Introduction
Diabetic nephropathy is one of the most serious
microvascular complications of diabetes and is the main cause of
end-stage renal disease (ESRD). Strict control of glucose and blood
pressure may delay the progression of diabetic nephropathy;
however, many patients still progress to ESRD (1). The pathophysiological mechanism(s) of
diabetic nephropathy is complex and has not been fully elucidated.
Hyperglycemia status, the activation of the
renin-angiotensin-aldosterone system (RAAS), oxidative stress,
inflammation and dysregulation of various angiogenic factors have
been shown to be involved. In high glucose conditions, advanced
glycation end products (AGEs) and receptor for advanced glycation
end products (RAGE) accelerated diabetic nephropathy via a complex
reaction. In addition, the other endogenous ligand for RAGE, high
mobility group box 1 (HMGB1) protein, also proved to play a role in
diabetic nephropathy. HMGB1 protein is a member of the high
mobility group nuclear protein family, and is one of the most
evolutionarily conserved proteins (2). Normally, HMGB1 is expressed in the
cell nuclei; occasionally, it is released from cells through
passive release, which occurs as a result of cellular necrosis in
most eukaryotic cells (3).
Extracellular HMGB1 is involved in several inflammatory diseases,
such as septic shock, systemic lupus erythematosus, autoimmune
hepatitis, rheumatoid arthritis and so on (4). Growing evidence shows that HMGB1 is
associated with diabetic nephropathy and is highly expressed in
both the cytoplasmic and nuclear compartments of renal glomerular
cells and tubular epithelial cells of diabetic animal models;
however, the exact mechanism is still unknown.
As an internal ligand for RAGE, HMGB1 has been shown
to interact with RAGE and to activate various intracellular signal
transduction processes, including inflammation, proliferation,
apoptosis, autophagy, and migration (5). Kim et al (3) first proved that HMGB1 is extensively
expressed in renal tissues of diabetic rats, and HMGB1 is released
from renal glomerular cells and renal tubular epithelial cells, and
it participates in the occurrence of diabetic nephropathy by
signaling through RAGE. RAGE expression and nuclear factor (NF)-κB
activity were also elevated in diabetic rats where the binding of
NF-κB to the RAGE promoter was increased. In addition, they also
found hyperglycemia-induced HMGB1 increased NF-κB activity.
Moreover, NF-κB activation is a major intracellular signaling
pathway of RAGE. Extracellular HMGB1 is able to induce a signaling
cascade that activates NF-κB, leading to the synthesis of
proinflammatory cytokines. Hyperglycemia-induced HMGB1 release may
induce renal injury in diabetic rats, and the pathogenic role of
HMGB1 might be dependent on RAGE through activation of NF-κB.
The kidney plays an important role in controlling
glucose homeostasis via gluconeogenesis and the reabsorption of
filtered glucose in the proximal tubules (6). Nearly 180 g of glucose is filtered in
the glomerular filtrate every day. The kidney absorbs ~90% of this
glucose through SGLT-2, which is located on the apical side of
proximal tubule cells and transports sodium and glucose
concurrently (7). In patients with
type 2 diabetes, SGLT-2 proteins can be upregulated, causing an
increase in the renal threshold for reabsorption of glucose, and
worsening hyperglycemia (8).
Increased glucose reabsorption results in renal hyperfiltration;
this is an important cause of renal damage in diabetic nephropathy.
The principal of pathological changes in diabetic nephropathy
include glomerular and tubulointerstitial lesions, while the latter
is more closely related to renal function decline. Reducing glucose
transport in human proximal tubular cells may lower the
inflammatory response and fibrosis of kidney tubules (7). Sodium-glucose co-transporter 2
(SGLT2) inhibitors are new recently developed oral hypoglycemic
agents that target the kidney and block the reabsorption of glucose
to achieve glycemic control. In addition, the hypoglycemic effect
is independent of insulin. Emerging data indicates that SGLT2
inhibitors have a protective effect on the kidney beyond the
glucose-lowering effects. Furthermore, SGLT-2 can achieve
hypotension and weight loss due to the drugs' diuretic effect, all
which are independent of its hypoglycemic effects (9).
Here, we propose the hypothesis that HMGB1 may bind
RAGE to activate NF-κB and lead to various intracellular
mechanisms, finally resulting in renal injury. Therefore,
activation of the HMGB1-RAGE-NF-κB signaling pathway could worsen
diabetic nephropathy; however, SGLT-2 inhibitors may block this
signaling pathway to achieve renoprotection. To test this
hypothesis, we designed a study that used dapagliflozin, a new
SGLT-2 inhibitor to treat HK-2 cells that were exposed to high
glucose to explore the effect of SGLT-2 inhibitors on diabetic
nephropathy.
