Introduction
Peroxiredoxins (Prxs) are antioxidant enzymes that
catalyse cellular hydrogen peroxide (H2O2)
production, exhibit a protective role in cells and are required for
cell metabolism and redox signalling (1–3); in
addition, Prxs play essential roles in neurodegenerative diseases,
cancer and inflammatory processes (4).
Peroxiredoxin I (Prx I), a member of the Prx family,
is a multifunctional protein originally identified as an
intracellular scavenger of H2O2. Prx I has
also been shown to act as a molecular chaperone with the ability to
modulate the actions of numerous molecules, as a regulator of
transcription, and as a signal modulator (5). Prx I knockout shortens the lifespan
of erythrocytes, resulting in haemolytic anaemia with increased
cellular ROS levels, protein oxidation, and Heinz body formation,
and several malignant cancers in mice (6), suggesting the important role of Prx I
in the defence against to oxidative stress. Recombinant Prx I can
also bind to Toll-like receptor 4 (TLR4) and stimulate
TLR4-dependent cytokine secretion in macrophages and dendritic
cells through CD14/MD2/MyD88-dependent signalling pathways
(7). Prx I knockdown can modulate
the secretion of inflammatory cytokines, such as interleukin
(IL)-10, IL-1β and tumour necrosis factor (TNF)-α, in
lipopolysaccharide (LPS)-stimulated RAW264.7 macrophages via the
STAT3 signalling pathway (8).
Furthermore, our previous study revealed that Prx I deficiency
attenuates the macrophage phagocytic capacity for clearing
erythrocytes in the mouse response to oxidative stimulation
(9). All these reports suggest the
involvement of Prx I in oxidation-related inflammatory
processes.
LPS has been reported to stimulate the innate immune
response via recognition of the TLR4 complex and triggering the
cellular signalling pathways that produce inflammatory cytokines
and organic septic shock (10,11).
It is well-known that intraperitoneal or intravenous injections of
high LPS concentrations into animals can induce fatal septic shock
and subsequently leads to tissue damage, body temperature
dysregulation, and lethality. However, the regulatory effect of Prx
I on LPS-stimulated mouse death is not understood.
In this study, we performed experiments to
understand the protective function of Prx I on LPS-induced lethal
shock in wild-type and Prx I knockout mice. Our results revealed
that Prx I deficiency increased LPS-induced mouse death, with more
liver damage, TNF-α tissue accumulation and oxidative stress, which
was marked by higher expression levels of the antioxidant enzymes
superoxide dismutase 2 (SOD2), catalase and glutathione peroxidase
(GPx).
Materials and methods
Mice and genotype analysis
C57BL/6J (wild-type and Prx I knockout)
pathogen-free mice were kindly provided by the laboratory of Dr.
Dae-Yeul Yu, Korea Research Institute of Bioscience and
Biotechnology (KRIBB). The mice used for the LPS challenge were
8–10 weeks of age. The experimental groups were age and sex
matched. Escherichia coli O26:B6 LPS (Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany) was diluted in sterile PBS and injected
intraperitoneally (i.p) into the animals. The primers used for the
mouse genotype analysis were 5′-gct tgg gtg gag agg cta ttcg-3′ and
5′-gta aag cac gag gaa gcg gtc agcc-3′ for the neo gene and 5′-ctg
gaa acc tgg cag tga ta-3′ and 5′-ctg tga ctg ata gaa gat tggt-3′
for the wild-type gene. All animals were housed in microisolator
cages in laminar flow units under ambient light. The Institutional
Animal Care and Use Committee (Heilong Bayi Agricultural
University, Daqing, China) approved both the animal care and
experiments.
ELISA and nitric oxide (NO) production
assay
TNF-α, IL-6 and IL-10 ELISA assay kits were
purchased from R&D Systems, Inc. (Minneapolis, MN, USA), and
assays were completed according to the manufacturer's instructions.
To determine the serum cytokine level in LPS-injected mice, the
wild-type and Prx I knockout mice were i.p. injected with 20 mg/kg
LPS, and the serum and tissue cytokine levels were detected at
different time points post injection. NO production was assessed
based on the accumulation of nitrite in the serum using a
colorimetric reaction with Griess reagent [0.1% N-(1-naphthyl)
ethylenediamine dihydrochloride, 0.1% sulfanilamide, and 2.5%
H3PO4]. The absorbance at 540 nm was measured
with a UV MAX kinetic microplate reader (Molecular Devices, Menlo
Park, CA, USA).
