Introduction
The incidence of obesity and aging has been
associated with Type 2 diabetes, a disease of increasing importance
in social health due to its prevalence. (1) Type 2 diabetes affects the homeostasis
of glucose metabolism and leads to serious complications, including
diabetic cardiomyopathy and diabetic nephropathy (2). At present, a cure for type 2 diabetes
is unavailable as the underlying mechanisms of the pathogenesis of
type 2 diabetes is not completely understood (3). Hyperglycaemia is characteristic of
type 2 diabetes (4). Hepatic
insulin resistance is a predominant cause of type 2 diabetes. In
the liver, glucose metabolism homeostasis is regulated via two
processes, gluconeogenesis and glycogenesis (5). Impaired insulin sensitivity in the
liver leads to increased gluconeogenesis and decreased glycogenesis
(6).
In the liver cells, insulin can decrease glucose
production and increase uptake of blood glucose into glycogen via
the phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K)/protein
kinase B (AKT) pathway (7).
Additionally, activated AKT promotes the phosphorylation of
forkhead box O1 (FOXO1), which can translocate into nuclear and
inhibit the expression of gluconeogenetic enzymes and
gluconeogenesis (8). Peroxisome
proliferator-activated receptor γ coactivator (PGC-1α) is another
important regulator of gluconeogenesis in liver cells. PGC-1α can
co-activate FOXO1 and enhance the transcription of
phosphoenolpyruvate carboxykinase (PEPCK) and glucose-6-phosphatase
(G6Pase). PGC-1α, PEPCK and G6Pase are key regulators of
gluconeogenesis in liver cells (9).
MicroRNAs (miRs) are a group of small, non-coding
RNAs. miRs negatively modulate gene expression by directly binding
to a partially complementary sequence in the 3′-untranslated region
(UTR) of their target mRNA (10,11).
Tumour necrosis factor (TNF)-α is an important mediator of insulin
resistance that functions by inhibiting the PI3K/AKT pathway.
Furthermore, in vitro and in vivo studies have
suggested that TNF-α treatment leads to increased gluconeogenesis
and decreased glycogenesis in hepatocytes (12). Our previous study demonstrated that
miR-338-3p overexpression restores impaired glycogenesis induced by
TNF-α by directly inhibiting protein phosphatase 4 regulatory
subunit 1 (PP4R1) expression (13). However, the association between
miR-338-3p and increased levels of gluconeogenesis induced by TNF-α
remains unknown. In the present study, the effects of miR-338-3p on
gluconeogenesis and the associated underlying mechanisms were
investigated. The results suggested that miR-338-3p may have an
important role in TNF-α-induced gluconeogenesis via targeting of
PP4R1.
Materials and methods
Animals
In order to establish a TNF-α-induced insulin
resistance model, 16-week-old male C57BL/6J mice (n=5; 30 g,
Beijing HFK Bioscience Co., Ltd., Beijing, China) were fed with a
standard laboratory diet and injected with 15 µg/ml TNF-α via Alzet
osmotic pumps (DURECT Corporation, Cupertino, CA, USA) for 2 weeks
continuously at an infusion rate of 0.5 µl/h. For the control
group, 16-week-old male C57BL/6J mice (n=5) were injected with 0.9%
NaCl via Alzet osmotic pumps for 2 weeks at the aforementioned
infusion rate. Following fasting for 8 h, mice were subsequently
anaesthetized via intrapertioneal injection with sodium
pentobarbital (40 mg/kg body weight), and liver samples were then
collected. All mice were housed in an experimental animal facility
of Beijing Normal University (Beijing, China) at a temperature of
20–24°C and humidity-controlled (45–55%) environment. A 12 h
light/dark cycle was maintained in the animal housing rooms. The
food and water were supplied for all days. All animal experiments
performed in the present study were approved by the Animal Use and
Care Committee of Beijing Normal University and were performed in
accordance with the Guide for Care and Use of Laboratory Animals
(14).
Adenoviral vector injection
For animal experiments, 6 mice were used in each
group. To investigate the effect of miR-338-3p on TNF-α-induced
gluconeogenesis, after 7 days following the implantation of the
TNF-α pumps, 6 C57BL/6J mice were injected with adenoviral vectors
expressing miR-338-3p mimics (Ad-338m) (2×109
plaque-forming unit/25 g body weight). A total of 6 C57BL/6J mice
were injected with adenoviral vectors expressing negative control
microRNA (Ad-NC) (2×109 plaque-forming unit/25 g body
weight). In addition, adenoviral vectors expressing miR-338-3p
inhibitors (Ad-338i) or negative control inhibitors (Ad-NCI) were
injected into 6 C57BL/6J mice (2×109 plaque-forming
unit/25 g body weight). All the adenoviral vectors, including
Ad-338m, Ad-NC, Ad-338i and Ad-NCI were purchased from Shanghai
GeneChem Co., Ltd. (Shanghai, China). After 7 days following
injection, mice were used for following experiments. The sequences
employed in the present study were as follows (5′-3′): miR-338-3p
mimic, UCCAGCAUCAGUGAUUUUGU and negative control mimics,
UUCUCCGAACGUGUCACGUTT; miR-338-3p inhibitor, CAACAAAAUCACUGAUGCUGGA
and negative control inhibitors, CAGUACUUUUGUGUAGUACAA.
Pyruvate tolerance testing
After 7 days following injection, pyruvate tolerance
analysis was performed. Following fasting for 14 h, mice were
intraperitoneally injected with pyruvate (2 g/kg of body weight) in
order to perform pyruvate tolerance testing. Blood glucose levels
were measured using an ACCU-CHEK Advantage glucometer (Roche
Diagnostics GmbH, Mannheim, Germany) at specific time
intervals.
Cell culture
HEPA1-6 murine liver cells (American Type Culture
Collection, Manassas, VA, USA) were cultured in low-glucose
Dulbecco's modified Eagle's medium (DMEM; Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) supplemented with 10% fetal
serum (HyClone; GE Healthcare Life Sciences, Logan, UT, USA), 100
units/ml penicillin (Invitrogen; Thermo Fisher Scientific, Inc.)
and 0.1 mg/ml streptomycin (Hyclone; GE Healthcare Life Sciences)
at 37°C with humidified air and 5% CO2. In order to
determine the effect of TNF-α on gluconeogenesis, the HEPA1-6 cells
were treated with 10 ng/ml TNF-α for 24 h at 37°C with humidified
air and 5% CO2.
