Introduction
Atherosclerosis can lead to vascular stenosis,
thrombosis and vascular occlusion, which are characterized by
chronic inflammatory disease and can cause ischemic damage to vital
organs (1,2). During the development of
atherosclerosis, numerous inflammatory cells and cytokines are
mediated by specific receptors, intracellular signal transduction
or gene transfer modifications, which can affect functional protein
expression and the development of inflammatory responses (3). Adhesion molecules are considered to
represent important factors in the initiation of inflammatory
reactions associated with atherosclerosis, and chemokines have
important roles in linking inflammation and atherosclerosis
(4).
The expression levels of the previously discovered
C-X-C motif chemokine ligand 16 (CXCL16) are significantly
increased in atherosclerosis, and CXCL16 is involved in the
occurrence of inflammation and atherosclerosis (5,6).
CXCL16 is produced by numerous inflammatory cells and is
preferentially expressed within atherosclerotic plaques, including
macrophages, vascular endothelial cell and smooth muscle cells, and
possesses the functions of chemokines, adhesion molecules and
scavenger receptors (7,8). Previous studies have demonstrated
that increased serum CXCL16 levels represent an independent risk
factor in ischemic stroke with atherosclerosis, and may also
represent a novel biomarker for the prediction of ischemic stroke
incidence (9,10). Studies have revealed that CXCL16
expression levels in atherosclerotic lesions are markedly increased
following induction with bacterial lipopolysaccharide (LPS),
endotoxin and nuclear factor (NF)-κB, which improves the absorption
capacity of oxidized low-density lipoprotein and aggravates the
development of atherosclerosis (11,12).
Toll like receptors (TLRs) have important roles in
the initiation of inflammatory responses (13). LPS is a common inflammatory
stimulus of cells, and can be recognized by TLR4. Myeloid
differentiation factor 88 functions as an important link between
TLR4 and interleukin-1 receptor associated kinase 1 (IRAK1), which
binds with tumor necrosis factor receptor-associated factor 6
(TRAF6) and subsequently activates downstream NF-κB, which results
in an increased expression of immune response genes (14). The NF-κB pathway is activated by
TLR4 and represents the main signaling pathway associated with the
regulation of inflammatory responses, as well as an intermediate
link in LPS-induced increased CXCL16 expression (15). Thus, it can be suggested that NF-κB
has an important role in the development of inflammation. LPS is
recognized by TRL4, resulting in NF-κB activation. Activated NF-κB
subsequently triggers target gene transcription and inflammatory
responses by inducing CXCL16, tumor necrosis factor (TNF)-α,
interleukin (IL)-6 and other inflammatory factors, which promote
the formation and progression of atherosclerotic plaques (11). Therefore, the inhibition of
TRL4/NF-κB/CXCL16 pathway and the suppression of inflammatory
reactions may represent novel therapeutic targets for the treatment
of patients with atherosclerotic diseases.
Long-term or exaggerated inflammatory responses are
harmful, and it is therefore important to regulate cellular
negative feedback loops to suppress or inhibit the inflammatory
response (16). Micro (mi)RNAs are
small, noncoding RNA molecules of 21–24 nucleotides in length that
function as negative regulators to mediate complex biological
reactions via binding to the 3′-untranslated region (UTR) of mRNAs,
which subsequently inhibits the translation or affects the
stability of the target gene (17,18).
miR-146a was the first miRNA revealed to be associated with the
inflammatory response, and is rapidly induced by inflammatory
stimulation (19). Studies have
demonstrated that miR-146a has an important negative effect on the
inflammatory response (20–22).
miR-146b is an additional member of the miR-146 family, only
differing from miR-146a by 2 nucleotides at the 3′UTR in its mature
sequence (23). It has been
demonstrated that miR-146a is a NF-κB-dependent gene that can
activate innate immune signaling pathways (19). miR-146a targets adaptor proteins of
TRAF6 and IRAK1, and subsequently inhibits NF-κB activation, which
suggests that miR-146a represents a negative feedback mechanism for
the regulation of the NF-κB pathway in monocytes (24,25).
It has been previously revealed that miR-146a inhibits the
expression levels of proteins associated with the NF-κB pathway,
such as IL-6, IL-8 and TNF-α (19,26).
miR-146b is an IL-10 reactive miRNA, which ameliorates inflammatory
reactions by targeting the TLR4 pathway (23). Associations between miR-146b and
the TLR4/NF-κB signaling pathway remain unclear. One study
demonstrated that miR-146b may mediate the TLR4 signaling pathway
via direct regulation of numerous proteins, such as TLR4, IRAK1 and
TRAF6; rather than via the NF-κB signaling pathway (23). The same study also revealed that
elevated miR-146b expression may lead to a marked reduction in the
LPS-dependent production of several inflammatory cytokines, such as
IL-6, TNF-α and CXCL10 (23).
However, a further study demonstrated that miR-146a and miR-146b
regulate apoptosis and inflammatory cytokines in human dendritic
cells via regulation of the TRAF6/IRAK1/NF-κB pathway (27). Therefore, it is important to
investigate the associations between CXCL16, the NF-kB pathway,
miR-146a and miR-146b.
To the best of the author's knowledge, there are no
reports regarding the associations of CXCL16, miR-146a and miR-146b
in atherosclerotic disease in vivo. The present study
investigated the associations between CXCL16, miR-146a and miR146b
in atherosclerotic disease in vivo. Atherosclerotic mouse
models were established, in which a perivascular collar was placed
around the carotid artery of ApoE−/− mice to form atherosclerotic
vascular lesions. The results of the present study may further the
understanding of the mechanisms underlying the associations between
CXCL16, miR-146a, miR-146b and the TLR4/NF-κB signaling pathway
during inflammatory responses associated with atherosclerosis.
Materials and methods
Mouse experiments
A total of 24 male ApoE−/− C57BL/6J mice
(8-week-old; weight 18–22 g) were obtained from Beijing HFK
Bioscience Co., Ltd. (Beijing, China). All mice were housed under a
12-h light/dark cycle at a room temperature of 22°C and 50–60%
relative humidity. All mice had free access to food and water. The
study protocol was approved by the Animal Ethics Committee of
Qingdao University prior to experimentation.