Materials and methods
Cell culture and treatment
The human proximal tubular cell line (HK-2) was
bought from the Shanghai Institute for Biological Sciences
(Shanghai, China) and was cultured in Dulbecco's modified Eagle's
medium/Nutrient Mixture F-12 (DMEM/F-12 medium; GE Healthcare Life
Sciences; Logan, UT, USA). Cells were cultured in DMEM/F12 with 25
and 5 mmol/l glucose, respectively, and supplemented with 100 U/ml
penicillin and 100 mg/ml streptomycin plus 10% fetal bovine serum
(FBS; Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA).
Cells were incubated at 37°C, with 5% CO2. Dapagliflozin
(Selleck Chemicals, Houston, TX, USA) was added to the culture
medium (10 or 100 µM final concentration) for 48 h to reconfirm the
promotion of oxidative stress makers and inflammatory
cytokines.
RNA isolate and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was isolated from HK-2 cells using TRIzol
regent (Invitrogen; Thermo Fisher Scientific, Inc.) according to
the manufacturer's instructions, reverse transcribed into cDNA
using the Prime Script® RT reagent kit (Takara Bio,
Inc., Otsu, Japan). RT-qPCR was performed using the SYBR Premix Ex
Taq TM kit and the ABI prism 7000 sequence detection system
(Applied Biosystems; Thermo Fisher Scientific, Inc.). The
thermocycling conditions were as follows: 95°C for 30 sec, followed
by 40 cycles of 95°C for 5 sec and 60°C for 30 sec. Relative levels
of gene expression were determined using the housekeeping gene
GAPDH. RT-qPCR data were normalized to the expression of GAPDH, and
relative expressions were calculated using the 2−∆∆Cq
method (10). The primer sequences
are presented in Table I.
 | Table I.Primers for reverse
transcription-quantitative polymerase chain reaction. |
Table I.
Primers for reverse
transcription-quantitative polymerase chain reaction.
| Target | Primer (5′-3′) |
|---|
| MDA | Forward,
TTGCCTGGGTTTTACCCTGC |
|
| Reverse,
AAGGCTTCCCACAGTTTCTGG |
| SOD | Forward,
GGTGGGCCAAAGGATGAAGAG |
|
| Reverse,
CCACAAGCCAAACGACTTCC |
| MCP-1 | Forward,
CAGCCAGATGCAATCAATGCC |
|
| Reverse,
TGGAATCCTGAACCCACTTCT |
| ICAM-1 | Forward,
ATGCCCAGACATCTGTGTCC |
|
| Reverse,
GGGGTCTCTATGCCCAACAA |
| Collagen I | Forward,
CCTGCTGGGATATTAGCTCCA |
|
| Reverse,
CAGCGGTAGGTGTCGAAGC |
| Fibronectin | Forward,
CGGTGGCTGTCAGTCAAAG |
|
| Reverse,
AAACCTCGGCTTCCTCCATAA |
| GAPDH | Forward,
CGGAGTCAACGGATTTGGTCGTAT |
|
| Reverse,
AGCCTTCTCCATGGTGGTGAAGAC |
Antibodies and western blot
analysis
HK-2 cells were collected and lysed in Radio
Immunoprecipitation Assay lysis buffer and protease inhibitor
mixture (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany). The
supernatant of HK-2 cells was harvested and lysed in ice-cold radio
immunoprecipitation assay buffer (RIPA), then separated by SDS-PAGE
and blotted to polyvinylidene fluoride membranes (PVDF; Bio-Rad
Laboratories, Inc., Shanghai, China). The blots were blocked with
5% skimmed milk, followed by incubation with antibodies against
malondialdehyde (MDA), superoxide dismutase (SOD), monocyte
chemoattractant protein-1 (MCP-1), intercellular adhesion
molecule-1 (ICAM-1; all Santa Cruz Biotechnology, Inc., Dallas, TX,
USA), fibronectin (FN), collagenase type 1 (COL-1) and HMGB1, RAGE
NF-κB and GAPDH (all Abcam, Cambridge, MA, USA) antibody,
horseradish peroxidase (HRP) labeled sheep anti-rabbit or sheep
anti-mouse were bought from Santa Cruz Biotechnology, Inc. Blots
were visualized using the enhanced chemiluminescent (ECL) detection
system. Blots were then incubated with species-specific horseradish
peroxidase-conjugated secondary antibodies (Santa Cruz
Biotechnology, Inc.), and visualized using Scion 146 Image software
(Scion Corporation, Frederick, MD, USA).
Statistical analysis
SPSS version 20.0 statistical software (IBM Corp.,
Armonk, NY, USA) was used for all data normality tests. All values
were expressed as the mean ± standard deviation. One-way analysis
of variance with Tukey's post hoc test was performed for
statistical analysis of differences between groups. P<0.05 was
considered to indicate a statistically significant difference.