Transfection
miR-338-3p mimic and inhibitor sequences used for
transfection were as follows (5′-3′): miR-338-3p mimic sense,
UCCAGCAUCAGUGAUUUUGUUG and antisense, ACAAAAUCACUGAUGCUGGAUU; NC
sense, UUCUCCGAACGUGUCACGUTT and antisense, ACGUGACACGUUCGGAGAATT;
miR-338-3p inhibitor, CAACAAAAUCACUGAUGCUGGA; NCI,
CAGUACUUUUGUGUAGUACAA. All miR oligos were purchased from
Genepharm, Inc. (Sunnyvale, CA, USA). A day before transfection,
3.6×105 cells were seeded in each well of 6-well plate
in 1.6 ml DMEM medium. miR-338-3p mimic and inhibitor (3.75 µl, 20
µM) were transfected into HEPA1-6 by using HiPerFect transfection
reagent (Qiagen, Inc., Valencia, CA, USA). Subsequent
experimentation was performed 48 h following transfection.
The sequences of PP4R1-specific small interfering
(siRNAs) used for transfection were as follows (5′-3′): PP4R1
sense, GAACCAACTGTGAGAGCGGA; and antisense, TGGGGCAGTCAGGTCTATGA.
All siRNA oligos were purchased from Genepharm, Inc. A day before
transfection, 3.6×105 cells were planted in each well of
6-well plate in 1.6 ml DMEM. PP4R1-specific siRNAs (3.75 µl, 20 µM)
were transfected into HEPA1-6 cells using HiPerFect transfection
reagent (Qiagen, Inc.). Subsequent assays were performed 48 h
following transfection.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA of HEPA1-6 cells and mouse liver cells was
extracted using TRIzol (Invitrogen; Thermo Fisher Scientific,
Inc.). A total of 1 µg RNA was reverse transcribed into cDNA using
the EasyScript First-strand cDNA Synthesis kit (Beijing Transgen
Biotech Co., Ltd., Beijing, China). The procedures of RT were
described as follow: 70°C for 10 min, 42°C for 60 min, 95°C for 5
min and 4°C hold. Expression levels of PGC-1α, PEPCK and G6Pase
were determined via qPCR using the SYBR Green II kit (Takara
Biotechnology Co., Ltd., Dalian, China) in accordance with the
manufacturer's protocol. cDNA (1 µg), 10 µl SYBR-Green mixture, 0.8
µl primers (10 µm) and 8.2 µl ddH2O were mixed. The
procedures of qPCR were as follows: 95°C for 30 sec, followed by 40
cycles at 95°C for 5 sec and 60°C for 20 sec. The sequences of
primers used for PCR were as follows (5′-3′): G6Pase forward,
TCTGTCCCGGATCTACCTTG and reverse, GTAGAATCCAAGCGCGAAA; PEPCK
forward, TCTGAGATCTCTGATCCAGACC and reverse,
GAAGTCCAGACCGTTATGCAGC; PGC-1α forward, GCCTATGAGCACGAAAGGC and
reverse, TCACACGGCGCTCTTCAATT; 18s forward, GGAAGGGCACCACCAGGAGT
and reverse, TGCAGCCCCGGACATCTAAG. The gene expression level was
calculated using 2−ΔΔCq method (15).
Western blotting
Total protein from liver and cells was extracted
with radioimmunoprecipitation (Beijing Solarbio Science &
Technology Co., Ltd., Beijing, China). And the concentration of
protein sample was measure by BCA kit (Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany). Cell protein samples (15 µg) were separated
via 10% SDS-PAGE, transferred to polyvinylidene fluoride membranes
(EMD Millipore, Billerica, MA, USA) and then blocked using 8%
non-fat dry milk for 2 h at room temperature. Following this, the
membranes incubated with 1:1,000 diluted primary antibodies for 12
h at 4°C. The blots were then incubated with 1:5,000 diluted
horseradish peroxidase-conjugated anti-IgG (cat. no. 7074; Cell
Signaling Technology, Inc. Danvers, MA, USA) for 2 h at room
temperature. Proteins were then visualized using an enhanced
chemiluminescent kit (EMD Millipore). Antibodies against FOXO1
(cat. no. 2880), phosphorylated FOXO1 (cat. no. 9461), PEPCK (cat.
no. 6924) and GAPDH (cat. no. 5174) were purchased from Cell
Signaling Technology, Inc. Antibodies against PGC-1α (ab54481) and
G6Pase (ab83690) were purchased from Abcam (Cambridge, UK).
Luciferase reporter assay
TargetScan (http://www.targetscan.org/), Pictar (http://www.pictar.org/) and miRanda (http://microrna.org/) databases were used to predict
that PP4R1 was a target of miR-338-3p. The miR-338-3p binding site
with in the PP4R1 3′-UTR was verified and in previously study
(13). The PP4R1 3′-UTR fragment,
including the miR-338-3p binding site was cloned from mouse liver
cells and inserted into pmirGLO vector (Promega Corporation,
Madison, WI, USA). A day prior to transfection, HEPA1-6 cells were
seeded at 3.6×105 cells per well of 6-well plate in 1.6
ml DMEM. pmirGLO-3′UTR PP4R1 with miR-338-3p mimic or inhibitor
(3.75 µl, 20 µM) were transfected into HEPA1-6 by using HiPerFect
transfection reagent. The subsequent assays were performed after 48
h post-transfection. Luciferase activity was analyzed by using the
Dual-Glo Luciferase assay system (Promega Corporation). Luciferase
activity was normalized to Renilla luciferase activity. All
the assays were repeat for 4 times.
Glucose production assay
After 24 h of TNF-α treatment, a glucose production
assay was performed. Cells were washed five times with PBS and then
stimulated with 10 ng/ml TNF-α, 2 mmol/l sodium pyruvate and 20
mmol/l sodium lactate in glucose- and serum-free DMEM medium for 18
h. The glucose concentration in the medium was subsequently
determined using a glucose assay kit (Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany) and subsequently normalized to the previously
determined total protein content via a BCA kit (Sigma-Aldrich;
Merck KGaA) in whole cell extracts.
