Introduction
The endoplasmic reticulum (ER) is a common organelle
demonstrated in eukaryotic cells, which is an important site for
the synthesis and modification of proteins, lipids and
carbohydrates (1,2). The ER is also involved in the
regulation of the intracellular calcium ion concentration through
the storage and release of calcium (3,4). The
ER in eukaryotic cells has four main physiological functions: i)
The synthesis of membrane proteins and secretory proteins; ii) the
formation of the correct three-dimensional conformation of proteins
by folding; iii) the storage of Ca2+; and iv) the
biology synthesis of lipid and cholesterol. The correct synthesis
and secretion of proteins in the ER is regulated by a variety of
mechanisms, including the mechanisms by which the oxidative
environment, the calcium ion concentration, ATP, protein disulphide
isomerase (PDI), heavy-chain binding protein and calprotectin are
maintained (1,2,4).
When the ER homeostastic balance is disrupted by a
variety of physiological and pathological factors, ER stress (ERS)
can be induced in the ER with increased amounts of unfolded and
misfolded proteins being formed, calcium depletion and disorder of
lipid synthesis (5,6). ERS involves three pathways, namely
the unfolded protein response (UPR), Ca2+ signaling and
ER-related degradation (5–7). They are the main reactionary
processes of ERS. ER homeostasis is ultimately achieved through the
UPR to reduce the synthesis of novel proteins, to promote folding
of unfolded proteins and to increase the degradation of misfolded
proteins (1,2,8). In
mammalian cells, UPR is mediated by an ER chaperone protein
glucose-regulated protein-78/binding immunoglobulin protein
(Grp78/Bip) and three ERS-sensing proteins: Protein kinase R-like
ER kinase (PERK), inositol-requiring kinase-I (IRE-1) and
activating transcription factor 6 (ATF6) (9,10).
Bip, which belongs to the family of heat shock protein 70 (HSP70),
is a molecular chaperone of the ER, also known as Grp78 (9,10).
It serves an important role in the regulation of ERS, and its
activation can be used as a marker of the ERS response (11). Both PERK and IRE-1 are ER type I
transmembrane protein kinases and belong to UPR proximal receptors
(1,10).
TF6, an ER type II transmembrane protein kinase, is
located on the outside of the ER (12). When the ER is in a state of stress,
a large number of unfolded or misfolded proteins accumulate in the
ER, while GRP78 dissociates from ATF-6 and PERK-induced proteins
and binds to unfolded proteins (12,13).
The activation of IRE-1 is unclear, and studies had demonstrated
that IRE-1 can be directly activated by unfolded proteins (14). UPR is then simulated by activated
free PERK, IRE-1 and ATF6 via their specific pathways, thereby
reducing the synthesis of novel proteins and decreasing the
accumulation of unfolded and misfolded proteins in the ER to
restore the stability of the environment within the ER (12–14).
However, when ERS is too intense or too long, the
steady state of ER cannot be restored, UPR can activate the
apoptosis signaling pathway to induce apoptosis (15,16).
ERS has been previously demonstrated to be a novel way to initiate
apoptosis (16). In the early
stage of the ERS response, UPR helps to promote cell survival, but
if the ERS is too great, the internal environment cannot be
restored in time and this leads to apoptosis (16,17).
ERS-induced apoptosis is achieved mainly through the following
three ways: The activation of transcription factors
C/EBP-homologous protein (CHOP)/growth inhibition and DNA
damage-inducible gene 153 (GADD153), activation of ASK1/JNK kinase
pathway and activation of caspase 12 (18–20).
The UPR pathway promotes cell survival and induces apoptosis,
however the mechanisms of the transition from pro-survival to
pro-apoptotic are unclear (18,20).
Previous studies had demonstrated that autophagy can
be induced by ERS through a variety of ways (21,22).