Haematoxylin and eosin (H&E)
staining
The livers and lungs were collected from wild-type
and Prx I-deficient mice at the indicated times after the
LPS-injections; then, the tissues were trans-cardiac
perfusion-fixed with heparinized saline containing 3.7%
formaldehyde. The tissue sections (4 µm in thickness) were stained
with H&E. The experiments were performed with 10 wild-type and
10 Prx I-deficient mice.
Western blotting analysis
Tissues protein lysates were separated on 12% sodium
dodecyl sulfate-polyacrylamide gels and transferred onto
nitrocellulose membranes (EMD Millipore, Billerica, MA, USA). The
membranes were blotted with primary antibodies against Prx I,
cleaved caspase-3, Bcl-2, SOD2, GPx, catalase (Santa Cruz
Biotechnology, Inc., Dallas, TX, USA), and β-actin (Sigma-Aldrich;
Merck KGaA) at 4°C overnight. The membranes were washed five times
with 10 mM Tris-HCl (pH 7.5) containing 150 mM NaCl (Tris-buffered
saline, TBS) and 0.2% Tween-20 and subsequently incubated with
horseradish peroxidase-conjugated goat anti-rabbit IgG
(Sigma-Aldrich; Merck KGaA) or anti-mouse IgG (Sigma-Aldrich; Merck
KGaA) for 1 h at room temperature. After the removal of excess
antibodies by washing with TBS, specific binding was detected using