All mice were randomly divided into two groups (n=12
per group): A control group and a model group. In the first week,
all mice were fed with normal food. However, from the second week
onwards, the ApoE−/− mice in the model group were fed a high-fat
diet, which constituted 15% cocoa butter, 0.25% cholesterol and
normal food (28,29). The ApoE−/− mice in the control
group remained on a diet of normal food. From the fifteenth day
onwards, each mouse in the model group had a perivascular collar
placed around the right common carotid artery (28–30).
Each of the mice in the control group underwent sham surgery. A
total of 8 weeks post-surgery, all mice were sacrificed and the
blood and right common carotid arteries were collected for the
subsequent experiments (28).
Lipid analysis
Levels of total cholesterol (TC), triglycerides
(TG), high-density lipoprotein cholesterol (HDL-c) and low-density
lipoprotein cholesterol (LDL-c) were determined in the blood
gathered from the femoral artery of the mice. The lipid levels were
measured by an Olympus AU640 automatic biochemical analyzer
(Olympus Corporation, Tokyo, Japan) at the clinical laboratory of
The Affiliated Hospital of Qingdao University.
Histopathological analysis
The common carotid artery were fixed in 10% neutral
formalin at 4°C for 24 h, dehydrated and then embedded in paraffin.
Continuous transverse paraffin sections of 5 µm were obtained.
Selective staining was performed with hematoxylin and eosin (HE) at
50-µm intervals at 25°C for 20 min. A pathological image analyzer
was used to measure the size of the staining patch area. Image-Pro
Plus 6.0 software (Media Cybernetics, Inc., Rockville, MD, USA) was
used to determine the proportion of atherosclerotic plaque area to
the luminal area (31,32).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
Total RNA was extracted from the right common
carotid arteries using TRIzol® (Thermo Fisher
Scientific, Inc., Waltham, MA, USA). The purity and concentration
of the total RNA were determined using a spectrophotometer, and
samples exhibiting a range of A260/280 between 1.8 and 2.0 were
regarded suitable for further experimentation. RT-qPCR was used to
determine the expression levels of miR-146a, miR-146b, TLR4, IRAK1,
TRAF6, NF-κB, CXCL16, TNF-α and IL-1β. A PrimeScript TM miRNA qPCR
Starter Kit Ver.2.0 (Takara Bio, Inc., Otsu, Japan) was used.
Primer sequences used for qPCR of miRNAs were: miR-146a-5p:
5′-GCCGTGAGAACTGAATTCCATG-3′; miR-146b-5p:
5′-GAGCTGAGAACTGAATTCCATAG-3′; the internal reference of miR-146a
and miR-146b was U6 (Takara Bio, Inc.). The primer sequences for U6
was: Forward, 5′-CTCGCTTCGGCAGCACA-3′ and reverse,
5′-AACGCTTCACGAATTTGCGT-3′. The primer sequences used for qPCR of
proteins were: TLR4 forward, 5′-TCAGAGCCGTTGGTGTATCTT-3′ and
reverse, 5′-TGTCCTCCCATTCCAGGTAG-3′; IRAK1 forward,
5′-ATAAGGCAGGCAATGTGAGG-3′ and reverse,
5′-TCATACCCACTGAGCCATCTC-3′; TRAF6 forward,
5′-TCGGAGTGCCGTGTATGTAG-3′ and reverse, 5′-CACCTTCTTCTGGCTTTCGT-3′;
NF-κB forward, 5′-TGGACGACTCTTGGGAGAAG-3′ and reverse,
5′-CACAGGCTCATACGGTTTCC-3′; CXCL16 forward,
5′-CAGGCTCGTCTCCATCAGT-3′ and reverse, 5′-GTAGAGGCAAAGGGTCAGCA-3′;
TNF-α forward, 5′-TCTGGGCAGGTCTACTTTGG-3′ and reverse,
5′-GGTTGAGGGTGTCTGAAGGA-3′; IL-1β forward,
5′-CATCAGCACCTCTCAAGCAG-3′ and reverse, 5′-AGTCCACATTCAGCACAGGA-3′.
The relative expression levels were normalized to the internal
reference gene GAPDH. The primer sequences of GAPDH were: Forward,
5′-AACAGCCTCAAGATCATCAGCAA-3′ and reverse,
5′-GACTGTGGTCATGAGTCCTTCCA-3′.
The reaction conditions for miR-146a and miR-146b
were: Initial denaturation step for 30 sec at 95°C, denaturing at
95°C for 10 sec, annealing and elongation 30 sec at 60°C for 45
cycles. The reaction conditions of TRL4, IRAK1, TRAF6, NF-kB and
CXCL16 were: Initial denaturation step for 30 sec at 95°C,
denaturing at 95°C for 10 sec, annealing 20 sec at 55°C and
elongation for 30 sec at 72°C for 55 cycles. The experiments were
repeated under the above experimental conditions 3 times. The
2−∆∆Cq method was used to compare the relative
expression results (33).
Western blot analysis
Total protein was extracted from the right common
carotid artery in each group using a mixture of tissue lysate and
protease inhibitor (Beijing Solarbio Science & Technology Co.,
Ltd., Beijing, China; 1:100), and the bicinchoninic acid method was
used to determine total protein concentration. Then, 20 µg
protein/lane was separated via SDS-PAGE at the following
percentages: IRAK (6%), IL-1β (12%), TNF-α (12%), CXCL16 (12%),
TRAF6 (8%), TLR4 (8%) and NF-κB (8%). The separated proteins were
subsequently transferred onto a polyvinylidene difluoride membrane
(0.2 µm for IL-1β and TNF-α and 0.45 µm for the other proteins).