From yeasts to mammals, the mutual regulation of ERS and autophagy
is a highly conserved process (23). Cell autophagy, also known as type
II programmed cell death, had been demonstrated to induce autophagy
under conditions of starvation, ERS, hypoxia, and radiation
(21,22). In mammalian cells, autophagy is
induced by increased levels of PERK, IRE-I and Ca2+
(24). During autophagy, a
autophagosome bilayer is formed, which separates the cytosol and
proteins from the cytoplasm (25).
The outer membrane of the autophagosome then fuses with lysosomes
to dissolve the endolyses and their solutes (25). Up to now, >30
autophagy-associated genes have been identified (26,27).
These genes are named autophagy-related genes (ATG). The ATG12-ATG5
and LC3-modified ubiquitin-like protein binding systems serve an
important role in the formation of autophagosomes in mammals
(28,29).
An increasing number of studies have demonstrated
that autophagy has an inhibitory effect on tumors and mutations or
deletions of related autophagy genes are involved in this process
(30,31). A single allelic deletion or
mutation of Beclinl was observed in 40–75% of human breast, ovarian
and prostate cancer, and but only a low level of expression occurs
in brain tumors (31). In
addition, investigations have demonstrated that inhibition of the
accumulation of autophagy substrate-p62 can prevent the damage
caused by autophagy defects, indicating that autophagy can also
inhibit the occurrence of tumors by reducing p62 accumulation
(32,33). Other studies revealed that cell
autophagy can be induced by high expression of ATG1/4, resulting in
a significant reduction in cell volume and inhibition of tumor
growth (34–36).
It has been demonstrated that nutritional
deficiencies, ERS and other factors can cause cell autophagy
(37). Previous investigations
have demonstrated that autophagy caused by starvation causes
degradation of a large number of misfolded proteins, but is not
specific (37,38). Autophagy caused by antineoplastic
agents and oxidative stress may be different from starvation
because they degrade misfolded proteins through the ubiquitination
process (28,29). Therefore, the mechanisms of
autophagy induced by drugs or oxidative stress, such as protection
or cytotoxicity, and autophagy-regulating cell death, need to be
further investigated.
Materials and methods
Cell culture
Human hepatoblastoma cell line HepG2 (39) were purchased from Chinese Academy
of Sciences Cell Bank (Shanghai, China). All cells were cultured in
Dulbecco's modified Eagle medium (DMEM; Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany) containing 10% fetal bovine serum
(Sigma-Aldrich; Merck KGaA), 100 U/ml penicillin and 100 µg/ml
streptomycin and placed in a cell culture chamber at 37°C and under
a humidified atmosphere with 5% CO2. After the cells
adhered and were grown to 80% confluence, the culture medium was
removed and washed with appropriate amount of PBS and digested with
0.25% trypsin (Sigma-Aldrich; Merck KGaA). Cells in the logarithmic
phase of growth were selected for following experiments.
Cell Counting Kit-8 (CCK-8) assay
The liver cancer cells in the logarithmic growth
phase were seeded in 96-well plates at 2×105 cells per
well and cultured overnight at 37°C. After the cells were
completely adhered and grown to 80% confluence, the culture medium
was removed and replaced with culture medium containing different
concentrations of H2O2 (0, 100, 200, 400, 800
and 1,000 µmol; Sigma-Aldrich; Merck KGaA). After 6, 12, 24
and 48 h with H2O2 treatment, 20 µl
CCK-8 (Dojindo Molecular Technologies, Inc., Kumamoto, Japan) was
added and the cells were cultured at 37°C for another 4 h. The
absorption of cell solution was measured at a wavelength of 450
nm.