Results
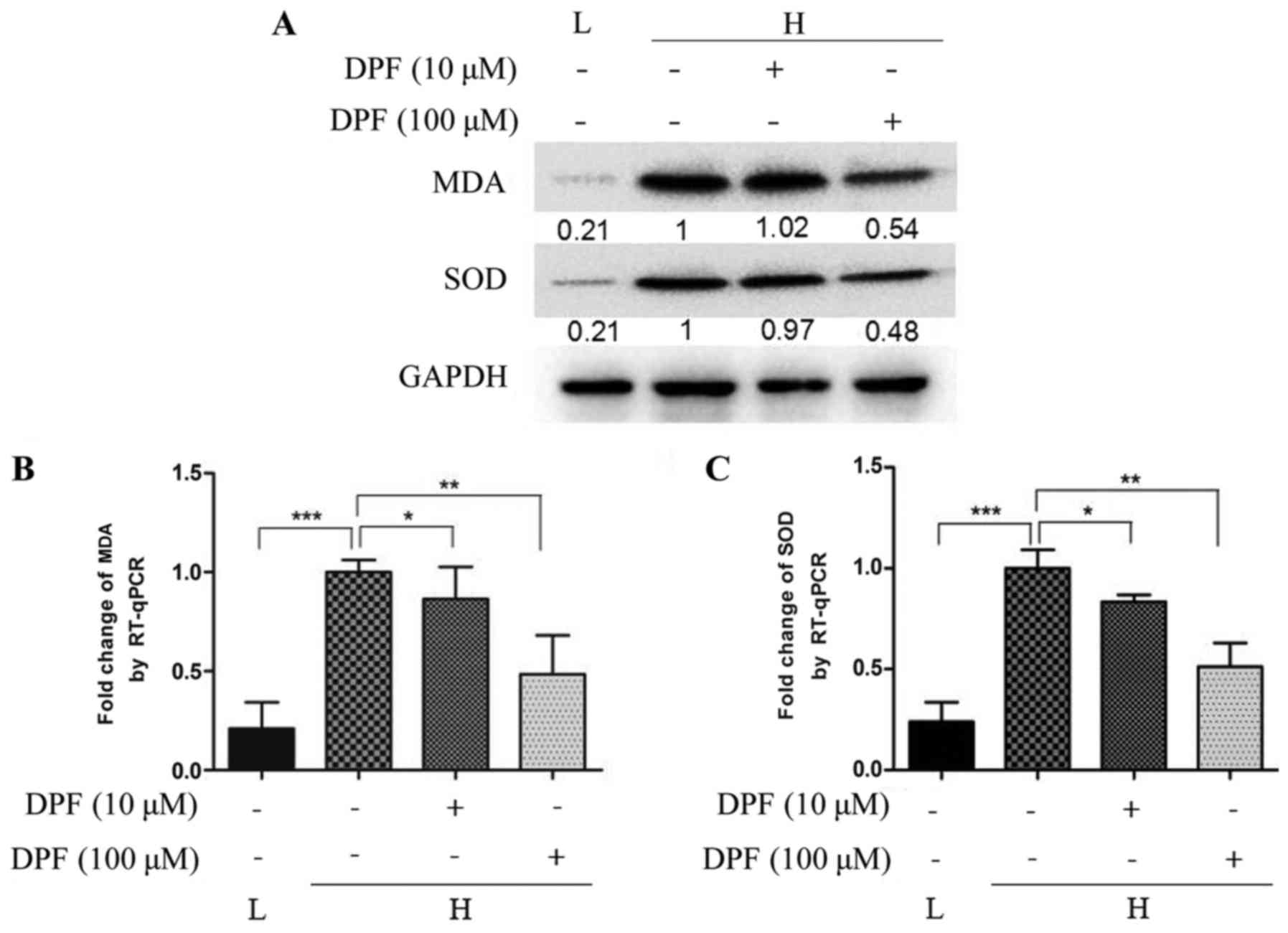
Dapagliflozin inhibits oxidative
stress induced by high glucose in HK-2 cells
As shown in Fig. 1,
mRNA and protein levels of the oxidative stress makers MDA and SOD
were higher in high glucose conditions compared to the control
group. This effect was inhibited by dapagliflozin; however, the
expression of MDA and SOD was lowest with 100 µM dapagliflozin,
suggesting that the inhibitory effect is positively correlated with
the concentration of dapagliflozin.