a chemiluminescence detection system (Amersham, Berkshire, UK)
according to the manufacturer's instructions.
Statistical analysis
The data are depicted as the means ± SEM. Student's
t-tests were performed using GraphPad Prism 4.0 software (GraphPad
Software, Inc., La Jolla, CA, USA), and P<0.05 was considered to
indicate a statistically significant difference.
Results
Prx I deletion increased LPS-induced
lethality in mice
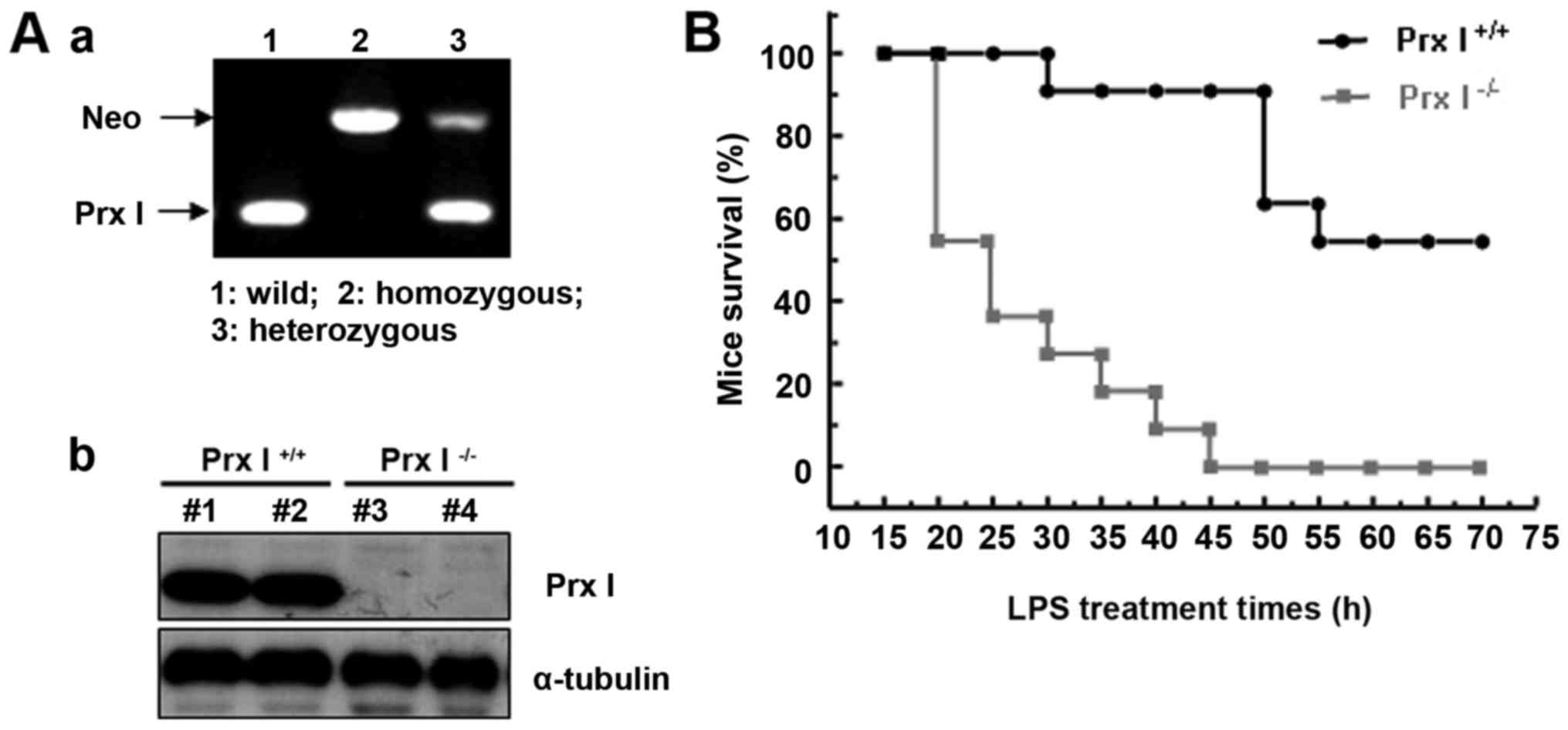
The homozygous knockout mice used in the experiments
were generated by heterozygous mice, the genotype was confirmed by
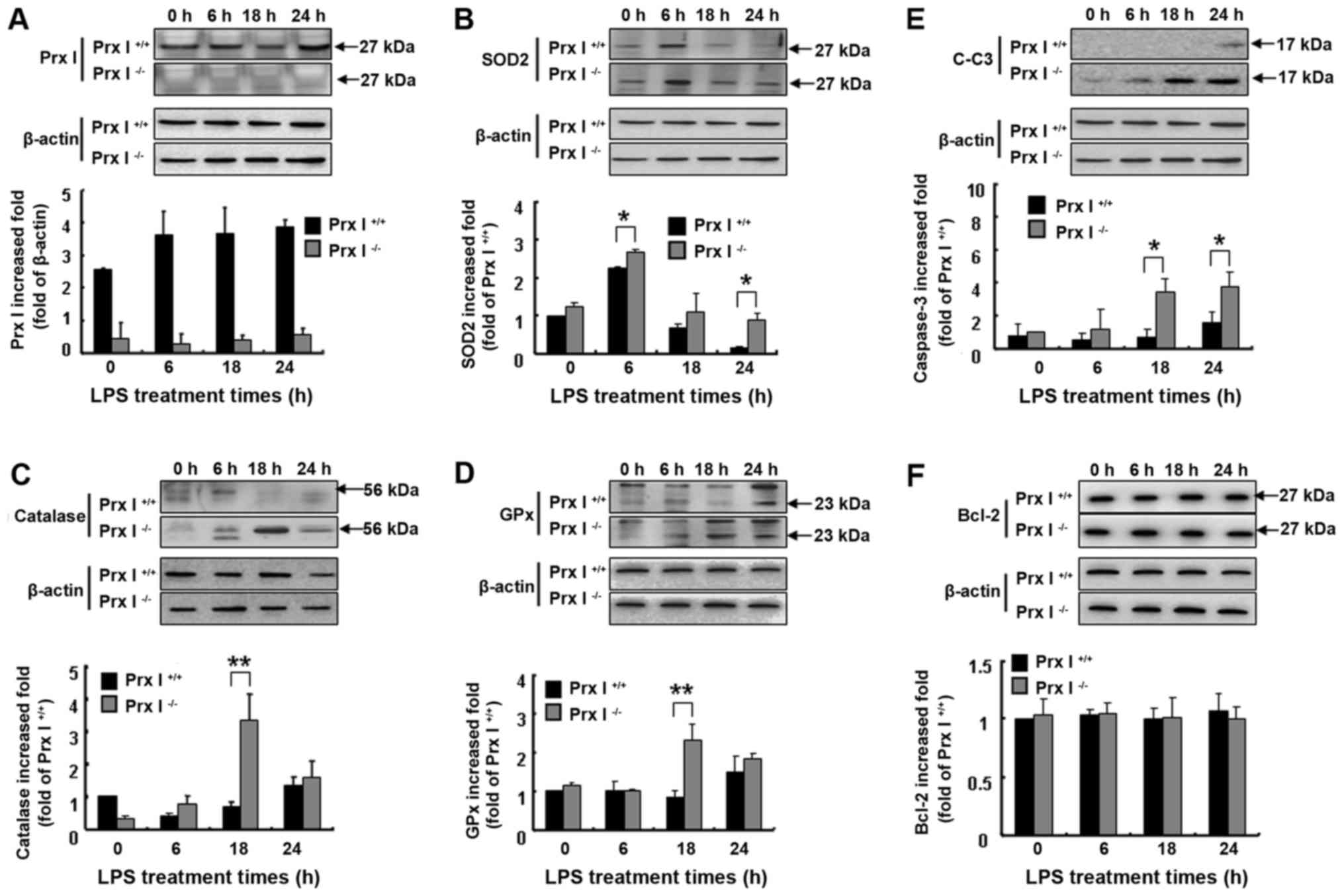
PCR (Fig. 1Aa), and the Prx I
protein level was confirmed by western blotting (Fig. 1Ab). To investigate the role of Prx
I on LPS-induced mouse lethal shock, we performed experiments to
assess whether Prx I deletion affected mouse susceptibility to
endotoxic shock. Prx I+/+ and Prx I-/- mice (11 mice
were used for each group) were injected i.p. with LPS (20 mg/kg
body weight), and mouse survival was observed for 4 d. As shown in
Fig. 1B, lethality was higher in
the LPS-stimulated Prx I−/− mice than in the Prx
I+/+ mice (approximately 54% for Prx I+/+ and
90% for Prx I−/−mice).