Statistical analysis
All data are presented as the mean ± standard error
of the mean. The two-tailed unpaired Student's t-test was used for
comparisons between two groups. In addition, One-way analysis of
variance followed by Tukey's post hoc test was used for comparisons
between more than two groups. Statistical analyses were performed
using SPSS 3.0 software (SPSS, Inc., Chicago, IL, USA). P<0.05
was considered to indicate a statistically significant difference.
All the experiments were repeated for four times.
Results
TNF-α treatment induces
gluconeogenesis in mouse livers and HEPA1-6 cells
In our previous study, it was demonstrated that
treatment with TNF-α decreased miR-338-3p expression levels and
suppressed the activation of the AKT/GSK pathway in the livers of
mice (13). In the present study,
treatment with TNF-α significantly decreased the level of
p-FOXO1/FOXO1 and significantly increased the protein and mRNA
expression levels of genes associated with gluconeogenesis in the
livers of mice, including PGC-1α, G6Pase and PEPCK (Fig. 1A and B). Consistently, treatment of
10 ng/ml TNF-α enhanced glucose production in HEPA1-6 cells
(Fig. 1C). Furthermore, in HEPA1-6
cells following treatment with 10 ng/ml TNF-α for 24 h, the levels
of p-FOXO1/FOXO1 were reduced, and the protein and mRNA expression
of PGC-1α, G6Pase and PEPCK were increased (Fig. 1D and E).
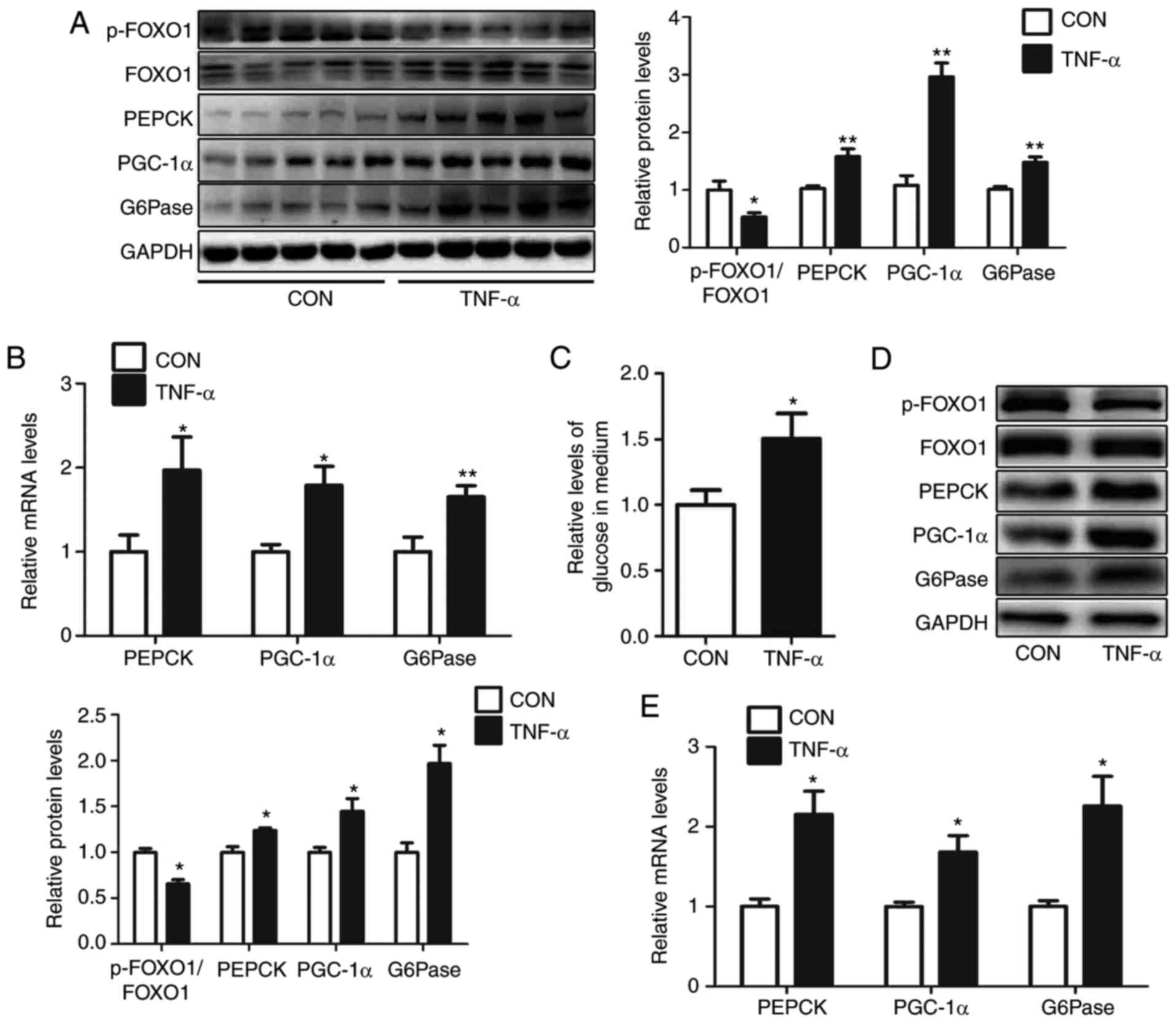 | Figure 1.TNF-α treatment induces
gluconeogenesis in mouse livers and HEPA1-6 hepatocytes. Levels of
p-FOXO1/FOXO1, PEPCK, PGC-1α and G6Pase in the livers of
TNF-α-treated mice were analyzed via (A) western blot and (B)
reverse transcription-quantitative polymerase chain reaction. (C)
Relative levels of glucose in culture medium were determined, as
well as (D) protein and (E) mRNA levels of p-FOXO1/FOXO1, PEPCK,
PGC-1α and G6Pase, in HEPA1-6 cells treated with TNF-α for 24 h.