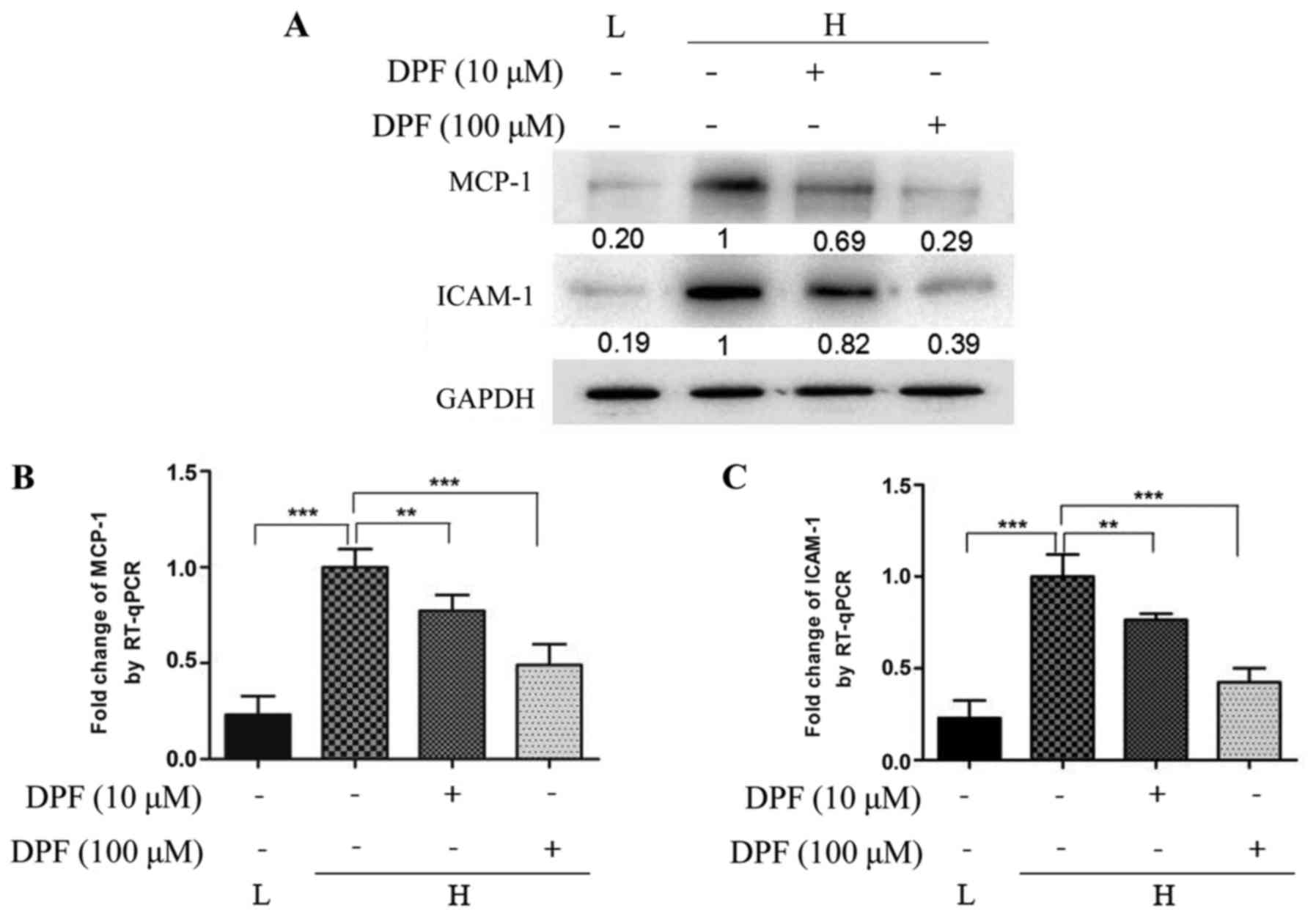
Dapagliflozin inhibits inflammation
induced by high glucose in HK-2 cells
Inflammation mediated by high glucose plays an
important role in the development of diabetic nephropathy. As shown
in Fig. 2, we detected the level
of the inflammatory factors MCP-1 and ICAM-1 in cultured HK-2 cells
by RT-qPCR and western blot. The mRNA and protein levels of MCP-1
and ICAM-1 were increased in high glucose conditions, and could be
reduced by dapagliflozin. The effect of inhibition was more
noticeable with the high concentration of dapagliflozin.