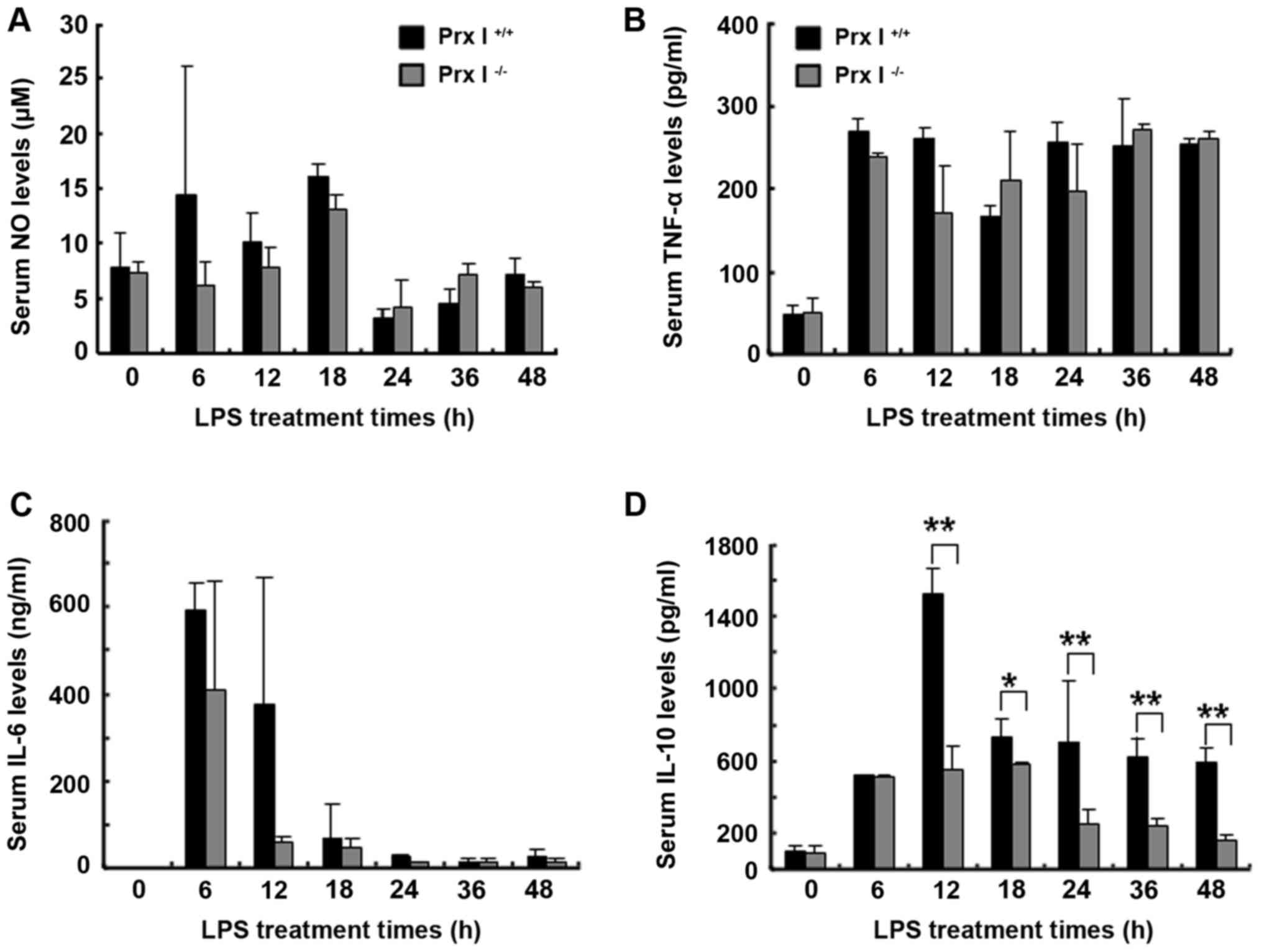
Decreased serum IL-10 production in
Prx I−/− mice after LPS injection
To examine the effect of Prx I deletion on serum
cytokine production after LPS stimulation, Prx I+/+ and
Prx I−/− mice were injected i.p. with LPS (20 mg/kg body
weight), and at the indicated times, the peripheral serum was
collected for analysing the NO, TNF-α, IL-6 and IL-10 levels with
Griess reagent (for NO determination) and ELISA kits. As shown in
Fig. 2, there was no remarkable
difference in NO, IL-6 and TNF-α production between Prx
I+/+ and Prx I−/− mice after the LPS
injection, but IL-10 production was decreased in Prx
I−/− mice after the LPS injection.