The membranes were blocked with 5% skimmed milk (Beijing Solarbio
Science & Technology Co., Ltd.) at 4°C for 2 h. The Primary
antibodies against the following proteins were used: β-actin (cat.
no. BM0627; 1:200; Wuhan Boster Biological Technology, Ltd., Wuhan,
China), TLR4 (cat. no. ab13867; 1:250; Abcam, Cambridge, UK), IRAK1
(cat. no. 4504; 1:1,000; CST Biological Reagents Co., Ltd.,
Shanghai, China), TRAF6 (cat. no. ab33915; 1:6,000; Abcam), NF-κB
(cat. no. 8242; 1:2,000; CST Biological Reagents Co., Ltd.), CXCL16
(cat. no. ab119350; 1:1,000; Abcam), TNF-α (cat. no. 3707; 1:1,000;
CST Biological Reagents Co., Ltd.) and IL-1β (cat. no. 12242;
1:1,000; CST Biological Reagents Co., Ltd.). The membranes were
washed in TBST buffer (Beijing Solarbio Science & Technology
Co., Ltd.) 4 times for 8 min at a time at 25°C and then incubated
at 4°C for 1 h with Goat anti-Mouse IgG (cat. no. BA1050; 1:3,500;
Wuhan Boster Biological Technology, Ltd.) and Goat anti-Rabbit IgG
(cat. no. 1:1,500; BA1054; Wuhan Boster Biological Technology,
Ltd.). Protein bands were visualized using an ECL kit (Wuhan Boster
Biological Technology, Ltd.). Fusion FX7 (Vilber Lourmat,
Marne-la-Vallée, France) was used to acquire images, and the
results were analyzed with ImageJ version 1.38 (National Institutes
of Health, Bethesda, MD, USA).
Statistical analysis
All experiments were performed in triplicate and
SPSS software version 19.0 (IBM Corp., Armonk, NY, USA) was used
for statistical analysis. All data are expressed as the mean ±
standard deviation. Statistically significant differences between
the two groups were determined using the Student's t-test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Body weight and lipid levels of
ApoE−/− mice
No significant differences in body weight were
observed between the two groups on the 1 day and 70 day time
intervals (P>0.05; Table I).
Levels of TG, TC and LDL-c in the model group were significantly
increased compared with the control group (P<0.05; Table I); however, no significant
differences were observed between the model and control groups
regarding HDL-c levels (P>0.05; Table I).
 | Table I.Body weight and lipid levels of
ApoE−/− mice. |
Table I.
Body weight and lipid levels of
ApoE−/− mice.
|
| Body weight at the
1 day time interval (g) | Body weight at the
70 day time interval (g) | TC (mmol/l) | TG (mmol/l) | LDL-c (mmol/l) | HDL-c (mmol/l) |
|---|
| Control group | 19.20±1.30 | 27.75±1.89 | 13.06±1.72 | 2.19±0.11 | 1.03±0.12 | 1.77±0.37 |
| Model group | 21.00±1.00 | 28.67±0.58 |
24.72±2.30a |
2.73±0.23a |
4.62±0.14a | 1.34±0.18 |
| P-value |
0.066 | 0.073 |
0.002 | 0.022 | <0.0001 | 0.105 |
Pathological analysis of common
carotid artery tissues obtained from ApoE−/− mice
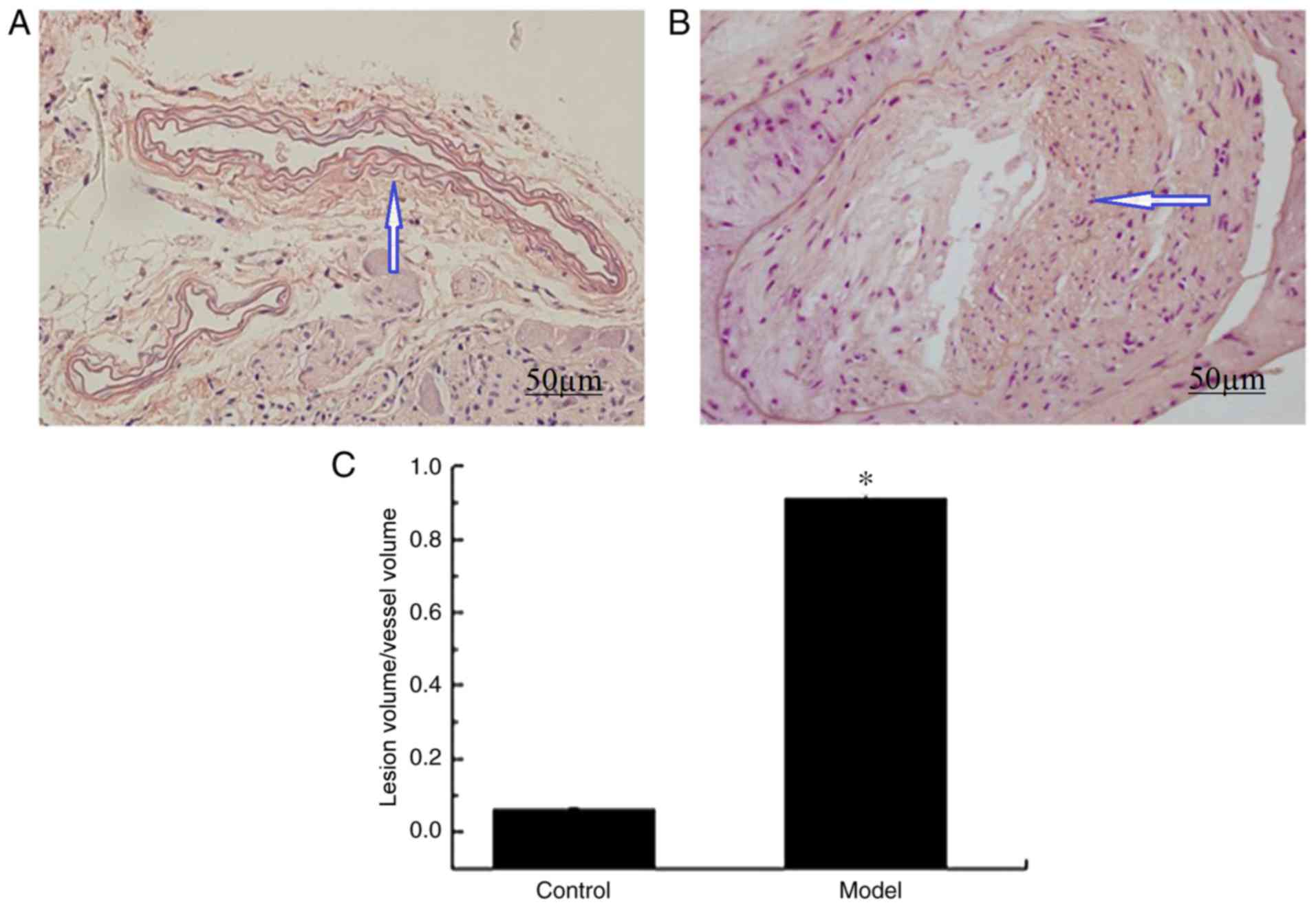
H&E staining results revealed that there were no
marked pathological symptoms were observed in the vascular lumen
and intima of ApoE−/− mice belonging to the control group (Fig. 1A); however, significant plaques and
lumen stenosis were observed in ApoE−/− mice belonging to the model
group (Fig. 1B). In addition, the
ratio of the lesion volume to the vessel volume was significantly
increased in the model group compared with the control group
(P<0.01; Fig. 1C). Therefore,
the results demonstrated that ApoE−/− mice in the model group
exhibited significantly increased formation of atherosclerotic
plaques compared with ApoE−/− mice in the control group.