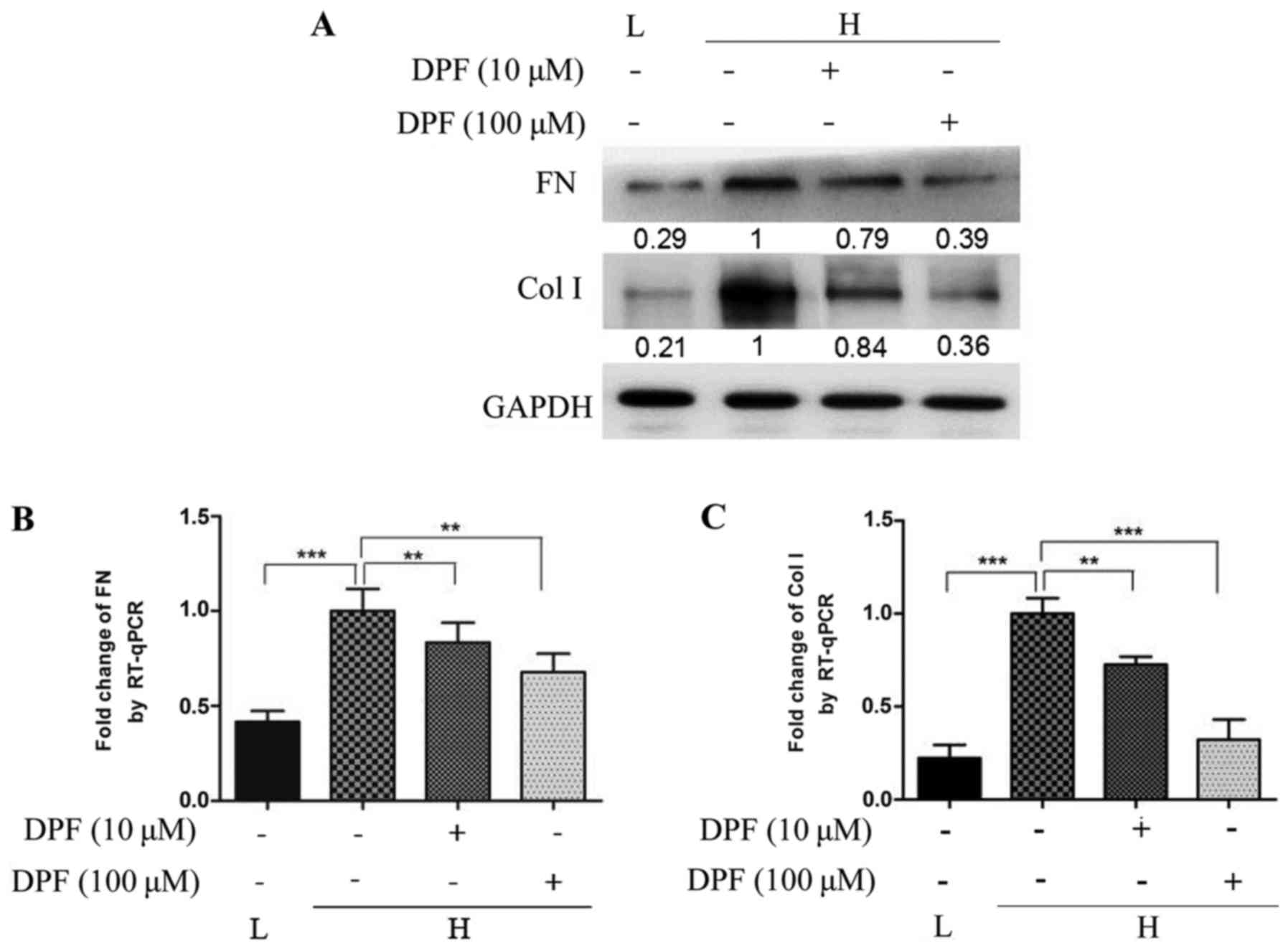
Dapagliflozin inhibits fibrosis
induced by high glucose in HK-2 cells
Tubulointerstitial fibrosis correlates more closely
with deterioration of renal function than histological changes in
the glomerulus. Interstitial fibrosis accompanies proximal tubular
cell basement membrane thickening, hyperplasia and hypertrophy and
ultimately correlates with the demise of renal function. As shown
in Fig. 3, We evaluated FN and Col
1, as indicators of fibrosis, and found that high glucose increased
the level of FN and Col 1 protein and mRNA compared to the control
group. However, the expression of FN and Col 1 in HK-2 cells
treated with dapagliflozin was significantly decreased and lowest
in the high concentration dapagliflozin group.