Data are presented as the mean ± standard error. For animal
experiments, 5 mice were used for each group. For cell experiments,
n=4 independent experiments. *P<0.05 and **P<0.01 vs.
control. p-, phosphorylated; FOXO1, forkhead box O1; PEPCK,
phosphoenolpyruvate carboxykinase; G6Pase, glucose-6-phosphatase;
PGC-1α, peroxisome proliferator-activated receptor γ
coactivator-1α; TNF-α, tumor necrosis factor-α; CON, control. |
Overexpression of miR-338-3p
attenuates TNF-α-induced gluconeogenesis
To determine the importance of miR-338-3p in the
regultion of gluconeogenesis, an adenovirus expressing miR-338-3p
mimic was injected into TNF-α-treated mice via the tail vein for 7
days. The results revealed that levels of p-FOXO1/FOXO1 were
significantly increased, whereas mRNA and protein expression levels
of PGC-1α, G6Pase and PEPCK were significantly decreased, in the
livers of mice injected with Ad-338m compared with the control
group accompanied by ameliorated pyruvate tolerance (Fig. 2A-C). In HEPA1-6 cells, transfection
with miR-338-3p mimics was demonstrated to significantly attenuate
TNF-α-induced glucose production (Fig.
2D). In addition, upregulation of miR-338-3p was revealed to
significantly enhanced levels of p-FOXO1/FOXO1, and significantly
decreased the expression levels of PGC-1α, G6Pase and PEPCK,
compared with the NC group (Fig. 2E
and F). In conclusion, these results suggested that miR-338-3p
may have an important role in TNF-α-induced gluconeogenesis.
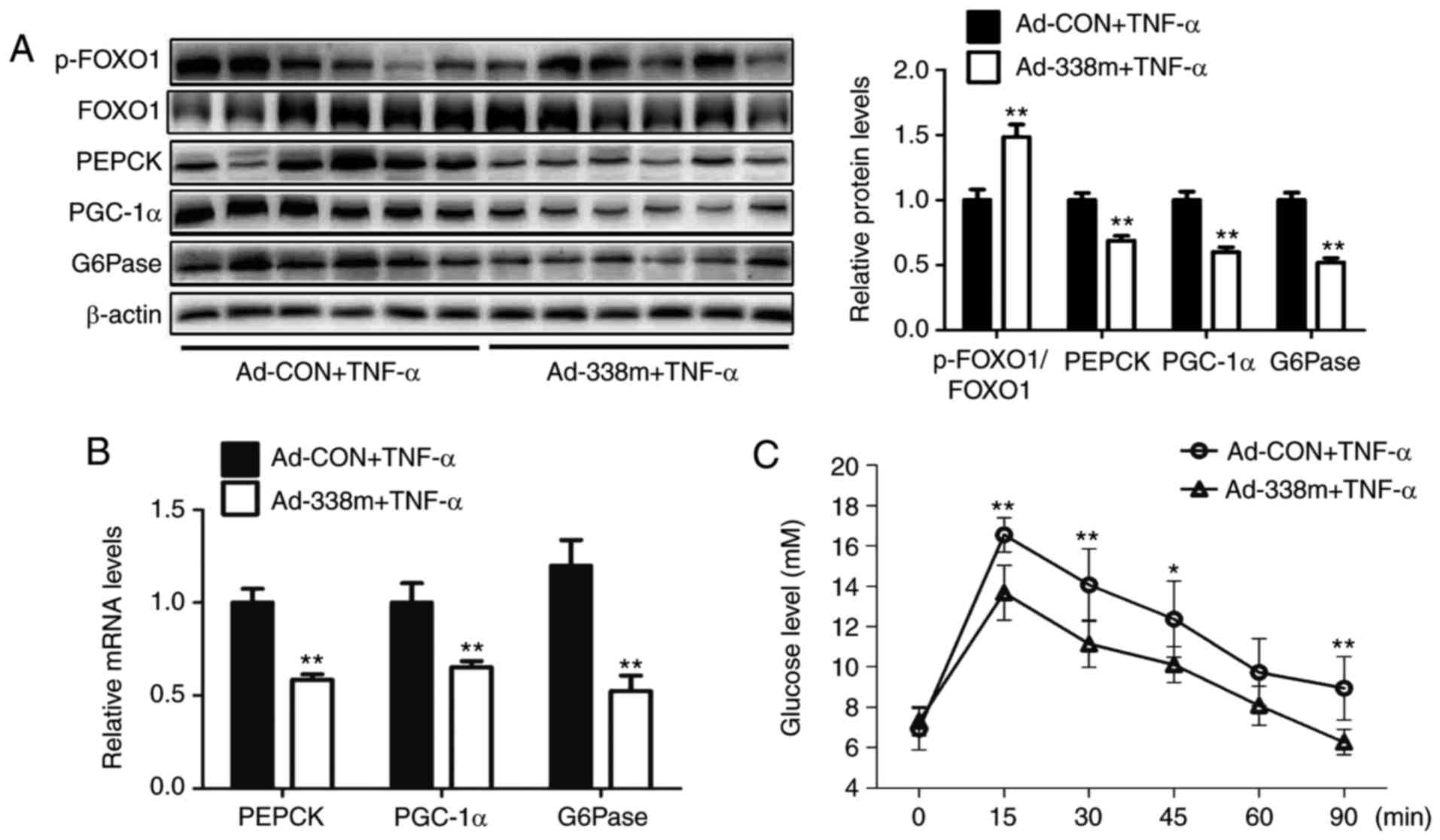 | Figure 2.Overexpression of miR-338-3p
attenuates TNF-α-induced gluconeogenesis. Levels of p-FOXO1/FOXO1,
PEPCK, PGC-1α and G6Pase in the livers of TNF-α-treated mice
injected with Ad-338m via the tail vein for 7 days were analyzed by
(A) western blot and (B) reverse transcription-quantitative
polymerase chain reaction. (C) Pyruvate tolerance testing using
mice was also performed. In cells, (D) relative levels of glucose
in medium were determined, as well as (E) relative mRNA levels of
PEPCK, PGC-1α and G6Pase, and (F) protein levels of p-FOXO1/FOXO1,
PEPCK, PGC-1α and G6Pase, in TNF-α-treated HEPA1-6 cells
transfected with miR-338-3p mimics. Data are presented as the mean
± standard error of the mean. For animal experiments, 6 mice were
used. For cell experiments, n=4 independent experiments. *P<0.05
and **P<0.01 vs. control or as indicated. miR, microRNA; p-,
phosphorylated; FOXO1, forkhead box O1; PEPCK, phosphoenolpyruvate
carboxykinase; PGC-1α, peroxisome proliferator-activated receptor γ
coactivator-1α; G6Pase, glucose-6-phosphatase; Ad, adenovirus; CON,
control; TNF-α, tumor necrosis factor-α; 338m, miR-338-3p mimic;
NC, mimic negative control. |
Treatment with miR-338-3p inhibitors
increases glucose production in hepatocytes
The effect of treatment with miR-338-3p inhibitors
on glucose production in hepatocytes was investigated in the
present study. An adenovirus expressing miR-338-3p inhibitor
(Ad-338i) was injected into mice via the tail vein over 7 days.
