomes in the cytoplasm were stained orange
by PI in Fig. 2. This abundance of
apoptotic cells, as indicated by arrows, was more prominent as the
incubation time increased. 3-MA markedly increased the number
reddish-orange stained nuclei in 100 µm CoCl2-treated cells
(Fig. 2D). These results indicated
that autophagy was associated with apoptosis in RLE-6TN cells in
hypoxic conditions and that autophagy may have a protective role in
a hypoxic environment.
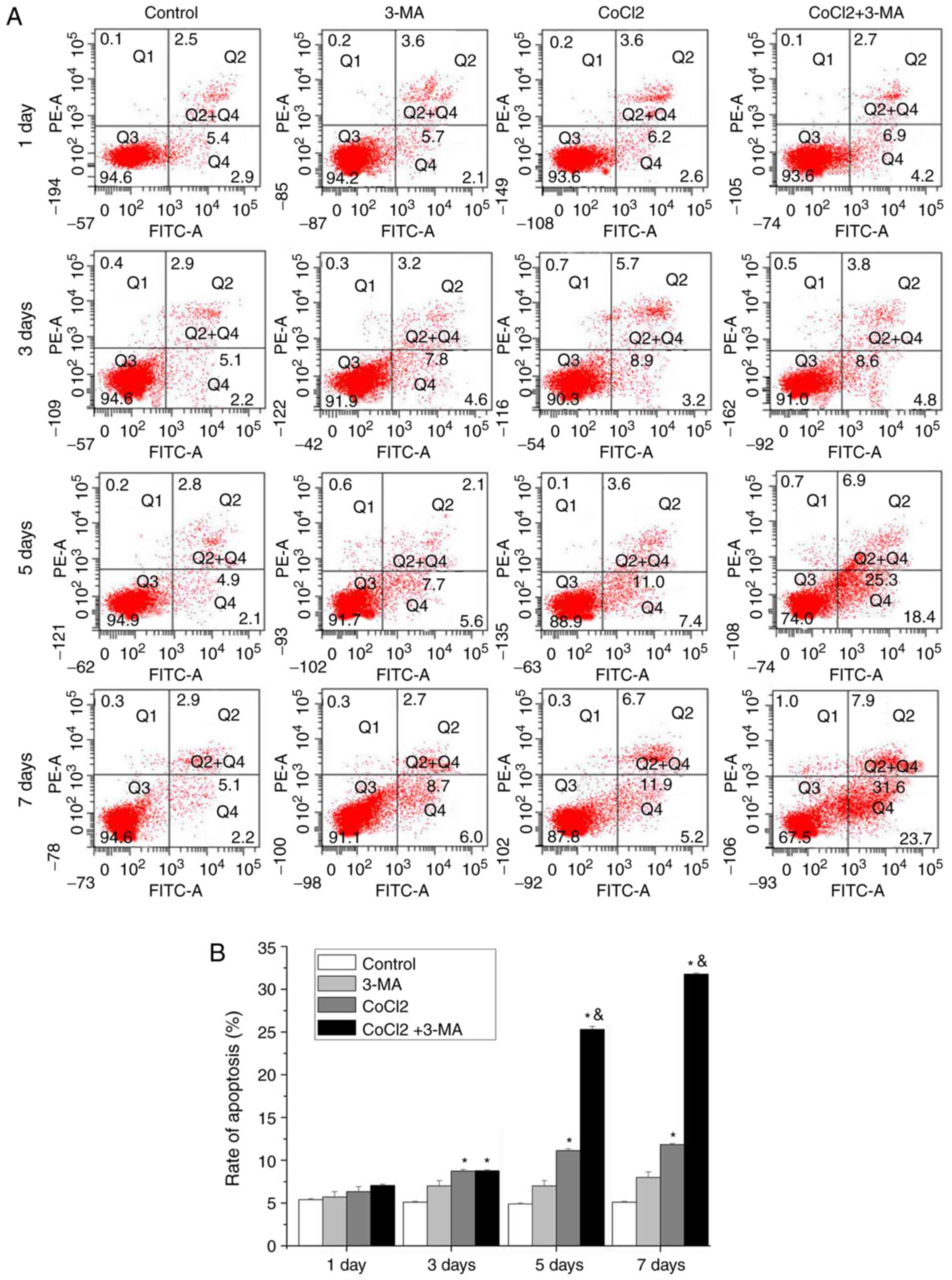
The apoptotic effect induced by CoCl2 was further
confirmed by flow cytometry (Fig.
3A). The results demonstrated that CoCl2 treatment
significantly increased in the percentage of cells in upper right
and lower right quadrants (early and late apoptotic cells,
respectively) after 3 days of treatment. 3-MA significantly
increased CoCl2-induced apoptosis in RLE-6TN cells at days 5 and 7
(P<0.05; Fig. 3B).
Taken together, these results suggested that the
inhibition of autophagy strongly accelerated the process of
apoptosis.