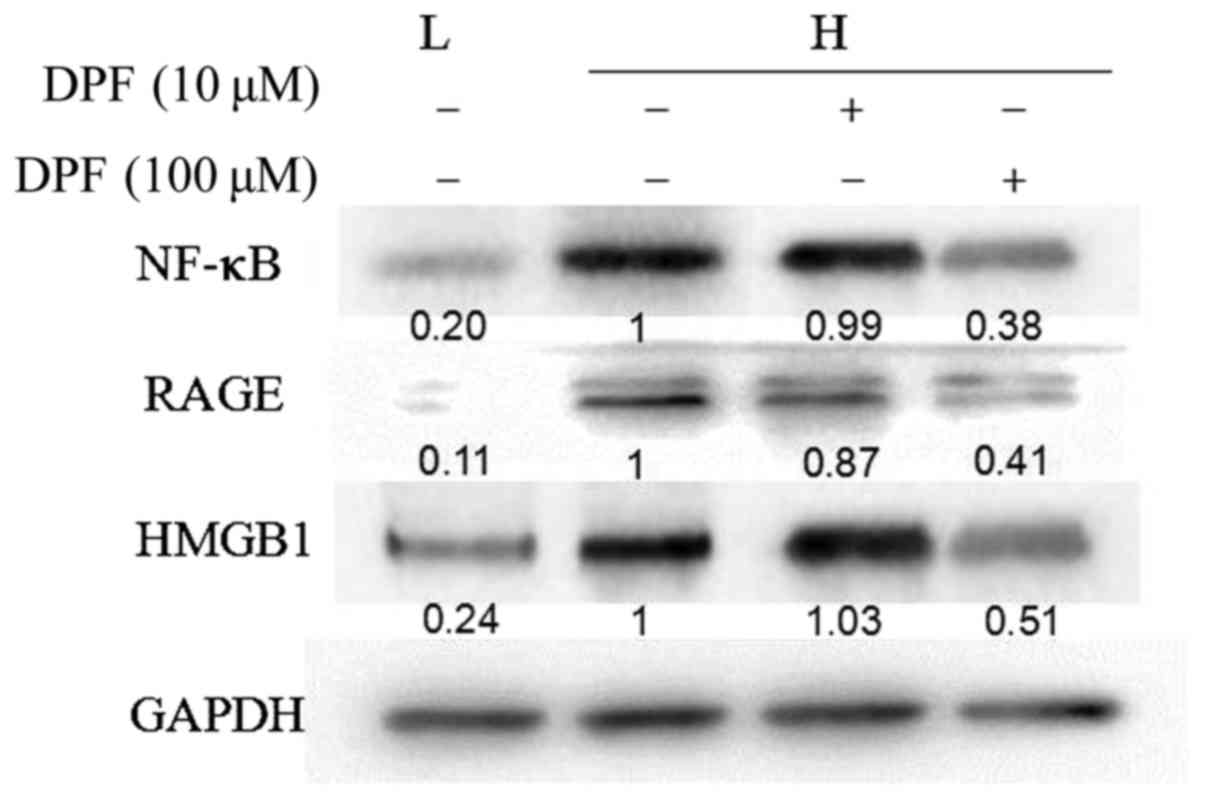
Dapagliflozin inhibits the activation
of the HMGB1-RAGE-NF-κB signal pathway induced by high glucose in
HK-2 cells
As an indicator of inflammation, HMGB1 is proven to
be involved in the development of diabetic nephropathy though
multiple intracellular reactions, and RAGE and NF-κB were key to
these processes.
As shown in Fig. 4,
the protein expression of HMGB1, RAGE and NF-κB was significantly
increased in HK-2 cells treated with high glucose, while they were
decreased in dapagliflozin groups, and lowest in the high
concentration group.
Discussion
SGLT-2 inhibitors are the latest class of
hypoglycemic agents and have a unique mechanism of lowering glucose
independent of insulin. SGLT-2 inhibitors are used for treating
type 2 diabetes at present, and numerous studies have demonstrated
that SGLT-2 inhibitors can decrease the level of serum glucose,
HbA1c, and hypertension, and increase weight loss. However, the
effect and mechanism of SGLT-2 inhibitors on diabetic nephropathy
is not fully understood.
Oxidative stress, inflammation, and fibrosis
mediated by high glucose is directly responsible for pathological
changes in diabetic nephropathy. Together, this results in the
classic structural and functional alterations of diabetic kidney
disease with alterations in glomerular permeability, glomerular
hyperfiltration, glomerular basement membrane thickening, mesangial
matrix synthesis, and ultimately the development of
glomerulosclerosis and interstitial fibrosis (11).
HMGB1 expression is upregulated in high glucose
conditions; the expression of RAGE, a potential receptor for HMGB1,
and NF-κB activity also play important roles in diabetic
nephropathy. In addition, diabetes enhances the binding of NF-κB to
the RAGE promoter. HMGB1 has been proposed as a potential causative
factor of renal damage (12). RAGE
is found on podocytes and tubular epithelial cells, while HMGB1
acts directly on RAGE and participates in the induction of diabetic
nephropathy. NF-κB is an important transcription factor involved in
the regulation of inflammation, immune responses, cell survival and
proliferation (13). In addition,
it is reported that HMGB1 increases NF-κB activity (3), and NF-κB activation is a main
intracellular signaling pathway of RAGE (14). Moreover, NF-κB signaling pathways
are essential for the destruction of tubular epithelial cells
(15). In the present study, we
used dapagliflozin, a novel selective SGLT-2 inhibitor to treat
HK-2 cells that were exposed to high glucose. We then detected the
gene and protein expression of MDA, SOD, MCP-1, ICAM-1, FN and
COL-1 to observe the renoprotective effect of the SGLT-2 inhibitor.