Inhibition of miR-338-3p decreased the levels of p-FOXO1/FOXO1 in
livers of mice; the protein and mRNA expression levels of PGC-1α,
G6Pase and PEPCK were increased in the liver, and pyruvate
tolerance was impaired in mice injected with Ad-338i (Fig. 3A-C). Furthermore, HEPA1-6
hepatocytes transfected with a miR-338-3p inhibitor exhibited
significantly reduced levels of p-FOXO1, increased levels of
glucose production, and increased mRNA and protein expression of
PGC-1α, G6Pase and PEPCK, compared with the NC group (Fig. 3D-F). These results suggested that
miR-338-3p may regulate glucose production via the AKT/FOXO1
pathway.
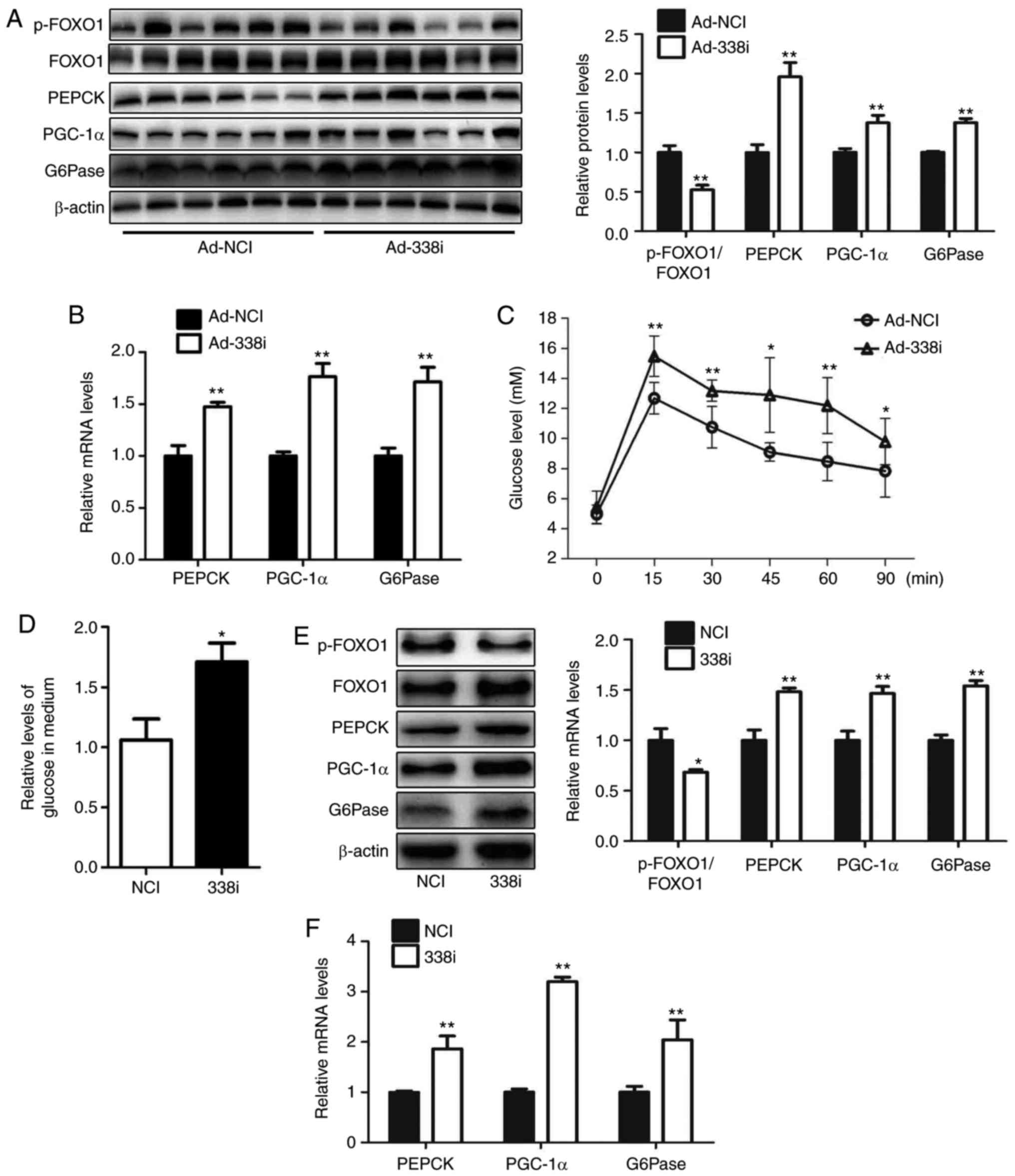 | Figure 3.Downregulation of miR-338-3p enhances
gluconeogenesis in hepatocytes. Levels of p-FOXO1/FOXO1, PEPCK,
PGC-1α and G6Pase in the livers of mice injected with Ad-338i via
the tail vein for 7 days were analyzed by (A) western blot and (B)
reverse transcription-quantitative polymerase chain reaction. (C)
Pyruvate tolerance testing using mice was performed. (D) Relative
levels of glucose in culture medium were also determined as well as
(E) levels of p-FOXO1, FOXO1, PEPCK, PGC-1α and G6Pase protein, and
(F) PEPCK, PGC-1α and G6Pase mRNA in HEPA1-6 cells transfected with
miR-338-3p inhibitors were determined. Data are presented as the
mean ± standard error. For animal experiments, 6 mice were used.
For cell experiments, n=4 independent experiments. *P<0.05 and
**P<0.01 vs. control. miR, microRNA; p-, phosphorylated; FOXO1,
forkhead box O1; PEPCK, phosphoenolpyruvate carboxykinase; PGC-1α,
peroxisome proliferator-activated receptor γ coactivator-1α;
G6Pase, glucose-6-phosphatase; Ad, adenovirus; NCI, negative
control inhibitor; 338i, miR-338-3p inhibitor. |
miR-338-3p is involved in
TNF-α-mediated gluconeogenesis via targeting of PP4R1
In our previous study, PP4R1 was revealed to
represent a target gene of miR-338-3p. The 3′-UTR of PP4R1 was
cloned and luciferase reporter vectors were subsequently
constructed (13). The relative
luciferase activity was significantly decreased following
transfection with miR-338-3p mimics (Fig. 4A); however, relative luciferase
activity was not markedly affected following transfection with
miR-338-3p inhibitors (Fig. 4B).