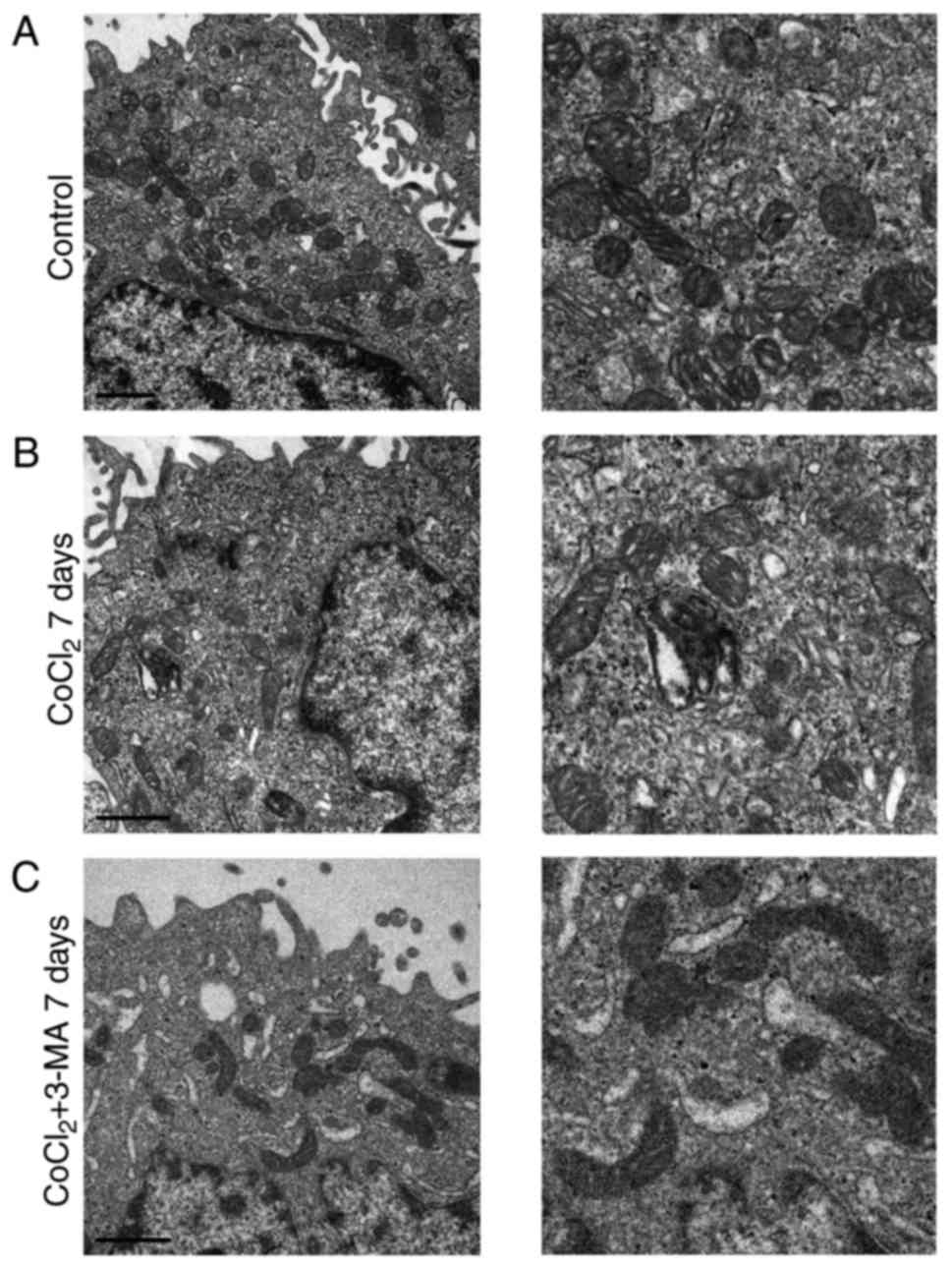
Inhibiting autophagy increases
mitochondrial damage
Mitochondria have a double-membrane structure and
are key organelles for the production of energy (20). In addition, mitochondria regulate
cellular redox signaling pathways and programmed cell death
(20). In order to explore the
effect of 3-MA in CoCl2-treated mitochondria, TEM was used to
examine the mitochondrial ultrastructure. As presented in Fig. 4, swollen mitochondria were observed
following CoCl2 treatment at day 7, compared with the control.
Mitochondrial damage was not notably observed on day 3; however,
more notable damage was reported on day 5 (data not shown).
Notably, more marked mitochondrial damage was observed in the CoCl2
+ 3-MA group, including severely swollen mitochondria,
double-membrane destruction and loss of normal morphology,
suggesting that the inhibition of autophagy increased the extent of
mitochondrial damage in RLE-6TN cells under hypoxic conditions.
Inhibiting autophagy increases the
production of ROS
ROS are critical regulators in various cellular
processes, including autophagy and apoptosis (21,22).