The involvement of RAGE and the NF-κB signaling pathway was also
studied.
Lowering of glucose levels can slow nephropathy
progression but takes at least 10 years to achieve clinically
relevant outcomes (16). However,
the hypoglycemic effect of SGLT-2 inhibitors is independent of
insulin and directly acts on renal proximal tubules. The SGLT2
inhibitor has also been associated with a 10–15% reduction in
plasma uric acid levels as a result of enhanced glycosuria, leading
to the secretion of uric acid in exchange for glucose reabsorption
through the GLUT9 transporter (17), and the effect of lowering serum
uric acid by SGLT2 inhibition might ameliorate endothelial
dysfunction, hypertension, and microvascular injury in diabetes
(18,19).
Moreover, SGLT2 inhibitors were effective in
decreasing the albuminuria in patients with type 2 diabetes and
hypertension through RAAS inhibition after adjusting for changes in
HbA1C, blood pressure, body weight, and estimated glomerular
filtration rate (GFR) (20).
Investigations into the extra effects of SGLT2 inhibitors beyond
glucose reduction have been established, and the mechanisms
underlying the renoprotection induced by SGLT2 inhibitors are
gradually becoming the focus of research. Oxidative stress is the
initial stage of diabetic nephropathy and activates a variety of
pathological pathways in virtually all types of kidney cells
(21).
In our study, we found an increase in MDA and SOD
production in HK-2 cells in the high glucose group compared to the
control group. Dapagliflozin inhibited the production of MDA and
SOD induced by hyperglycaemia, indicating that dapagliflozin could
prevent the oxidative stress process in vitro, and this
effect is concentration-dependent since the levels of MDA and SOD
were lowest in the high concentration dapagliflozin (100 µM) group.
The development of diabetic nephropathy is associated with
significant inflammatory cell infiltration and increasing plasma
levels of inflammatory cytokines. These cytokines act in a
paracrine or autocrine manner and induce a variety of effects on
different renal structures, playing a significant role in the
development and progression of several renal disorders (22). MCP-1 and ICAM-1 are recognized as
proinflammatory cytokines that participate in cytokine-associated
signaling pathways, and engage in the pathogenesis of diabetic
nephropathy through different actions, including intrarenal
hemodynamic alterations, modifications of the renal structure,
abnormalities in the expression of diverse molecules, cellular
necrosis and apoptosis, modification in the permeability of
glomerular endothelium, and increment in the production of ROS
(23). Our data showed that MCP-1
and ICAM-1 were highly expressed in the high glucose group but
expression could be decreased by dapagliflozin, which suggests that
the SGLT-2 inhibitor may achieve its renal protection through
inhibiting the inflammatory process. Similar results were reported
by Panchapakesan et al (9).
Numerous studies have shown that oxidative stress and inflammation
promote renal tubule and interstitial fibrosis, resulting in
irreversible renal damage, eventually leading to end-stage
nephropathy. In our study, we examined the expression of FN and
COL1 which act as makers of renal fibrosis, and we observed that
dapagliflozin reduced the level of FN and COL1 induced by
hyperglycaemia in a dose-dependent manner. These results suggest
that dapagliflozin ameliorates the renal injury of diabetic
nephropathy by reducing oxidative stress, inflammation and fibrosis
in diabetic kidneys. However, the exact mechanism of renoprotection
is unknown. In addition, we also observed the changes of HMGB1,
RAGE and NF-κB in the presence or absence of dapagliflozin. HMGB1,
RAGE and NF-κB were highly expressed in the high glucose group and
were inhibited by dapagliflozin. We propose the hypothesis that
hyperglycaemia promotes the progression of oxidative stress,
inflammation and fibrosis, and results in the occurrence of
diabetic nephropathy via activating the HMGB1-RAGE-NF-κB signaling
pathway, and this effect could be prevented by SGLT-2
inhibition.
In conclusion, dapagliflozin has renoprotective
effects through its anti-oxidative stress and anti-inflammatory
action, and our results suggest the effects might be achieved via
inhibition of the HMGB1-RAGE- NF-κB signaling pathway. However,
further studies are also needed.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Jiangsu
Provincial Health and Family Planning Commission (grant no.
H201458) and the Nanjing Medical University School Fund (grant no.
2016NJM140).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
DY conceived the study, designed the experiments and
wrote the manuscript. SW performed the experiments and analyzed the
data. MY performed the experiments and organized the figures. WL
designed the experiments, contributed to the writing of the
manuscript and revised the manuscript. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Xue R, Gui D, Zheng L, Zhai R, Wang F and
Wang N: Mechanistic insight and management of diabetic nephropathy:
Recent progress and future perspective. J Diabetes Res.