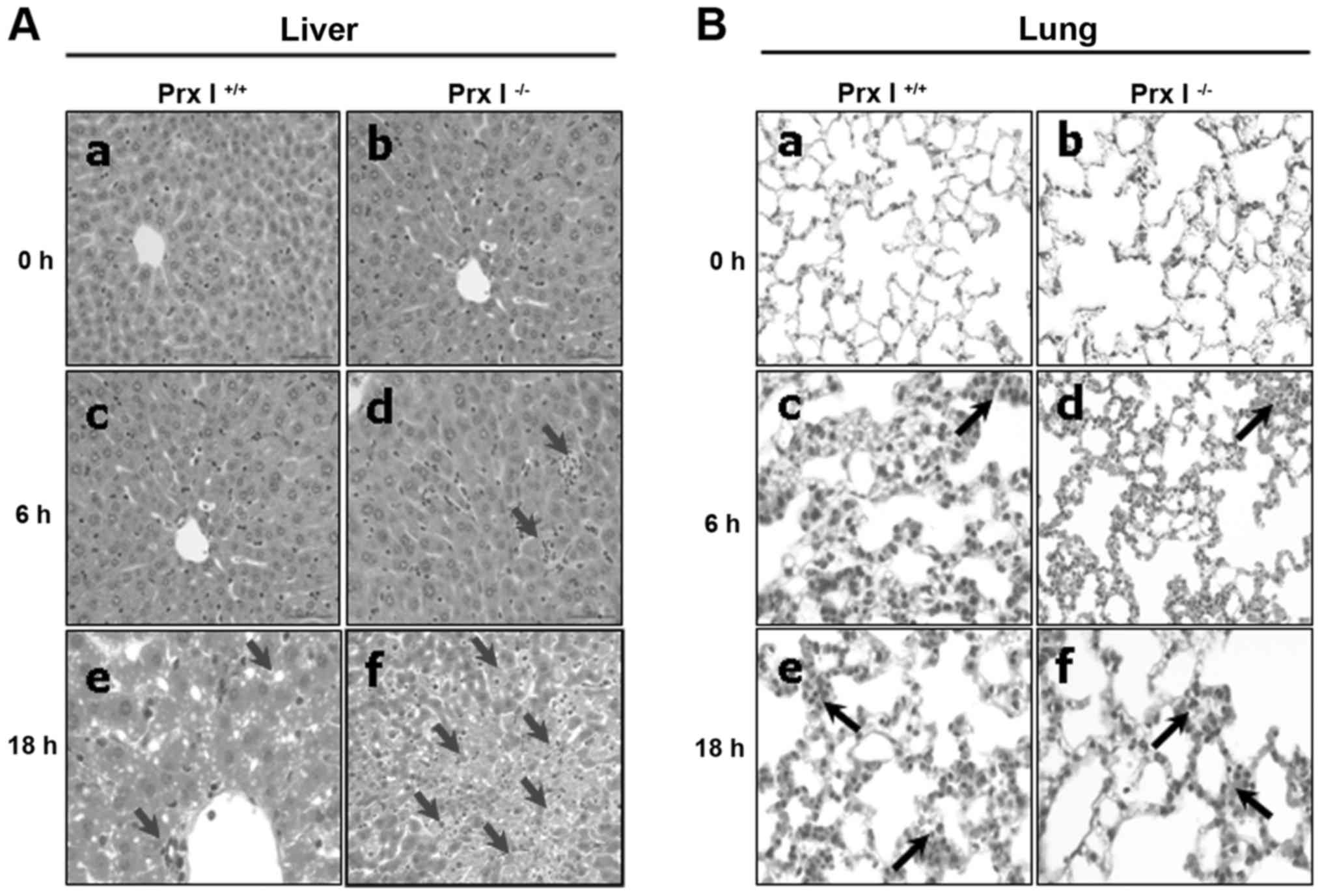
Increased liver damage and apoptosis
in Prx I-deficient mice after LPS injection
To assess the effect of Prx I deletion on
LPS-induced tissue damage, we performed histological experiments
after the LPS injection in Prx I+/+ and Prx
I−/− mice. LPS stimulation increased the liver and lung
damage, with increasing inflammatory cell infiltration, liver cell
death and diffuse alveolar haemorrhages in both Prx I+/+
and Prx I−/− mice (Fig.