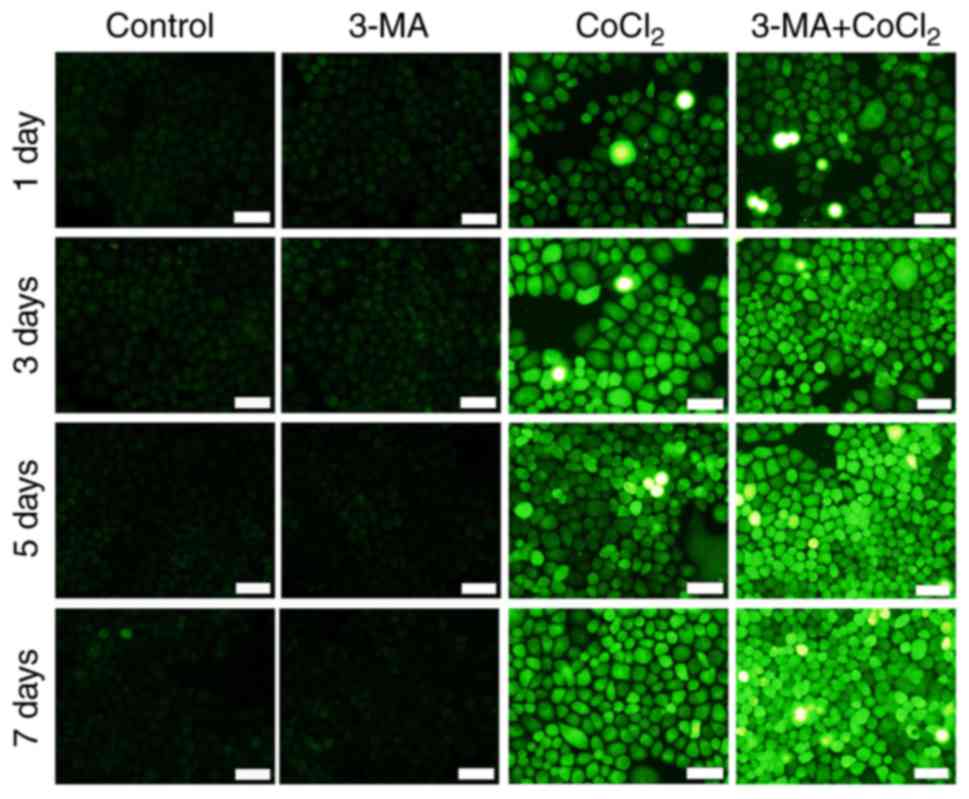
Mitochondria are a major source of ROS in cells (23). A fluorescent probe, DCFH-DA, was
used to detect the generation of ROS. As presented in Fig. 5, DCFH-DA fluorescence was markedly
increased following CoCl2 and 3-MA treatment, compared with CoCl2
treatment alone. The results revealed that the inhibition of
autophagy with 3-MA increased ROS generation and increased the rate
of apoptosis in RLE-6TN cells. These results indicated that
autophagy may prevent cell impairment in a hypoxic environment.
3-MA upregulates the expression of
caspases in hypoxic conditions
Autophagy and apoptosis are two important catabolic
processes (24) and the
relationship between them remains unclear. In order to explore the
complex crosstalk between autophagy and apoptosis, the expression
of several proteins associated with these processes in response to
hypoxia was investigated. As presented in Fig. 6, HIF-1α expression was
significantly upregulated on days 2, 3, 5 and 7 after treatment
with CoCl2. This data indicated that the hypoxia model was
successfully conducted in our study. In addition,
cleaved-caspase-9, cleaved-caspase-8, cleaved-caspase-3, LC3II/I
exhibited increasing trend; the expression levels were
significantly higher on days 5 and 7 compared with in the control.
Notably, 3 days following CoCl2 treatment, cleaved-caspase-8 also
significantly increased. Following 3-MA treatment (Fig. 7), the expression levels of
LC3II/LCI, key proteins in autophagy, did not exhibit significant
alterations under hypoxic conditions compared with in the control.
By contrast, expression levels of cleaved caspase-9 and cleaved
caspase-3 were significantly increased, particularly on days 5 and
7. Compared with cleaved-caspase-9 and cleaved-caspase-3, the
expression level of cleaved caspase-8 appeared to be upregulated to
a lesser degree. These results indicated that autophagy and
apoptosis occurred in RLE-6TN cells under hypoxic conditions.
Inhibition of autophagy may have accelerated apoptosis,
predominantly through caspase-9-mediated intrinsic pathways in
RLE-6TN cells in a chronic hypoxic environment.