Reactive oxygen species (ROS)
assay
The cells in the logarithmic growth phase were
seeded in 96-well plates at 2×105 cells per well and
cultured overnight at 37°C. After the cells were grown to 80%
confluence, the culture medium was removed and the cells were
divided into several groups with medium containing different
concentrations of H2O2 (0, 200, 400, 600 and
1,000 µmol), respectively. After cultured for another 24 h, cells
were harvested, and washed with PBS. Cells were incubated in 20 µM
DCFH-DA solution (Sigma-Aldrich; Merck KGaA) at 37°C for 1 h
according to manufacturer's protocol. The cells were washed with
PBS, ROS production in cells was determined using a flow cytometer
(BD Biosciences, Franklin Lakes, NJ, USA). The data was analyzed
using FlowJo software version 7.6.1 (FlowJo LLC, Ashland, OR,
USA).
Experimental design
The liver cancer cells in logarithmic growth phase
were seeded in 96-well plates at 2×105 cells per well
and cultured overnight at 37°C. After the cells were completely
adhered and grown to 80% confluence, the culture medium was removed
and then the cells were divided into four groups as follows: i)
Cells cultured in culture medium without drugs and used as a
control; ii), cells cultured in medium containing 600 µmol
H2O2 and named the H2O2
group; iii), cells cultured in medium containing 600 µmol of
H2O2 and ERS inhibitor Salubrinal (SAL;
Sigma-Aldrich; Merck KGaA), and called the
H2O2 + SAL group; iv), cells cultured in
medium containing 600 µmol H2O2 and ERS
inducer Tunicamycin (TM; Sigma-Aldrich; Merck KGaA), and expressed
as H2O2 + TM group. After the cells were
cultured for 24 h, they were used for the following assays.
Apoptosis detection by flow
cytometry
The cells from all groups were harvested and
digested, and then were washed twice with PBS and collected at
1×105 cells per tube. Apoptosis was detected using a
Fluorescein isothiocyanate (FITC) Annexin V Apoptosis Detection kit
(BD Biosciences). After 5 µl Annexin V-FITC and 5 µl propidium
iodide dyes were added to each well, the cells were incubated at
room temperature for 15 min. Then apoptosis was detected by flow
cytometry within 1 h. The upper left quadrant represents a
mechanically injured cell and the upper right quadrant is a late
apoptotic cell. The lower left quadrant is a normal cell and the
lower right quadrant is an early apoptotic cell. In this
experiment, the proportion of the upper right quadrant + the lower
right quadrant was used as the percentage of apoptotic cells. The
data was analyzed using FlowJo software version 7.6.1.
Autophagy rate detection by flow
cytometry assay
Minimum essential medium (MEM; Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) containing 2 ml
monodansylcadaverine (MDC; final concentration 50 NM) was added to
each group and incubated for 30 min in a constant temperature
incubator at 37°C, 5% CO2. The supernatant was collected
at 500 × g for 5 min at 4°C. Then the obtained supernatant was
discarded and 500 µl MEM containing MDC (final concentration of 50
NM) was added to each group and then incubated at 37°C for 30 min.
Cells were washed twice with PBS and digested with 0.25% trypsin
for 2 min, then 1 ml PBS was added and mixed. The cells obtained
were then collected in 1.5 ml centrifuge tubes and centrifuged at
500 × g for 5 min at 4°C. The supernatants were discarded, 1 ml PBS
was added to re-suspend the supernatant. The solutions were then
centrifuged at 500 × g for 5 min at 4°C. A total of 800 µl
supernatant was sucked out and the remaining 200 µl was mixed. The
fluorescence intensity of MDC staining of the solution obtained was
measured on a flow cytometer at an excitation wavelength of 488 nm
after passing a 300 mesh copper mesh. The data was analyzed using
FlowJo software version 7.6.1.