To verify whether miR-338-3p regulates glucose production via
targeting of PP4R1, miR-338-3p inhibitors and PP4R1 siRNA were
co-transfected into HEPA1-6 cells. The results revealed that
downregulation of PP4R1 significantly attenuated miR-338-3p-induced
increased levels of glucose production (Fig. 4C), and expression levels of genes
associated with gluconeogenesis. However, transfection of PP4R1
siRNA reversed the effect of miR-338-3p inhibitor on the level of
p-FOXO1/FOXO1 (Fig. 4D and E).
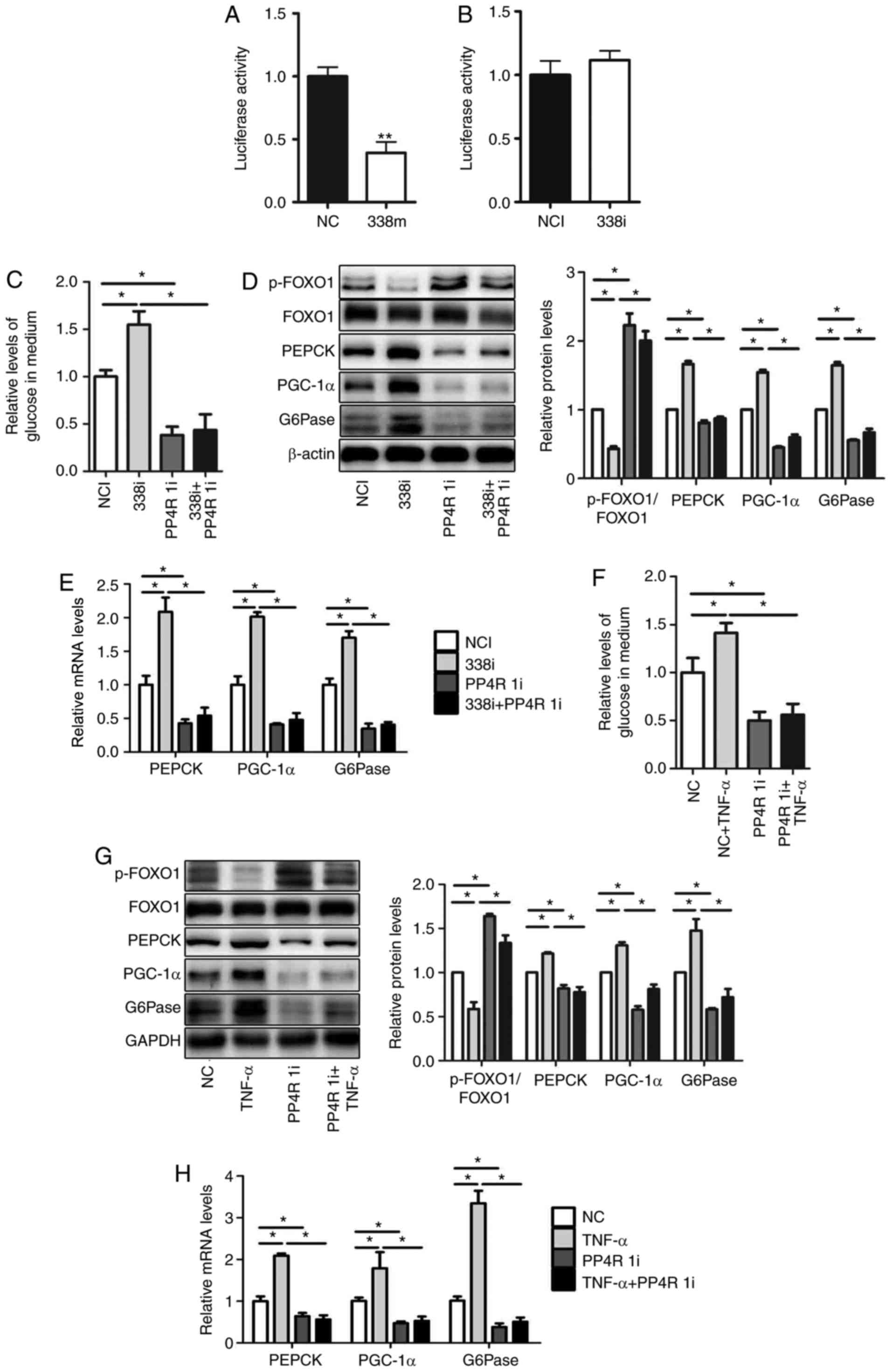 | Figure 4.miR-338-3p is involved in
TNF-α-mediated gluconeogenesis via targeting of PP4R1. PP4R1 is a
direct target of miR-338-3p. Luciferase activity was analyzed in
HEPA1-6 cells transfected with (A) miR-338 mimics (B) or
inhibitors. (C) Relative levels of glucose in medium, as well as
(D) levels of p-FOXO1/FOXO1, PEPCK, PGC-1α and G6Pase protein, and
(E) PEPCK, PGC-1α and G6Pase mRNA were determined in HEPA1-6 cells
co-transfected with miR-338-3p inhibitors and PP4R1 small
interfering RNA. (F) Relative levels of glucose in medium, as well
as (G) protein levels of p-FOXO1/FOXO1, PEPCK, PGC-1α and G6Pase,
and (H) PEPCK, PGC-1α and G6Pase mRNA were determined in HEPA1-6
cells treated with 10 ng/ml TNF-α for 24 h followed by transfection
with PP4R1 small interfering RNA for 48 h. Data are presented as
the mean ± standard error of the mean. Experiments were repeated
four times. *P<0.05 and **P<0.01 vs. control or as indicated.