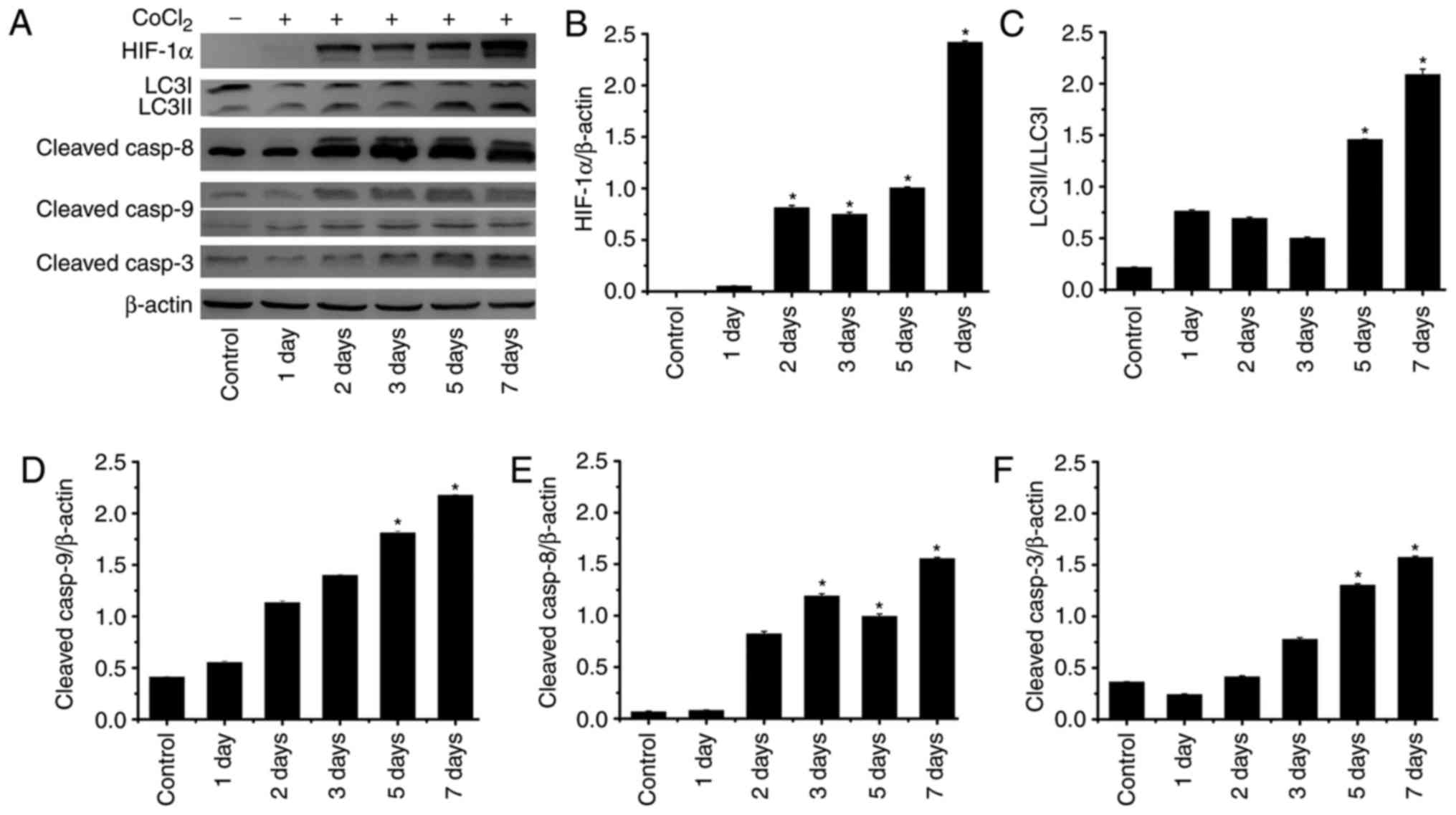 | Figure 6.Expression levels of proteins
associated with autophagy and apoptosis in RLE-6TN cells induced by
CoCl2. (A) Western blot analysis of HIF-1α, LC3I/II, cleaved
caspase-8, cleaved caspase-9 (the upper band=37 kDa, the lower
band=17 kDa) and cleaved caspase-3 expression in RLE-6TN cells of
each group. Quantification of (B) HIF-1α, (C) LC3I/II, (D) cleaved
caspase-8, (E) cleaved caspase-9 and (F) cleaved caspase-3 by
densitometry. *P<0.05 vs. control. HIF-1α, hypoxia-inducible
factor 1α; LCI/II, microtubule associated proteins 1A/1B light
chain 3B; casp, caspase; CoCl2, cobalt (II) chloride. |
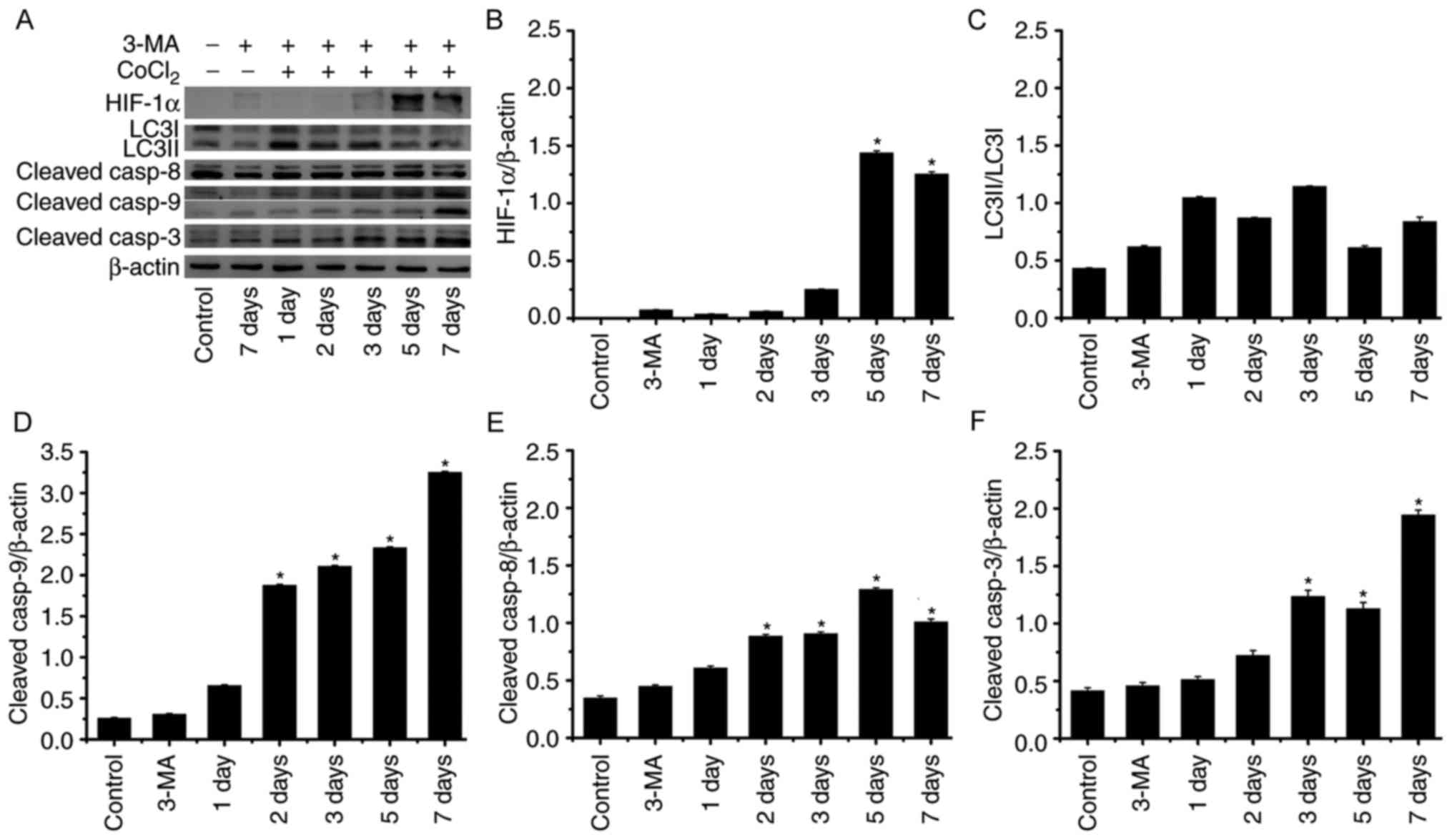 | Figure 7.Expression levels of proteins
associated with autophagy and apoptosis in RLE-6TN cells induced by
CoCl2 and treated with 3-MA. (A) Western blot analysis of HIF-1α,
LC3I/II, cleaved caspase-8, cleaved caspase-9 (the upper band=37
kDa, the below band=17 kDa) and cleaved caspase-3 expression in
RLE-6TN cells of each group. Quantification of (B) HIF-1α, (C)
LC3I/II, (D) cleaved caspase-8, (E) cleaved caspase-9 and (F)
cleaved caspase-3 by densitometry. *P<0.05 vs. control. HIF-1α,
hypoxia-inducible factor 1α; LCI/II, microtubule associated
proteins 1A/1B light chain 3B; asp, caspase; CoCl2, cobalt (II)
chloride; 3-MA, 3-methyladenine. |
Discussion
Cellular stress stimuli, such as hypoxia, may induce
cells to exhibit their dual role (25). Cells may activate cytoprotective
pathways, such as autophagy, to favor survival until the stress is
resolved. Conversely, cells also may undergo programmed cell death.
CoCl2 has been extensively used as a reagent to establish hypoxia
in vitro (26–29). In the present study, autophagy and
apoptosis occurred in RLE-6TN cells following exposure to CoCl2.
The association between these two processes is complex.
Autophagy serves an essential role in cellular
catabolism (30). Autophagy is a
process which involves the engulfment of cytoplasmic materials and
intracellular organelles within autophagosomes, which are
subsequently delivered to lysosomes (31). The materials within
autophagolysosomes provide energy in order to maintain cell
metabolism. Cell survival may be impeded if autophagy is
interrupted (32). By contrast,
excessive autophagy may lead to a type of cell death known as
autophagic apoptosis (33).