3). However, severe liver damage was observed in Prx
I−/− mice compared with the Prx I+/+ mice
(Fig. 3A), but no differences in
the lungs (Fig. 3B) and kidneys
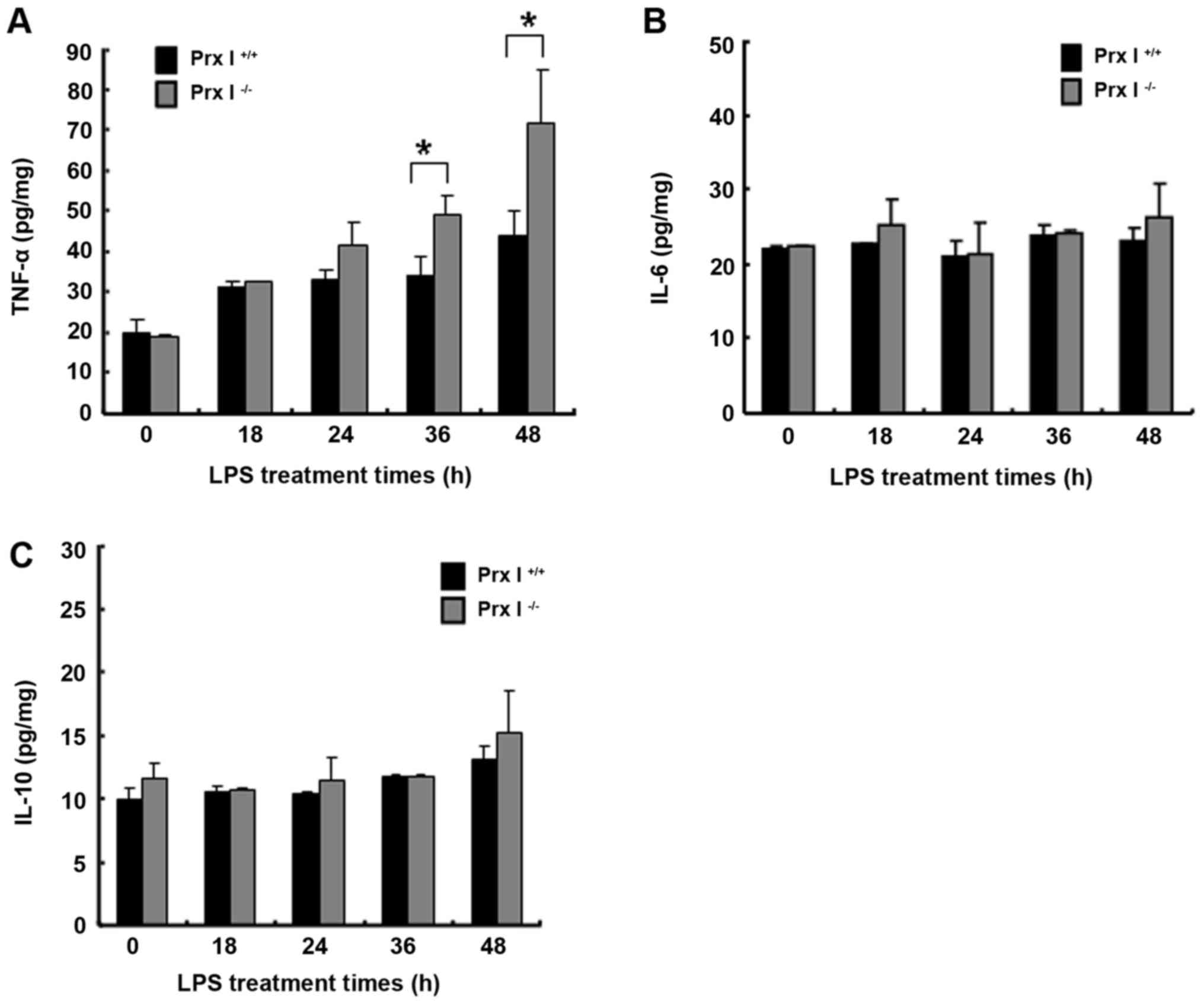
(data not shown) were observed. Furthermore, we also examined the
tissue cytokine levels at the indicated times after the LPS
injection. The results revealed that the liver tissue TNF-α levels
were increased in the Prx I−/− mice compared with the
Prx I+/+ mice, but there were no differences in IL-6 and
IL-10 production levels (Fig.
4A-C). In addition, the pro-apoptotic cleaved caspase-3 protein
levels were more up-regulated in the Prx I−/− mouse
livers than in the Prx+/+ mouse livers, but Bcl-2
protein expression levels were not different between the mouse
genotypes (Fig. 5E and F).
Prx I deletion increased the liver
antioxidant protein expression level in response to LPS
injection
To verify whether Prx I deficiency affected the
liver oxidative stress response to LPS stimulation, we also
observed the liver antioxidant enzyme protein expression levels at
the indicated times after the LPS injection in both Prx
I+/+ and Prx I−/− mice. Compared with the Prx
I+/+ mice, SOD2, catalase and GPx protein levels were
up-regulated in Prx I−/− mice after the LPS injection
(Fig. 5A-D), suggesting more
oxidative stress accumulation in Prx I−/− mice.
Discussion
In the present study, we showed that Prx I is a key
regulator of LPS-induced mouse lethal shock, as Prx I deficiency
leads to a higher mortality, caused by the acute liver damage that
accompanies the increased TNF-α tissue accumulation and apoptosis,
rapid immune cell accumulation and decreased serum IL-10 levels.