miR, microRNA; NC, mimic negative control; 338m, miR-338-3p mimic;
NCI, negative control inhibitor; 338i, miR-338-3p inhibitor; PPR41,
protein phosphatase 4 regulatory subunit 1; PP4R 1i, PP4R siRNA;
p-, phosphorylated-; FOXO1, forkhead box O1; PEPCK,
phosphoenolpyruvate carboxykinase; PGC-1α, peroxisome
proliferator-activated receptor γ coactivator-1α; G6Pase,
glucose-6-phosphatase; NC, negative siRNA control; siRNA, small
interfering RNA; TNF-α, tumor necrosis factor-α. |
Furthermore, in order to determine whether PP4R1 is
involved in the regulation of glucose production by TNF-α, PP4R1
siRNA was transfected into HEPA1-6 cells for 48 h, which was
followed by treatment with TNF-α for 24 h. The results demonstrated
that downregulation of PP4R1 significantly attenuated increased
levels of glucose production following treatment with TNF-α
(Fig. 4F), as well as PGC-1α,
G6Pase and PEPCK mRNA and protein expression levels However,
transfection of PP4R1 siRNA reversed the effect of TNF-α on the
level of p-FOXO1/FOXO1 (Fig 4G and
H). In conclusion, these results suggested that miR-338-3p is
involved in TNF-α-mediated glucose production via targeting of
PP4R1.
Discussion
Chronic inflammation is a major factor resulting in
the development of pathogenesis associated with insulin resistance
(16). In addition, TNF-α promotes
inflammation and attenuates insulin resistance in hepatocytes.
Furthermore, in vivo and in vitro studies have
suggested that treatment with TNF-α blocks the insulin signalling
pathway and results in insulin resistance (13).
In our previous study, it was demonstrated that
treatment with TNF-α decreased glycogen synthesis and decreased the
level of miR-338-3p in hepatocytes (13). It has been previously suggested
that miR-338-3p has an important role in TNF-α-mediated
glycogenesis via regulation of PP4R1 (13). However, the effect of miR-338-3p on
TNF-α-mediated gluconeogenesis remains unknown. In the present
study, whether miR-338-3p modulates gluconeogenesis via regulation
of PP4R1 expression was investigated. In our previous study, we
validated the effect of miR-338-3p mimics, miR-338-3p inhibitor,
Ad-338m, Ad-338i and PP4R1-targeting siRNA in HEPA1-6 cells
(13). Additionally, hepatic
miR-338-3p level was increased >10 fold at 7 days after the
Ad-338m injection; hepatic miR-338-3p level was decreased to ~30%
at 7 days after the AD-338i injection. The results revealed that
treatment with TNF-α significantly induced gluconeogenesis in mouse
liver and HEPA1-6 cells. In addition, it was demonstrated that
miR-338-3p overexpression significantly suppressed glucose
production in the livers of TNF-α-treated mice and TNF-α-treated
HEPA1-6 cells. Furthermore, the results revealed that
downregulation of miR-338-3p induced gluconeogenesis in the livers
of TNF-α-treated mice and TNF-α-treated HEPA1-6 cells. Finally, it
was demonstrated that miR-338-3p was involved in TNF-α-mediated
glucose production via targeting of PP4R1.
Hepatic insulin resistance leads to increased
gluconeogenesis in the liver, which may impair glucose metabolism
homeostasis (17). miRs are a
cluster of small, non-coding RNAs. Numerous miRs have important
roles in the regulation of the activation of the insulin signalling
pathway, including miR-291b-3p (18), miR-200s (19), miR-152 (20), miR-20a-5p (21), miR-19a (22) and miR-301a (23). The results of our previous study
suggested that miR-338-3p regulates the AKT/GSK pathway via
targeting of PP4R1, which subsequently regulates protein
phosphatase 4 (PP4) expression (11). The miR-338-3p gene is located
within intron 8 of the apoptosis-associated tyrosine kinase gene
(24). In the present study, it
was demonstrated that miR-338-3p modulates gluconeogenesis via
regulation of AKT/FOXO1 pathway activation. Gluconeogenesis is an
important metabolic pathway for the conversion of non-carbohydrate
metabolites (lactate, glycerol and amino acids) to glucose, which
provides energy for other organs during a prolonged fast;
gluconeogenesis predominantly occurs in the liver and is regulated
by insulin (25). Gluconeogenesis
is regulated by two key enzymes in the liver: PEPCK and G6Pase.
Hepatic expression of PEPCK and G6Pase is inhibited by insulin in
the fed state (26). In the
present study, it was revealed that hepatic overexpression of
miR-338-3p induces the insulin signalling pathway, which suppresses
PEPCK and G6Pase expression levels in gluconeogenesis both in
vivo and in vitro.
The mechanism underlying the miR-338-3p role in
TNF-α-induced gluconeogenesis was also investigated in the present
study. FOXO1 co-activates PGC1α, which subsequently induces PEPCK
and G6Pase transcription. FOXO1 is the predominant modulator of
insulin activity (27). Insulin
suppresses FOXO1 activity via activation of the PI3K/AKT signalling
pathway. Activated AKT phosphorylates FOXO1, which leads to nuclear
exclusion of FOXO and subsequent suppression of FOXO-induced
transcription (9). Furthermore,
PGC1α is an additional transcriptional co-activator that is
activated under fasting conditions (28). Insulin suppresses PGC1α activity
via increased phosphorylation at Ser570 by activated AKT. In
addition, the results of the present study revealed that miR-338-3p
regulates FOXO1 phosphorylation via the PI3K/AKT pathway.
In a previous study, PP4R1 was revealed to be a
direct target of miR-338-3p, which regulates AKT/glycogen synthase
kinase pathway activation. PP4R1 is a regulatory subunit of PP4.
PP4 has been previously reported to be an important regulator of
TNF-α-induced hepatic insulin resistance (12). In the present study, it was
demonstrated that PP4R1 was involved in miR-338-3p-mediated
gluconeogenesis.
In conclusion, the results of the present study
suggested that miR-338-3p is involved in TNF-α-induced hepatic
gluconeogenesis in vivo and in vitro. miR-338-3p may
be associated with TNF-α-induced gluconeogenesis via targeting of
PP4R1.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81570789, 81600618
and 81700709), the Beijing Natural Science Foundation (grant no.
7182144) and the Beijing Hospital Nova Project (grant no.