Apoptosis is a highly controlled genetic program of cell death,
which also serves a critical role in determining cell fate.
Apoptosis is triggered by multiple signaling pathways, including
the extrinsic and intrinsic signaling pathways (34). Zhang et al (35) demonstrated that the inhibition of
autophagy with 3-MA increases apoptosis in a mouse model of middle
cerebral artery occlusion (MCAO). In addition, hypoxia-induced
autophagy decreases the production of cytochrome c and the
activation of caspase-mediated apoptotic pathways (35). Therefore, the ischemia-induced
neuronal injury is depressed in an MCAO model following treatment
with 3-MA. Yan et al (36)
demonstrated that chronic ischemia induces autophagy in the heart,
and the areas of damaged heart have fewer apoptotic cells due to an
increase in autophagy. By contrast, Cheng et al (14) revealed that pre-treatment of human
malignant glioma U87-MG cells in hypoxic conditions with autophagy
inhibitors suppresses cell apoptosis and caspase-3 activation.
These studies were performed in different experimental models and
suggest that there is a complex interaction between autophagy and
apoptosis. Examining the potential underlying mechanisms of
interaction in various environments of cellular stress will aid in
the understanding of the relationship between autophagy and
apoptosis.
Evidence suggests that mitochondria are the main
intracellular source of ROS in cells (37,38).
ROS participate in various cellular processes, including autophagy
and apoptosis (39). Accumulation
of ROS impairs mitochondrial function (40). In response to diverse pathological
conditions, including oxidative stress, autophagy serves a dual
role by exhibiting protective and harmful effects, which promote
cell survival and apoptosis, respectively (12,41).
Dewaele et al (42)
demonstrated that ROS-dependent accumulation of LC3-PE facilitates
autophagosome formation. Previous studies have demonstrated that
autophagy may inhibit apoptosis in hypoxic conditions through ROS
removal and inhibition of caspase activation (43,44).
Chien et al (45) revealed
that following ischemia/reperfusion in the kidney, autophagy serves
a central role in ROS clearance and prevents apoptosis. The results
of the present study were consistent with this finding: An increase
in mitochondrial damage and dysfunction was observed following
autophagy inhibition with 3-MA, resulting in increased ROS
production. In addition, the expression levels of
apoptosis-associated proteins were increased following autophagy
inhibition, including caspase-9, caspase-8 and caspase-3.
Furthermore, the number apoptotic cells increased. It is well
established that the intrinsic apoptotic pathway is predominantly
dependent on caspase-9, and the extrinsic apoptotic pathway is
predominantly mediated by caspase-8 (14). Notably, the expression level of
caspase-9 may have increased faster and to a greater degree
compared with caspase-8 following 3-MA treatment. Therefore, the
results of the current study demonstrated that 3-MA treatment in
hypoxic conditions increased the production of ROS and the
apoptotic rate, primarily through the caspase-9-dependent intrinsic
apoptotic pathway. Furthermore, these results suggested that
autophagy may serve a protective role in the early stages of lung
damage in a hypoxic environment.
In conclusion, autophagy served a protective role in
RLE-6TN cells, via inhibition of ROS production and prevention of
apoptosis in hypoxic conditions. The present study only
investigated the effects of one inhibitor in one cell line.
Therefore, further research is required to provide further
evidence, in order to explore the potential molecular mechanisms
between hypoxia-induced autophagy and apoptosis. This may provide
new insights into pulmonary disease surveillance, diagnosis and
treatment.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Natural
Science Foundation of Shandong Province (grant no. ZR2014HL005),
the Scientific Research Foundation of Fang Han Team, the National
Natural Science Foundation of Youth Fund (grant no. 81600069), the
Natural Science Foundation of Shandong Province (grant nos.
ZR2013HL005 and ZR2013CL003) and the Scientific Research Foundation
of Binzhou Medical University (grant no. BY2013KYOD12).
Availability of data and materials
Not applicable.
Author's contributions
YY made substantial contributions to the conception
and design of the present study. WL conducted the AO-PI staining,
western blotting, ROS detection and drafted the manuscript. LR
conducted western blotting, analyzed the data and critically
revised the manuscript for important intellectual content. CY
conducted TEM. DL and XH made substantial contributions to western
blotting. YS conducted flow cytometry. CL and FH analyzed the data
and critically revised the manuscript for important intellectual
content. FH revised the manuscript and gave final approval of the
version to be published.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Jeong JK, Gurunathan S, Kang MH, Han JW,
Das J, Choi YJ, Kwon DN, Cho SG, Park C, Seo HG, et al:
Hypoxia-mediated autophagic flux inhibits silver
nanoparticle-triggered apoptosis in human lung cancer cells. Sci
Rep. 6:216882016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Semenza GL: HIF-1, O(2), and the 3 PHDs:
How animal cells signal hypoxia to the nucleus. Cell. 107:1–3.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Huang LE and Bunn HF: Hypoxia-inducible
factor and its biomedical relevance. J Biol Chem. 278:19575–19578.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Haase VH: The VHL tumor suppressor: Master
regulator of HIF. Curr Pharm Des. 15:3895–3903. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bellot G, Garcia-Medina R, Gounon P,
Chiche J, Roux D, Pouysségur J and Mazure NM: Hypoxia-induced
autophagy is mediated through hypoxia-inducible factor induction of
BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol.