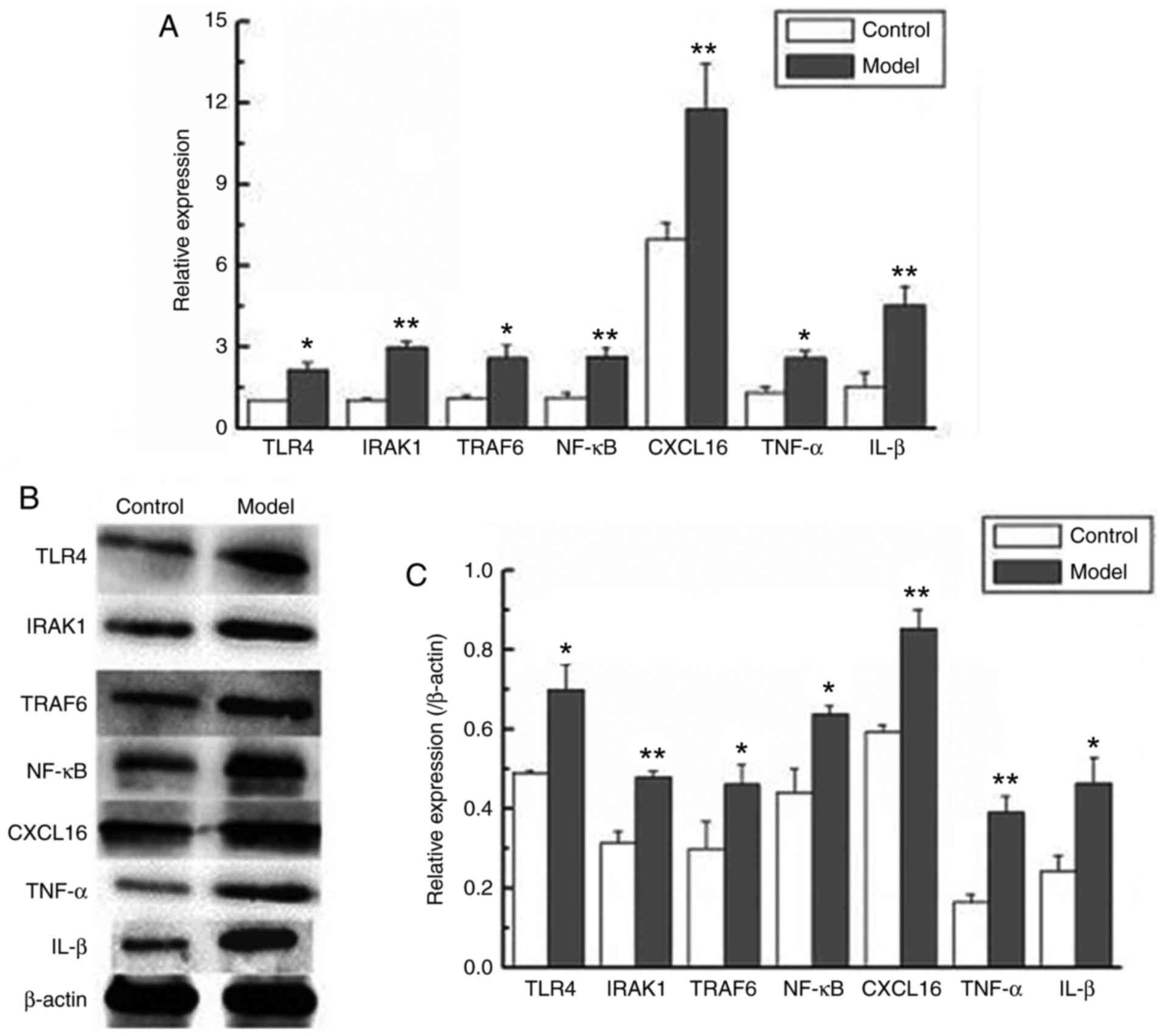
Expression levels of CXCL16 and
proteins associated with the TRL4/NF-κB signaling pathway are
increased in atherosclerotic ApoE−/− mice
The CXCL16 and TRL4/NF-κB signaling pathway serve
important roles in the inflammatory responses, and so their
expression in atherosclerotic ApoE−/− mice was investigated using
RT-qPCR and western blot analysis in the present study. The results
revealed that the mRNA and protein levels of CXCL16 were
significantly upregulated in the model group compared with the
control group (P<0.01; Fig. 2).
The results also demonstrated that the mRNA and protein levels of
TRL4, IRAK1, TRAF6, NF-κB, TNF-α and IL-1β were significantly
upregulated in the model group compared with the control group
(P<0.05 and P<0.01; Fig. 2).
These results revealed that the expression levels of CXCL16 and
proteins associated with the TRL4/NF-κB signaling pathway were
significantly upregulated in atherosclerotic ApoE−/− mice compared
with control ApoE−/− mice.