BJ-2018-138).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
SW, LL, XC and XH conducted the cell experiments, JL
and XS performed the animal experiments, YZ and TS constructed the
luciferase reporter vector, JG, YM and WT performed pyruvate
tolerance test and glucose production assay; LD and JL made
contributions to the design of the present study. All authors read
and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by Ethic and Animal
Welfare committee College of Life Science Beijing Normal
University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Shaw JE, Sicree RA and Zimmet PZ: Global
estimates of the prevalence of diabetes for 2010 and 2030. Diabetes
Res Clin Pract. 87:4–14. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gaggini M, Morelli M, Buzzigoli E,
Defronzo RA and Gastaldelli A: Non-alcoholic fatty liver disease
(NAFLD) and its connection with insulin resistance, dislipidemia,
atherosclerosis and coronary heart disease. Nutrients. 5:1544–1560.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Puig-Domingo M and Pellitero S: New
therapies for type 2 diabetes mellitus. Med Clin (Barc).
144:560–565. 2015.(In Spanish). View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Erol A: Insulin resistance is an
evolutionarily conserved physiological mechanism at the cellular
level for protection against increased oxidative stress. Bioessays.
29:811–818. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Nordie RC, Foster JD and Lange AJ:
Regulation of glucose production by the liver. Annu Rev Nutr.
19:379–406. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Leclercq IA, Da Silva Morais A, Schroyen
B, Van Hul N and Geerts A: Insulin resistance in hepatocytes and
sinusoidal liver cells: mechanisms and consequences. J Hepatol.
47:142–156. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bugianesi E, McCullough AJ and Marchesini
G: Insulin resistance: A metabolic pathway to chronic liver
disease. Hepatology. 42:987–1000. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lu M, Wan M, Leavens KF, Chu Q, Monks BR,
Fernandez S, Ahima RS, Ueki K, Kahn CR and Birnbaum MJ: Insulin
regulates liver metabolism in vivo in the abesence of hepatic akt
and foxo1. Nat Med. 18:388–395. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yoon JC, Puigserver P, Chen G, Donovan J,
Wu Z, Rhee J, Adelmant G, Stafford J, Kahn CR, Granner DK, et al:
Control of hepatic gluconeogenesis through the transcriptional
coactivator PGC-1. Nature. 413:131–138. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ono K: MicroRNA links obesity and impaired
glucose metabolism. Cell Res. 21:864–866. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhao H, Huang X, Jiao J, Zhang H, Liu J,
Qin W, Meng X, Shen T, Lin Y, Chu J and Li J: Protein phosphatase 4
(PP4) functions as a critical regulator in tumor necrosis factor
(TNF)-α-induced hepatic insulin resistance. Sci Rep. 5:180932015.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dou L, Wang S, Sun L, Huang X, Zhang Y,
Shen T, Guo J, Man Y, Tang W and Li J: Mir-338-3p mediates
Tnf-A-Induced hepatic insulin resistance by targeting PP4r1 to
regulate PP4 expression. Cell Physiol Biochem. 41:2419–2431. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
National Research Council (US) committee
for the update of the Guide for the Care and Use of Labortory
Animals: Guide for the care and use of laboratory animals. 8th.
Washington (DC): Nation Academies Press; 2011
|
|
15
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Mehtods. 25:402–408. 2001.
|
|
16
|
Kim JH, Bachmann RA and Chen J:
Interleukin-6 and insulin resistance. Vitam Horm. 80:613–633. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Fernandez-Valverde SL, Taft RJ and Mattick
JS: MicroRNAs in β-cell biology, insulin resistance, diabetes and
its complications. Diabetes. 60:1825–1831. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Guo J, Dou L, Meng X, Chen Z, Yang W, Fang
W, Yang C, Huang X, Tang W, Yang J and Li J: Hepatic MiR-291b-3p
mediated glucose metabolism by directly targeting p65 to upregulate
PTEN expression. Sci Rep. 7:398992017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dou L, Zhao T, Wang L, Huang X, Jiao J,
Gao D, Zhang H, Shen T, Man Y, Wang S and Li J: miR-200s contribute
to interleukin-6 (IL-6)-induced insulin resistance in hepatocytes.
J Biol Chem. 288:22596–22606. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang S, Wang L, Dou L, Guo J, Fang W, Li
M, Meng X, Man Y, Shen T, Huang X and Li J: MicroRNA 152 regulates
hepatic glycogenesis by targeting PTEN. FEBS J. 283:1935–1946.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fang W, Guo J, Cao Y, Wang S, Pang C, Li
M, Dou L, Man Y, Huang X, Shen T and Li J: MicroRNA-20a-5p
contributes to hepatic glycogen synthesis through targeting p63 to
regulate p53 and PTEN expression. J Cell Mol Med. 20:1467–1480.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dou L, Meng X, Sui X, Wang S, Shen T,
Huang X, Guo J, Fang W, Man Y, Xi J and Li J: MiR-19a regulates
PTEN expression to mediate glycogen synthesis in hepatocytes. Sci
Rep. 5:116022015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dou L, Wang S, Sui X, Meng X, Shen T,
Huang X, Guo J, Fang W, Man Y, Xi J and Li J: MiR-301a mediates the
effect of IL-6 on the AKT/GSK pathway and hepatic glycogenesis by
regulating PTEN expression. Cell Physiol Biochem. 35:1413–1424.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jacovetti C, Jimenez V, Ayuso E, Laybutt
R, Peyot ML, Prentki M, Bosch F and Regazzi R: Contribution of
intronic mir-338-3p and its hosting gene AATK to compensatory
beta-cell mass expansion. Mol Endocrinol. 29:693–702. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Radzuk J and Pye S: Hepatic glucose
uptake, gluconeogenesis and the regulation of glycogen synthesis.
Diabetes Metab Res Rev. 17:250–272. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Oh KJ, Han HS, Kim MJ and Koo SH:
Transcriptional regulators of hepatic gluconeogenesis. Arch Pharm
Res. 36:189–200. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kousteni S: FoxO1, the transcriptional
chief of staff of energy metabolism. Bone. 50:437–443. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Puigserver P, Rhee J, Donovan J, Walkey
CJ, Yoon JC, Oriente F, Kitamura Y, Altomonte J, Dong H, Accili D
and Spiegelman BM: Insulin-regulated hepatic gluconeogenesis
through FOXO1-PGC-1alpha interaction. Nature. 423:550–555. 2003.
View Article : Google Scholar : PubMed/NCBI
|