29:2570–2581. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ozpolat B and Benbrook DM: Targeting
autophagy in cancer management-strategies and developments. Cancer
Manag Res. 7:291–299. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gordy C and He YW: The crosstalk between
autophagy and apoptosis: Where does this lead? Protein Cell.
3:17–27. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Volm M and Koomägi R: Hypoxia-inducible
factor (HIF-1) and its relationship to apoptosis and proliferation
in lung cancer. Anticancer Res. 20:1527–1533. 2000.PubMed/NCBI
|
|
9
|
Piret J, Lecocl C, Toffoli S, Ninane N,
Raes M and Michiels C: Hypoxia and CoCl2 protect HepG2 cells
against serum deprivation- and t-BHP-induced apoptosis: A possible
anti-apoptotic role for HIF-1. Exp Cell Res. 295:340–349. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pan T, Rawal P, Wu Y, Xie W, Jankovic J
and Le W: Rapamycin protects against rotenone-induced apoptosis
through autophagy induction. Neuroscience. 164:541–551. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ghavami S, Shojaei S, Yeganeh B, Ande SR,
Jangamreddy JR, Mehrpour M, Christoffersson J, Chaabane W, Moghadam
AR, Kashani HH, et al: Autophagy and apoptosis dysfunction in
neurodegenerative disorders. Prog Neurobiol. 112:24–49. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li T, Jiao YR, Wang LH, Zhou YH and Yao
HC: Autophagy in myocardial ischemia reperfusion injury: Friend or
foe? Int J Cardiol. 239:102017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang G, Hazra TK, Mitra S, Lee HM and
Englander EW: Mitochondrial DNA damage and a hypoxic response are
induced by CoCl(2) in rat neuronal PC12 cells. Nucleic Acids Res.
28:2135–2140. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cheng BC, Chen JT, Yang ST, Chio CC, Liu
SH and Chen RM: Cobalt chloride treatment induces autophagic
apoptosis in human glioma cells via a p53-dependent pathway. Int J
Oncol. 50:964–974. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pouysségur J, Dayan F and Mazure NM:
Hypoxia signalling in cancer and approaches to enforce tumour
regression. Nature. 441:437–443. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kim J and Klionsky DJ: Autophagy,
cytoplasm-to-vacuole targeting pathway, and pexophagy in yeast and
mammalian cells. Annu Rev Biochem. 69:303–342. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Häcker G: The morphology of apoptosis.
Cell Tissue Res. 301:5–17. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ovadje P, Chatterjee S, Griffin C, Tran C,
Hamm C and Pandey S: Selective induction of apoptosis through
activation of caspase-8 in human leukemia cells (Jurkat) by
dandelion root extract. J Ethnopharmacol. 133:86–91. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Rajagopalan V and Yusuf AH: Hannun:
Sphingolipid metabolism and signaling as a target for cancer
treatmentCell death signaling in cancer biology and treatment.
Johnson DE: Humana Press; New York: pp. 205–229. 2013, View Article : Google Scholar
|
|
20
|
Heo JM and Rutter J: Ubiquitin-dependent
mitochondrial protein degradation. Int J Biochem Cell Biol.
43:1422–1426. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Simon HU, Haj-Yehia A and Levi-Schaffer F:
Role of reactive oxygen species (ROS) in apoptosis induction.
Apoptosis. 5:415–418. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Scherz-Shouval R and Elazar Z: Regulation
of autophagy by ROS: Physiology and pathology. Trends Biochem Sci.
36:30–38. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hasanain M, Bhattacharjee A, Pandey P,
Ashraf R, Singh N, Sharma S, Vishwakarma AL, Datta D, Mitra K and
Sarkar J: α-Solanine induces ROS-mediated autophagy through
activation of endoplasmic reticulum stress and inhibition of
Akt/mTOR pathway. Cell Death Dis. 6:e18602015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wu H, Che X, Zheng Q, Wu A, Pan K, Shao A,
Wu Q, Zhang J and Hong Y: Caspases: A molecular switch node in the
crosstalk between autophagy and apoptosis. Int J Biol Sci.