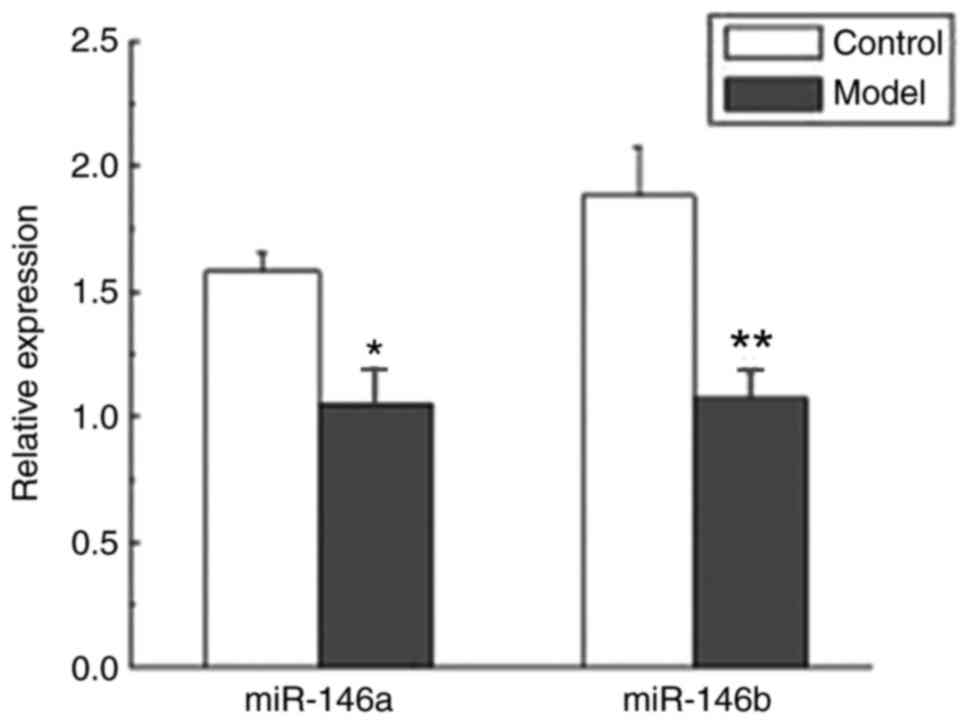
Expression levels of miR-146a and
miR-146b are decreased in atherosclerotic ApoE−/− mice
To investigate the expression levels of miR-146a and
miR146b in atherosclerotic disease, ApoE−/− mice were fitted with a
perivascular collar and administered a high-fat diet. The
expression levels of miR-146a and miR-146b were investigated using
RT-qPCR. Compared with the control group, the expression levels of
miR-146a and miR-146b were significantly suppressed in the ApoE−/−
mice belonging to the model group compared with ApoE−/− mice
belonging to the control group (P<0.05 and P<0.01; Fig. 3).
Discussion
In the present study, a perivascular collar was
placed around the carotid artery of ApoE−/− mice to induce
atherosclerotic pathological changes, as previously described
(29,30,34).
The results of the present study revealed a significant formation
of atherosclerotic plaques in the carotid artery of atherosclerotic
ApoE−/− mice compared with the control ApoE−/− mice. Recent studies
have demonstrated that CXCL16 expression is significantly increased
in patients suffering from acute ischemic stroke, and that enhanced
CXCL16 levels may represent a biomarker for ischemic stroke
incidence prediction (9,10). The present study demonstrated that
the mRNA and protein levels of CXCL16 were significantly
upregulated in the model group exhibiting atherosclerotic plaques.
Previous research has demonstrated that the level of CXCL16
increases in atherosclerotic ApoE−/− mice and may represent a
potential atherogenic biomarker (35). In studies of coronary heart disease
(CHD), a high level of CXCL16 has been revealed to be associated
with the severity of acute coronary syndrome, and may represent a
potential biomarker for epidemiological and clinical application in
CHD (36,37). Laugsand et al (38) demonstrated that CXCL16 is closely
associated with the risk of myocardial infarction and may aid in
assessing cardiovascular risk. These studies have revealed that
CXCL16 is involved in atherosclerotic disease. However, the
pathophysiological role of CXCL16 in atherosclerotic diseases in
vivo remains unclear.
Furthermore, the present study demonstrated that the
expression of NF-κB, TNF-α and IL-1β were significantly increased
in atherosclerotic ApoE−/− mice compared with control ApoE−/− mice.
NF-κB is activated by TLR4, which is associated with inflammation
(39,40). TNF-α and IL-1β are located
downstream of the NF-κB pathway and are activated by NF-κB
(41,42). In an in vitro study,
simulated cardiac ischemia/reperfusion injury in human umbilical
vein endothelial cells enhanced levels of CXCL16, TNF-α and
intercellular adhesion molecule-1 (ICAM-1), thus suggesting that
NF-κB aggravates the inflammatory response via the upregulation of
CXCL16, TNF-α and ICAM-1 (42).
Lehrke et al (43)
demonstrated that CXCL16 expression is significantly upregulated
following stimulation with LPS and downregulated following
treatment with NF-κB-targeting anti-inflammatory drugs in
vivo. In addition, CXCL16 expression is significantly
downregulated following treatment with the NF-κB inhibitor, SN50.
However, Lehrke et al (43)
also revealed that following activation of NF-κB, CXCL16 expression
is significantly upregulated by overexpression of activated IκB
kinase in vitro. In addition, Izquierdo et al
(44) demonstrated that TNF-like
weak inducer of apoptosis upregulated CXCL16 expression in
vivo and in vitro via activation of the NF-κB
transcription factor; however, expression of CXCL16 was revealed to
be downregulated via inhibition of NF-κB activation, which
demonstrated that the expression of CXCL16 is NF-κB-dependent. The
present study demonstrated that the expression levels of NF-κB and
CXCL16 were significantly increased in atherosclerotic ApoE−/− mice
compared with control ApoE−/− mice, which also suggested that
CXCL16 may represent a positive feedback mechanism to NF-κB in
atherosclerotic diseases in vivo. Thus, it was demonstrated
that the role of CXCL16 in atherosclerotic diseases may be closely
associated with NF-κB.
The present study also revealed that the expression
levels of TRL4, IRAK1 and TRAF6 were significantly increased in
atherosclerotic ApoE−/− mice compared with control ApoE−/− mice.
IRAK1 and TRAF6 are important adapter molecules in the TLR4/NF-κB
pathway that can be activated by TLR4, which subsequently activates
the NF-κB pathway (45). TLR4 can
activate NF-κB pathways and increase the expression levels of IL-6
and IL-8 (46). Overexpression of
TRAF6 and/or IRAK1 in mature dendritic cells by lentivirus
transduction has been revealed to increase the expression level of
NF-κB (27). The present study
demonstrated that the expression levels of upstream and downstream
factors of the NF-κB signaling pathway, such as TLR4, IRAK1, TRAF6,
CXCL16, TNF-α and IL-1β, were significantly increased in
atherosclerotic ApoE−/− mice compared with control ApoE−/− mice.