2017:18398092017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhu P, Xie L, Ding HS, Gong Q, Yang J and
Yang L: High mobility group box 1 and kidney diseases (Review). Int
J Mol Med. 31:763–768. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kim J, Sohn E, Kim CS, Jo K and Kim JS:
The role of high-mobility group box-1 protein in the development of
diabetic nephropathy. Am J Nephrol. 33:524–529. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Scaffidi P, Misteli T and Bianchi ME:
Release of chromatin protein HMGB1 by necrotic cells triggers
inflammation. Nature. 418:191–195. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Xie J, Méndez JD, Méndez-Valenzuela V and
Aguilar-Hernández MM: Cellular signalling of the receptor for
advanced glycation end products (RAGE). Cell Signal. 25:2185–2197.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kawanami D, Matoba K, Takeda Y, Nagai Y,
Akamine T, Yokota T, Sango K and Utsunomiya K: SGLT2 inhibitors as
a therapeutic option for diabetic nephropathy. Int J Mol Sci.
18(pii): E10832017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Komala MG, Panchapakesan U, Pollock C and
Mather A: Sodium glucose cotransporter 2 and the diabetic kidney.
Curr Opin Nephrol Hypertens. 22:113–119. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Moses RG, Colagiuri S and Pollock C: SGLT2
inhibitors: New medicines for addressing unmet needs in type 2
diabetes. Australas Med J. 7:405–415. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Panchapakesan U, Pegg K, Gross S, Komala
MG, Mudaliar H, Forbes J, Pollock C and Mather A: Effects of SGLT2
inhibition in human kidney proximal tubular cells-renoprotection in
diabetic nephropathy? PLoS One. 8:e544422013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gallagher H and Suckling RJ: Diabetic
nephropathy: Where are we on the journey from pathophysiology to
treatment? Diabetes Obes Metab. 18:641–647. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Penfold SA, Coughlan MT, Patel SK,
Srivastava PM, Sourris KC, Steer D, Webster DE, Thomas MC, MacIsaac
RJ, Jerums G, et al: Circulating high-molecular-weight RAGE ligands
activate pathways implicated in the development of diabetic
nephropathy. Kidney Int. 78:287–295. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Karin M and Ben-Neriah Y: Phosphorylation
meets ubiquitination: The control of NF-[kappa]B activity. Annu Rev
Immunol. 18:621–663. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yan SD, Schmidt AM, Anderson GM, Zhang J,
Brett J, Zou YS, Pinsky D and Stern D: Enhanced cellular oxidant
stress by the interaction of advanced glycation end products with
their receptors/binding proteins. J Biol Chem. 269:9889–9897.
1994.PubMed/NCBI
|
|
15
|
Morcos M, Sayed AA, Bierhaus A, Yard B,
Waldherr R, Merz W, Kloeting I, Schleicher E, Mentz S, Abd el Baki
RF, et al: Activation of tubular epithelial cells in diabetic
nephropathy. Diabetes. 51:3532–3544. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zoungas S, Chalmers J, Neal B, Billot L,
Li Q, Hirakawa Y, Arima H, Monaghan H, Joshi R, Colagiuri S, et al:
Follow-up of blood-pressure lowering and glucose control in type 2
diabetes. N Engl J Med. 371:1392–1406. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lytvyn Y, Škrtić M, Yang GK, Yip PM,
Perkins BA and Cherney DZ: Glycosuria-mediated urinary uric acid
excretion in patients with uncomplicated type 1 diabetes mellitus.
Am J Physiol Renal Physiol. 308:F77–F83. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hovind P, Rossing P, Johnson RJ and
Parving HH: Serum uric acid as a new player in the development of
diabetic nephropathy. J Ren Nutr. 21:124–127. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Satirapoj B, Supasyndh O, Chaiprasert A,
Ruangkanchanasetr P, Kanjanakul I, Phulsuksombuti D, Utainam D and
Choovichian P: Relationship between serum uric acid levels with
chronic kidney disease in a Southeast Asian population. Nephrology
(Carlton). 15:253–258. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Heerspink HJ, Johnsson E, Gause-Nilsson I,
Cain VA and Sjöström CD: Dapagliflozin reduces albuminuria in
patients with diabetes and hypertension receiving renin-angiotensin
blockers. Diabetes Obes Metab. 18:590–597. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Inoguchi T and Nawata H: NAD(P)H oxidase
activation: A potential target mechanism for diabetic vascular
complications, progressive beta-cell dysfunction and metabolic
syndrome. Curr Drug Targets. 6:495–501. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Noronha IL, Niemir Z, Stein H and Waldherr
R: Cytokines and growth factors in renal disease. Nephrol Dial
Transplant. 10:775–786. 1995.PubMed/NCBI
|
|
23
|
Navarro-González JF and Mora-Fernández C:
The role of inflammatory cytokines in diabetic nephropathy. J Am
Soc Nephrol. 19:433–442. 2008. View Article : Google Scholar : PubMed/NCBI
|