10:1072–1083. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Rubinstein AD and Kimchi A: Life in the
balance-a mechanistic view of the crosstalk between autophagy and
apoptosis. J Cell Sci. 125:5259–5268. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Merelli A, Caltana L, Girimonti P, Ramos
AJ, Lazarowski A and Brusco A: Recovery of motor spontaneous
activity after intranasal delivery of human recombinant
erythropoietin in a focal brain hypoxia model induced by CoCl2 in
rats. Neurotox Res. 20:182–192. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Badr GA, Zhang JZ, Tang J, Kern TS and
Ismail-Beigi F: Glut1 and glut3 expression, but not capillary
density, is increased by cobalt chloride in rat cerebrum and
retina. Brain Res Mol Brain Res. 64:24–33. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zou W, Yan M, Xu W, Huo H, Sun L, Zheng Z
and Liu X: Cobalt chloride induces PC12 cells apoptosis through
reactive oxygen species and accompanied by AP-1 activation. J
Neurosci Res. 64:646–653. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhu X, Zhou W, Cui Y, Zhu L, Li J, Feng X,
Shao B, Qi H, Zheng J, Wang H and Chen H: Pilocarpine protects
cobalt chloride-induced apoptosis of RGC-5 cells: Involvement of
muscarinic receptors and HIF-1 alpha pathway. Cell Mol Neurobiol.
30:427–435. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ouyang L, Shi Z, Zhao S, Wang FT, Zhou TT,
Liu B and Bao JK: Programmed cell death pathways in cancer: A
review of apoptosis, autophagy and programmed necrosis. Cell
Prolif. 45:487–498. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Levine B and Kroemer G: Autophagy in the
pathogenesis of disease. Cell. 132:27–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Boya P, González-Polo RA, Casares N,
Perfettini JL, Dessen P, Larochette N, Métivier D, Meley D,
Souquere S, Yoshimori T, et al: Inhibition of macroautophagy
triggers apoptosis. Mol Cell Biol. 25:1025–1040. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhu H, Tannous P, Johnstone JL, Kong Y,
Shelton JM, Richardson JA, Le V, Levine B, Rothermel BA and Hill
JA: Cardiac autophagy is a maladaptive response to hemodynamic
stress. J Clin Invest. 117:1782–1793. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhao Y, Li R, Xia W, Neuzil J, Lu Y, Zhang
H, Zhao X, Zhang X, Sun C and Wu K: Bid integrates intrinsic and
extrinsic signaling in apoptosis induced by alpha-tocopheryl
succinate in human gastric carcinoma cells. Cancer Lett. 288:42–49.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhang X, Yan H, Yuan Y, Gao J, Shen Z,
Cheng Y, Shen Y, Wang RR, Wang X, Hu WW, et al: Cerebral
ischemia-reperfusion-induced autophagy protects against neuronal
injury by mitochondrial clearance. Autophagy. 9:1321–1333. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yan L, Vatner DE, Kim SJ, Ge H, Masurekar
M, Massover WH, Yang G, Matsui Y, Sadoshima J and Vatner SF:
Autophagy in chronically ischemic myocardium. Proc Natl Acad Sci
USA. 102:13807–13812. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Park J, Min JS, Kim B, Yun JW, Choi MS,
Kong IK, Chang KT and Lee DS: Mitochondrial ROS govern the
LPS-induced pro-inflammatory response in microglia cells by
regulating MAPK and NF-κB pathways. Neurosci Lett. 584:191–196.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hou JT, Li K, Yang J, Yu KK, Liao YX, Ran
YZ, Liu YH, Zhou XD and Yu XQ: A ratiometric fluorescent probe for
in situ quantification of basal mitochondrial hypochlorite in
cancer cells. Chem Commun (Camb). 51:6781–6784. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ravikumar B, Sarkar S, Davies JE, Futter
M, Garcia-Arencibia M, Green-Thompson ZW, Jimenez-Sanchez M,
Korolchuk VI, Lichtenberg M, Luo S, et al: Regulation of mammalian
autophagy in physiology and pathophysiology. Physiol Rev.
90:1383–1435. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lemasters JJ, Nieminen AL, Qian T, Trost
LC, Elmore SP, Nishimura Y, Crowe RA, Cascio WE, Bradham CA,
Brenner DA and Herman B: The mitochondrial permeability transition
in cell death: A common mechanism in necrosis, apoptosis and
autophagy. Biochim Biophys Acta. 1366:177–196. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Shintani T and Klionsky DJ: Autophagy in
health and disease: A double-edged sword. Science. 306:990–995.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Dewaele M, Maes H and Agostinis P:
ROS-mediated mechanisms of autophagy stimulation and their
relevance in cancer therapy. Autophagy. 6:838–854. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Grishchuk Y, Ginet V, Truttmann AC, Clarke
PG and Puyal J: Beclin 1-independent autophagy contributes to
apoptosis in cortical neurons. Autophagy. 7:1115–1131. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Li M, Gao P and Zhang J: Crosstalk between
autophagy and apoptosis: Potential and emerging therapeutic targets
for cardiac diseases. Int J Mol Sci. 17:3322016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Chien CT, Shyue SK and Lai MK: Bcl-xL
augmentation potentially reduces ischemia/reperfusion induced
proximal and distal tubular apoptosis and autophagy.
Transplantation. 84:1183–1190. 2007. View Article : Google Scholar : PubMed/NCBI
|