These results demonstrated that the important role of CXCL16 in
atherosclerosis may be via the TRL4/NF-κB/CXCL16 pathway.
In recent years, numerous studies have suggested
that the expression levels of miRNAs are associated with human
diseases, particularly diseases involving an inflammatory response.
For example, decreased expression of let-7 in lung cancer tissues
may promote the expression of Ras, and therefore Ras may be
involved in the mechanism underlying the association between let-7
and lung cancer (47).
Furthermore, expression of miR-181 has been revealed to be
decreased in the aortic intima of ApoE−/− mice administered with
high fat diets, which may promote the formation of atherosclerosis
(48). Downregulation of miR-149
in osteoarthritism chondrocytes has been previously revealed be
correlated with increased expression of pro-inflammatory cytokines
(49). According to numerous
previous studies, miRNA networks have important roles in the
inhibition of inflammatory responses (50–52).
For example, decreased expression levels of miR-10a and miR-181b
have been revealed to be associated with inhibition of the
development of atherosclerosis (51,52).
Numerous studies have demonstrated that miR-146a and
miR-146b serve critical roles in the inflammatory response
(53–55). Cheng et al (54) revealed that miR-146a can inhibit
the NF-κB pathway, which may suppress the inflammatory response
in vitro. Cheng et al (54) also demonstrated that miR-146b may
regulate endothelial activation that represses the inflammatory
response via inhibition of the NF-κB pathway activation, which
suggested that increased expression levels of miR-146a or miR-146b
in the vasculature may represent an effective treatment for the
inhibition of the inflammatory response. Park et al
(27) demonstrated that miR-146a
and miR-146b regulate cell apoptosis and cytokine production via
TRAF6 and IRAK1, and thus function as negative feedback mechanisms.
Overexpression of miR-146a has been revealed to suppress NF-κB
activity via decreasing IRAK1 and TRAF6 expression levels in the
myocardium and attenuating the production of inflammatory
cytokines, such as TNF-α, IL-1β and ICAM-1 (56). Curtale et al (23) demonstrated that miR-146b represents
an IL-10-dependent regulator of the TLR4 signaling pathway that
directly targets multiple elements, including IRAK1 and TRAF6.
Furthermore, Curtale et al (23) revealed that increased expression of
miR-146b markedly suppresses levels of proinflammatory cytokines,
including CXCL10, TNF-α, IL-6 and IL-8 (23). The present study demonstrated that
the expression levels of miR-146a and miR-146b were significantly
downregulated in atherosclerotic ApoE−/− mice compared with control
ApoE−/− mice; however, NF-κB and its downstream products were
demonstrated to be significantly upregulated in atherosclerotic
ApoE−/− mice compared with control ApoE−/− mice. This suggested
that the downregulation of miR-146a and miR-146b expression levels
may reduce the inhibitory effect on CXCL16 via the TLR4/NF-κB
signaling pathway.
In conclusion, the present study revealed that the
expression levels of CXCL16 and proteins associated with the
TLR4/NF-κB signaling pathway were significantly upregulated, and
the expression levels of miR-146a and miR146b were significantly
downregulated, in atherosclerotic ApoE−/− mice compared with
control ApoE−/− mice in vivo. The results of the present
study suggested that CXCL16 may be associated with the
TRL4/NF-κB/CXCL16 signaling pathway, and that miR-146a and miR-146b
may negatively regulate CXCL16 via TLR4/NF-κB/CXCL16 signaling
pathway in atherosclerosis in vivo. Therefore, enhanced
expression of miR-146a and miR146b may represent an effective
approach for the reduction or prevention of the inflammatory
response in patients with atherosclerosis.
Acknowledgements
Not applicable.
Funding
The present study was funded by the National Natural
Sciences Foundation of China (grant no. 81571112).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
AM and XZ conceived, designed and performed the
experiments, analyzed the data and drafted the manuscript. SY and
TW performed the experiments and analyzed and interpreted the data.
YW and XX also performed the experiments. SL provided experimental
technical support. XP provided the concept and design of the study,
together with critical revision of the manuscript for content, and
obtained funding performed the administration.
Ethics approval and consent to
participate
The study protocol was approved by the Animal Ethics
Committee of Qingdao University prior to experimentation.
Patients consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Gimbrone MA Jr and Garcia-Cardeña G:
Vascular endothelium, hemodynamics, and the pathobiology of
atherosclerosis. Cardiovasc Pathol. 22:9–15. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Pober JS and Sessa WC: Evolving functions
of endothelial cells in inflammation. Nat Rev Immunol. 7:803–815.
2007. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Libby P: Inflammation in atherosclerosis.
Nature. 420:868–874. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Barlic J and Murphy PM: Chemokine
regulation of atherosclerosis. J Leukoc Biol. 82:226–236. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Borst O, Münzer P, Gatidis S, Schmidt EM,
Schönberger T, Schmid E, Towhid ST, Stellos K, Seizer P, May AE, et
al: The inflammatory chemokine CXC motif ligand 16 triggers
platelet activation and adhesion via CXC motif receptor 6-dependent
phosphatidylinositide 3-kinase/Akt signaling. Circ Res.
111:1297–1307. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Darash-Yahana M, Gillespie JW, Hewitt SM,
Chen YY, Maeda S, Stein I, Singh SP, Bedolla RB, Peled A, Troyer
DA, et al: The chemokine CXCL16 and its receptor, CXCR6, as markers
and promoters of inflammation-associated cancers. PLoS One.