Paradoxically, no expected proinflammatory cytokine outbursts have
been classically-associated with septic death. Our study revealed
the existence of an alternative mechanism for LPS-induced lethal
shock independent of the severe inflammatory response, which is
limited by Prx I.
LPS injections have been reported to markedly
increase both the serum and macrophage cytokine production, such as
TNF-α, IL-6 and NO, and accelerate mortality in Prx II (a member of
the Prx families) knockout mice (12), indicating that the Prxs may play
essential roles in endotoxin-induced septic lethality. On the other
hand, Prx I knockdown in RAW264.7 macrophage cells decreased the
IL-10 production stimulated by LPS (8), indicating that Prx I and Prx II have
different regulatory roles in macrophage cells. In our i.p. toxin
model (13,14), LPS-induced death in Prx
I−/− mice did not have a surge in serum cytokine
production but did decrease the serum IL-10 level (Fig. 2), suggesting that Prx I deletion
may down-regulate the macrophage anti-inflammatory response to LPS
stimulation. Furthermore, recombinant Prx I stimulated the
secretion of proinflammatory cytokines, such as TNF-α and IL-6, by
binding to toll-like receptor 4 (TLR4), and this binding was
dependent on Prx I chaperone activity and independent of its
peroxidase activities in macrophages and dendritic cells (7). These findings suggest that Prx I may
be a key regulator of LPS-induced macrophage cytokine production,
but the systemic mechanism by which Prx I affects LPS-stimulated
septic lethality requires further study.
LPS/TLR4 signalling is strongly associated with
chronic liver diseases, such as alcoholic/nonalcoholic fatty liver
diseases, hepatic fibrosis and hepatocarcinoma (15). In addition, several reports have
also shown that LPS-induced mouse lethal shock is related to acute
liver damage (16–18), suggesting that the liver is a major
target tissue in response to LPS-induced lethal shock. In our
studies, rapid immune cell accumulation and increased liver TNF-α
and cleaved caspase-3 protein expression levels were observed in
the Prx I−/− mice compared with the Prx I+/+
mice after LPS administration (Figs.
3, 4 and 5E-F). These results suggest that Prx I
deficiency leads to more damage in the liver in response to LPS
stimulation, indicating a protective role for Prx I against
endotoxin-induced injury.
Mammalian antioxidant enzymes, such as SOD, GPx and
catalase, play essential roles against oxidative stress and DNA
damage-induced cell death as well as endotoxin stimulation
(19–22). In response to LPS, the protein
expression levels of SOD2, GPx and catalase were up-regulated in
the Prx I−/− mouse livers compared with those of the Prx
I+/+ mice (Fig. 5A-D).
These results revealed that LPS stimulation increased oxidative
stress in the livers of Prx I−/− mice, resulting in the
up-regulation of liver antioxidant enzyme levels. However, the
potential mechanism by which antioxidant enzymes are up-regulated
requires further study.
In summary, our results reveal the role of Prx I in
response to LPS-induced mouse lethal shock. Prx I deletion was
associated with accelerated mouse death and marked hepatitis,
accompanied by a compensatory up-regulation of antioxidant enzyme
levels. These findings provide insight into the function of Prx I
against endotoxin-induced injury.
Acknowledgements
Not applicable.
Funding
This work was supported by the Natural Science
Foundation of Heilongjiang Province of China (grant no. C2015036)
and the Research Project of Heilongjiang Bayi Agricultural
University (grant no. XA2014-04).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HNS and LF constructed the model and wrote the
manuscript. AGW, JYW and LL performed the mouse care and handling.
MHJ, GNS, CHJ and DSL performed the data analysis. THK and YDC
performed the image analysis. DYY and YHH provided substantial
contributions to conception and design of the study.
Ethics approval and consent to
participate
The Institutional Animal Care and Use Committee
(Heilong Bayi Agricultural University, China) approved both the
animal care and experiments.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Wood ZA, Schröder E, Harris Robin J and
Poole LB: Structure, mechanism and regulation of peroxiredoxins.
Trends Biochem Sci. 28:32–40. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rhee SG, Kang SW, Chang TS, Jeong W and
Kim K: Peroxiredoxin, a novel family of peroxidases. IUBMB Life.