4:e66952009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Petit SJ, Wise EL, Chambers JC, Sehmi J,
Chayen NE, Kooner JS and Pease JE: The CXCL16 A181V mutation
selectively inhibits monocyte adhesion to CXCR6 but is not
associated with human coronary heart disease. Arterioscler Thromb
Vasc Biol. 31:914–920. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Minami M, Kume N, Shimaoka T, Kataoka H,
Hayashida K, Akiyama Y, Nagata I, Ando K, Nobuyoshi M, Hanyuu M, et
al: Expression of SR-PSOX, a novel cell-surface scavenger receptor
for phosphatidylserine and oxidized LDL in human atherosclerotic
lesions. Arterioscler Thromb Vasc Biol. 21:1796–1800. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ma A, Pan X, Xing Y, Wu M, Wang Y and Ma
C: Elevation of serum CXCL16 level correlates well with
atherosclerotic ischemic stroke. Arch Med Sci. 10:47–52. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ma A, Yang S, Wang Y, Wang X and Pan X:
Increase of serum CXCL16 level correlates well to microembolic
signals in acute stroke patients with carotid artery stenosis. Clin
Chim Acta. 460:67–71. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zeng M, Yan H, Chen Y, Zhao HJ, Lv Y, Liu
C, Zhou P and Zhao B: Suppression of NF-κB reduces myocardial
no-reflow. PLoS One. 7:e473062012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Altenburg JD and Siddiqui RA:
Docosahexaenoic acid downregulates interferon gamma-induced
expression of CXCL16 in human aortic smooth muscle cells. Biochem
Biophys Res Commun. 391:609–614. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
O'Neill LA: How Toll-like receptors
signal: What we know and what we don't know. Curr Opin Immunol.
18:3–9. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Akira S and Takeda K: Toll-like receptor
signalling. Nat Rev Immunol. 4:499–511. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lötzer K, Döpping S, Connert S, Gräbner R,
Spanbroek R, Lemser B, Beer M, Hildner M, Hehlgans T, van der Wall
M, et al: Mouse aorta smooth muscle cells differentiate into
lymphoid tissue organizer-like cells on combined tumor necrosis
factor receptor-1/lymphotoxin beta-receptor NF-kappaB signaling.
Arterioscler Thromb Vasc Biol. 30:395–402. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ruland J: Return to homeostasis:
Downregulation of NF-κB responses. Nat Immunol. 12:709–714. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ma X, Buscaglia Becker LE, Barker JR and
Li Y: MicroRNAs in NF-kappaB signaling. J Mol Cell Biol. 3:159–166.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Feinberg MW and Moore KJ: MicroRNA
regulation of atherosclerosis. Circ Res. 118:703–720. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Taganov KD, Boldin MP, Chang KJ and
Baltimore D: NF-kappaB-dependent induction of microRNA miR-146, an
inhibitor targeted to signaling proteins of innate immune
responses. Proc Natl Acad Sci USA. 103:12481–12486. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
O'Neill LA, Sheedy FJ and McCoy CE:
MicroRNAs: The fine-tuners of Toll-like receptor signalling. Nat
Rev Immunol. 11:163–175. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Etzrodt M, Cortez-Retamozo V, Newton A,
Zhao J, Ng A, Wildgruber M, Romero P, Wurdinger T, Xavier R,
Geissmann F, et al: Regulation of monocyte functional heterogeneity
by miR-146a and Relb. Cell Rep. 1:317–324. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jurkin J, Schichl YM, Koeffel R, Bauer T,
Richter S, Konradi S, Gesslbauer B and Strobl H: miR-146a is
differentially expressed by myeloid dendritic cell subsets and
desensitizes cells to TLR2-dependent activation. J Immunol.
184:4955–4965. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Curtale G, Mirolo M, Renzi TA, Rossato M,
Bazzoni F and Locati M: Negative regulation of Toll-like receptor 4
signaling by IL-10-dependent microRNA-146b. Proc Natl Acad Sci USA.
110:11499–11504. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nahid MA, Pauley KM, Satoh M and Chan EK:
miR-146a is critical for endotoxin-induced tolerance: Implication
in Innate Immunity. J Biol Chem. 284:34590–34599. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bhaumik D, Scott GK, Schokrpur S, Patil
CK, Campisi J and Benz CC: Expression of microRNA-146 suppresses
NF-kappaB activity with reduction of metastatic potential in breast
cancer cells. Oncogene. 27:5643–5647. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Boldin MP, Taganov KD, Rao DS, Yang L,
Zhao JL, Kalwani M, Garcia-Flores Y, Luong M, Devrekanli A, Xu J,
et al: miR-146a is a significant brake on autoimmunity,
myeloproliferation, and cancer in mice. J Exp Med. 208:1189–1201.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Park H, Huang X, Lu C, Cairo MS and Zhou
X: MicroRNA-146a and microRNA-146b regulate human dendritic cell
apoptosis and cytokine production by targeting TRAF6 and IRAK1
proteins. J Biol Chem. 290:2831–2841. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Pan X, Hou R, Ma A, Wang T, Wu M, Zhu X,
Yang S and Xiao X: Atorvastatin upregulates the expression of
miR-126 in Apolipoprote in E-knockout mice with carotid
atherosclerotic plaque. Cell Mol Neurobiol. 37:29–36. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cai X, Li X, Li L, Huang XZ, Liu YS, Chen
L, Zhang K, Wang L, Li X, Song J, et al: Adiponectin reduces
carotid atherosclerotic plaque formation in ApoE−/− mice: Roles of
oxidative and nitrosative stress and inducible nitricoxide
synthase. Mol Med Rep. 11:1715–1721. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
von der Thusen JH, van Berkel TJ and
Biessen EA: Induction of rapid atherogenesis by perivascular
carotid collar placement in apolipoprotein E-deficient and
low-density lipoprotein receptor-deficient mice. Circulation.
103:1164–1170. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Xue-Mei L, Jie C, Xuan D, Xiao-Xing L,
Chun-Lin H and Yu-Jie L: Changes in CD4+CD25+
Tregs in the pathogenesis of atherosclerosis in ApoE−/− mice. Exp
Biol Med (Maywood). 242:918–925. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hu XB, Zhang PF, Su HJ, Yi X, Chen L, Rong
YY, Zhang K, Li X, Wang L, Sun CL, et al: Intravascular ultrasound
area strain imaging used to characterize tissue components and
assess vulnerability of atherosclerotic plaques in a rabbit model.
Ultrasound Med Biol. 37:1579–1587. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Method. 25:402–408. 2001.
View Article : Google Scholar
|
|
34
|
Scalia R, Gooszen ME, Jones SP, Hoffmeyer
M, Rimmer DM III, Trocha SD, Huang PL, Smith MB, Lefer AM and Lefer
DJ: Simvastatin exerts both anti-inflammatory and cardioprotective
effects in apolipoprotein E-deficient mice. Circulation.
103:2598–2603. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yi GW, Zeng QT, Mao XB, Cheng M, Yang XF,
Liu HT, Mao Y, Guo M, Ji QW and Zhong YC: Overexpression of CXCL16
promotes a vulnerable plaque phenotype in Apolipoprotein E-Knockout
mice. Cytokine. 53:320–326. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yi GW and Zeng QT: Circulating CXCL16 is
related to the severity of coronary artery stenosis. Arch Med Res.
39:531–535. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Jansson AM, Aukrust P, Ueland T, Smith C,
Omland T, Hartford M and Caidahl K: Soluble CXCL16 predicts
long-term mortality in acute coronary syndromes. Circulation.
119:3181–3188. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Laugsand LE, Asvold BO, Vatten LJ, Janszky
I, Platou C, Michelsen AE, Arain F, Damås JK, Aukrust P and Ueland
T: Soluble CXCL16 and risk of myocardial infarction: The HUNT study
in Norway. Atherosclerosis. 244:188–194. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ogawa Y, Tasaka S, Yamada W, Saito F,
Hasegawa N, Miyasho T and Ishizaka A: Role of Toll-like 4 in
hyperoxia-induced lung inflammation in mice. Inflamm Res.
56:334–338. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Mudaliar H, Pollock C, Ma J, Wu H, Chadban
S and Panchapakesan U: The role of TLR2 and 4-mediated inflammatory
pathways in endothelial cells exposed to high glucose. PLoS One.
9:e1088442014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Campo GM, Avenoso A, Nastasi G, Micali A,
Prestipino V, Vaccaro M, D'Ascola A, Calatroni A and Campo S:
Hyaluronan reduces inflammation in experimental arthritis by
modulating TLR-2 and TLR-4 cartilage expression. Biochim Biophys
Acta. 1812:1170–1181. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Etemadi N, Chopin M, Anderton H, Tanzer
MC, Rickard JA, Abeysekera W, Hall C, Spall SK, Wang B, Xiong Y, et
al: TRAF2 regulates TNF and NF-κB signalling to suppress apoptosis
and skin inflammation independently of Sphingosine kinase 1. Elife.
4:pii: e10592. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Lehrke M, Millington SC, Lefterova M,
Cumaranatunge RG, Szapary P, Wilensky R, Rader DJ, Lazar MA and
Reilly MP: CXCL16 is a marker of inflammation, atherosclerosis, and
acute coronary syndromes in humans. J Am Coll Cardiol. 49:442–449.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Izquierdo MC, Sanz AB, Mezzano S, Blanco
J, Carrasco S, Sanchez-Niño MD, Benito-Martín A, Ruiz-Ortega M,
Egido J and Ortiz A: TWEAK (tumor necrosis factor-like weak inducer
of apoptosis) activates CXCL16 expression during renal
tubulointerstitial inflammation. Kidney Int. 81:1098–1107. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zhang G and Ghosh S: Toll-like
receptor-mediated NF-kappaB activation: A phylogenetically
conserved paradigm in innate immunity. J Clin Invest. 107:13–19.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Beutler B: Tlr4: Central component of the
sole mammalian LPS sensor. Curr Opin Immunol. 12:20–26. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Johnson SM, Grosshans H, Shingara J, Byrom
M, Jarvis R, Cheng A, Labourier E, Reinert KL, Brown D and Slack
FJ: RAS is regulated by the let-7 microRNA family. Cell.
120:635–647. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Sun X, He S, Wara AKM, Icli B, Shvartz E,
Tesmenitsky Y, Belkin N, Li D, Blackwell TS, Sukhova GK, et al:
Systemic delivery of microRNA-181b inhibits nuclear factor-κB
activation, vascular inflammation, and atherosclerosis in
apolipoprotein E-deficient mice. Circ Res. 114:32–40. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Santini P, Politi L, Vedova PD, Scandurra
R and d'Abusco Scotto A: The inflammatory circuitry of miR-149 as a
pathological mechanism in osteoarthritis. Rheumatol Int.
34:711–716. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Fish JE and Cybulsky MI: Taming
endothelial activation with a microRNA. J Clin Invest.
122:1967–1970. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Fang Y, Shi C, Manduchi E, Civelek M and
Davies PF: MicroRNA-10a regulation of pro-inflammatory phenotype in
athero-susceptible endothelium in vivo and in vitro. Proc Natl Acad
Sci USA. 107:13450–13455. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Sun X, Icli B, Wara AK, Belkin N, He S,
Kobzik L, Hunninghake GM, Vera MP; MICU Registry, ; Blackwell TS,
et al: MicroRNA-181b regulates NF-κB-mediated vascular
inflammation. J Clin Invest. 122:1973–1990. 2012.PubMed/NCBI
|
|
53
|
Li K, Ching D, Luk FS and Raffai RL:
Apolipoprotein E enhances microRNA-146a in monocytes and
macrophages to suppress nuclear factor-κB-driven inflammation and
atherosclerosis. Circ Res. 117:e1–e11. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Cheng HS, Sivachandran N, Lau A, Boudreau
E, Zhao JL, Baltimore D, Delgado-Olguin P, Cybulsky MI and Fish JE:
MicroRNA-146 represses endothelial activation by inhibiting
pro-inflammatory pathways. EMBO Mol Med. 5:1017–1034. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Zhang L, Chopp M, Liu X, Teng H, Tang T,
Kassis H and Zhang ZG: Combination therapy with VELCADE and tissue
plasminogen activator is neuroprotective in aged rats after stroke
and targets microRNA-146a and the toll-like receptor signaling
pathway. Arterioscler Thromb Vasc Biol. 32:1856–1864. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Gao M, Wang X, Zhang X, Ha T, Ma H, Liu L,
Kalbfleisch JH, Gao X, Kao RL, Williams DL, et al: Attenuation of
cardiac dysfunction in polymicrobial sepsis by MicroRNA-146a is
mediated via targeting of IRAK1 and TRAF6 expression. J Immunol.
195:672–682. 2015. View Article : Google Scholar : PubMed/NCBI
|