52:35–41. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rhee SG: Overview on Peroxiredoxin. Mol
Cells. 39:1–5. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Park MH, Jo M, Kim YR, Lee CK and Hong JT:
Roles of peroxiredoxins in cancer, neurodegenerative diseases and
inflammatory diseases. Pharmacol Ther. 163:1–23. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chae HZ, Oubrahim H, Park JW, Rhee SG and
Chock PB: Protein glutathionylation in the regulation of
peroxiredoxins: A family of thiol-specific peroxidases that
function as antioxidants, molecular chaperones, and signal
modulators. Antioxid Redox Signal. 16:506–523. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Neumann CA, Krause DS, Carman CV, Das S,
Dubey DP, Abraham JL, Bronson RT, Fujiwara Y, Orkin SH and Van
Etten RA: Essential role for the peroxiredoxin Prdx1 in erythrocyte
antioxidant defence and tumour suppression. Nature. 424:561–565.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Riddell JR, Wang XY, Minderman H and
Gollnick SO: Peroxiredoxin 1 stimulates secretion of
proinflammatory cytokines by binding to TLR4. J Immunol.
184:1022–1030. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lim Tae Y, Song Sup D, Won Joon T, Lee YJ,
Yoo JS, Hyung Eun K, Won Yoon J, Park SY and Hwang Woo K:
Peroxiredoxin-1, a possible target in modulating inflammatory
cytokine production in macrophage like cell line RAW264.7.
Microbiol Immunol. 56:411–419. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Han YH, Kwon T, Kim SU, Ha HL, Lee TH, Kim
JM, Jo EK, Kim BY, Yoon DY and Yu DY: Peroxiredoxin I deficiency
attenuates phagocytic capacity of macrophage in clearance of the
red blood cells damaged by oxidative stress. BMB Rep. 45:560–564.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Park BS and Lee JO: Recognition of
lipopolysaccharide pattern by TLR4 complexes. Exp Mol Med.
45:e662013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tan Y and Kagan JC: A cross-disciplinary
perspective on the innate immune responses to bacterial
lipopolysaccharide. Mol Cell. 54:212–223. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yang CS, Lee DS, Song CH, An SJ, Li S, Kim
JM, Kim CS, Yoo DG, Jeon BH, Yang HY, et al: Roles of peroxiredoxin
II in the regulation of proinflammatory responses to LPS and
protection against endotoxin-induced lethal shock. J Exp Med.
204:583–594. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ghosn EE, Cassado AA, Govoni GR, Fukuhara
T, Yang Y, Monack DM, Bortoluci KR, Almeida SR and Herzenberg LA
and Herzenberg LA: Two physically, functionally, and
developmentally distinct peritoneal macrophage subsets. Proc Natl
Acad Sci USA. 107:2568–2573. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Henderson RB, Hobbs JA, Mathies M and Hogg
N: Rapid recruitment of inflammatory monocytes is independent of
neutrophil migration. Blood. 102:328–335. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Soares JB, Pimentel-Nunes P,
Roncon-Albuquerque R and Leite-Moreira A: The role of
lipopolysaccharide/toll-like receptor 4 signaling in chronic liver
diseases. Hepatol Int. 4:659–672. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Alazawi W, Heath H, Waters JA, Woodfin A,
O'Brien AJ, Scarzello AJ, Ma B, Lopez-Otalora Y, Jacobs M, Petts G,
et al: Stat2 loss leads to cytokine-independent, cell-mediated
lethality in LPS-induced sepsis. Proc Natl Acad Sci USA.
110:8656–8661. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu H, Zhang W, Dong S, Song L, Zhao S, Wu
C, Wang X, Liu F, Xie J, Wang J and Wang Y: Protective effects of
sea buckthorn polysaccharide extracts against LPS/d-GalN-induced
acute liver failure in mice via suppressing TLR4-NF-κB signaling. J
Ethnopharmacol. 176:69–78. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tang J, Li L, Li CM, Wu J, Sun Y and Wang
GL: Upregulation of HO-1 with Haemin Alleviates LPS-stimulated
pro-inflammatory responses through downregulation of p38 signalling
pathways in rat liver. Scand J Immunol. 82:443–451. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fischer TW, Kleszczyński K, Hardkop LH,
Kruse N and Zillikens D: Melatonin enhances antioxidative enzyme
gene expression (CAT, GPx, SOD), prevents their UVR-induced
depletion, and protects against the formation of DNA damage
(8-hydroxy-2′-deoxyguanosine) in ex vivo human skin. J Pineal Res.
54:303–312. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yang S, Jensen MK, Rimm EB, Willett W and
Wu T: Erythrocyte superoxide dismutase, glutathione peroxidase, and
catalase activities and risk of coronary heart disease in generally
healthy women: A prospective study. Am J Epidemiol. 180:901–908.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Crawford A, Fassett RG, Coombes JS, Kunde
DA, Ahuja KD, Robertson IK, Ball MJ and Geraghty DP: Glutathione
peroxidase, superoxide dismutase and catalase genotypes and
activities and the progression of chronic kidney disease. Nephrol
Dial Transplant. 26:2806–2813. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kubota Y, Takahashi S and Sato H:
Significant contamination of superoxide dismutases and catalases
with lipopolysaccharide-like substances. Toxicol In Vitro.
18:711–718. 2004. View Article : Google Scholar : PubMed/NCBI
|