Western blotting assay
Buffer A cell lysates (Cell Signaling Technology
Inc., Danvers, MA, USA) containing a protease and phosphatase
inhibitor cocktail (Roche Diagnostics, Basel, Switzerland) were
added to the cells collected from all groups, and homogenized for
30 min at 4°C. Total protein extracts were quantified by
Bicinchoninic acid Protein Assay kit (Thermo Fisher Scientific,
Inc.). Proteins (30 µg) were separated by 10% SDS-PAGE and then
transferred to a nitrocellulose membrane. Subsequently, the
membranes were blocked with 5% fat-free dry milk at room
temperature for 1 h. Following this, the blots were incubated with
human anti-GRP78 (cat. no. ab181499; Abcam, Cambridge, MA, USA;
1:1,000), anti-IRE1 (cat. no. ab48187, Abcam; 1:2,000), anti-ATF6
(cat. no. ab37149; Abcam; 1:1,000), anti-CHOP (cat. no. ab11419;
Abcam; 1:1,000), anti-pro caspase-3 (cat. no. ab32150; Abcam;
1:1,000), anti-pro caspase-9 (cat. no. ab135544; Abcam; 1:500),
anti-caspase-12 (cat. no. ab62484; Abcam; 1:1,000), anti-p62 (cat.
no. ab56416; Abcam; 1:500), anti-β-actin (cat. no. ab8226; Abcam;
1:2,000) overnight at 4°C. The membranes were again washed with PBS
and incubated with horseradish peroxidase (HRP)-conjugated goat
anti-rabbit IgG secondary antibodies (cat. no. P0448; Dako; Agilent
Technologies, Inc., Santa Clara, CA, USA; 1:2,000) at room
temperature for 1 h. The proteins were finally examined by an
enhanced chemiluminescence system (ECL) (Bio-Rad Laboratories,
Inc., Hercules, CA, USA). Band intensities were quantified by
densitometry using ImageJ Software version 1.6 (National Institutes
of Health, Bethesda, MD, USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) assay
Total RNA from cells was extracted using the RNeasy
mini-kit (Qiagen GmbH, Hilden, Germany) according to manufacturer's
protocol. Total RNA (1 µg) was reverse-transcribed with an iScript
cDNA Synthesis kit (Bio-Rad Laboratories, Inc.), according to the
manufacturer's protocol. Primers were synthesized by Sangon Biotech
Co., Ltd., (Shanghai, China). Relative quantification of mRNA was
performed by qPCR using iQ SYBR-Green Supermix and iCycler iQ
thermal cycler (Bio-Rad Laboratories, Inc.). The thermocycling
conditions were as follows: 95°C for 10 min, followed by 40 cycles
of 95°C for 15 sec, 60°C for 60 sec, and 72°C for 60 sec. Each
sample was determined in duplicate. All PCR products were confirmed
by 1.5% agarose gel electrophoresis. Data were calculated using the
2−ΔΔCq method (40) and
relative expression was normalized to an endogenous reference
(β-actin). Primers were as follows: GRP78:
5′-GAGTAGGCGACGGTGAGGTC-3′ (forward) and 5′-GAGCACAGCGCAATTTCCGA-3′
(reverse); IRE1: 5′-GCTCCAGAGATGCTGAGCGA-3′ (forward) and
5′-GTGCTTCTCTGGGTGCAAGC-3′ (reverse); p50ATF6:
5′-AGAGGCAACCCACGTTGTCA-3′ (forward) and 5′-GCCACCAAGGCAGAAAGCAG-3′
(reverse); CHOP: 5′-TGCAGAGATGGCAGCTGAGT-3′ (forward) and
5′-CCAAGCCAGAGAAGCAGGGT-3′ (reverse); β-actin:
5′-GGCACTCTTCCAGCCTTCCT-3′ (forward) and 5′-GCACTGTGTTGGCGTACAGG-3′
(reverse).
Statistical analysis
The results are expressed as the mean ± standard
deviation of the mean of three independent experiments. Statistical
comparisons between different groups were conducted by with SPSS
software (version 20; IBM Corps., Armonk, NY, USA). Two-tailed
one-way analysis of variance followed by Bonferroni post hoc
pairwise comparison was used to assess the differences between
groups. P<0.05 was considered to indicate a statistically
significant difference.
Results
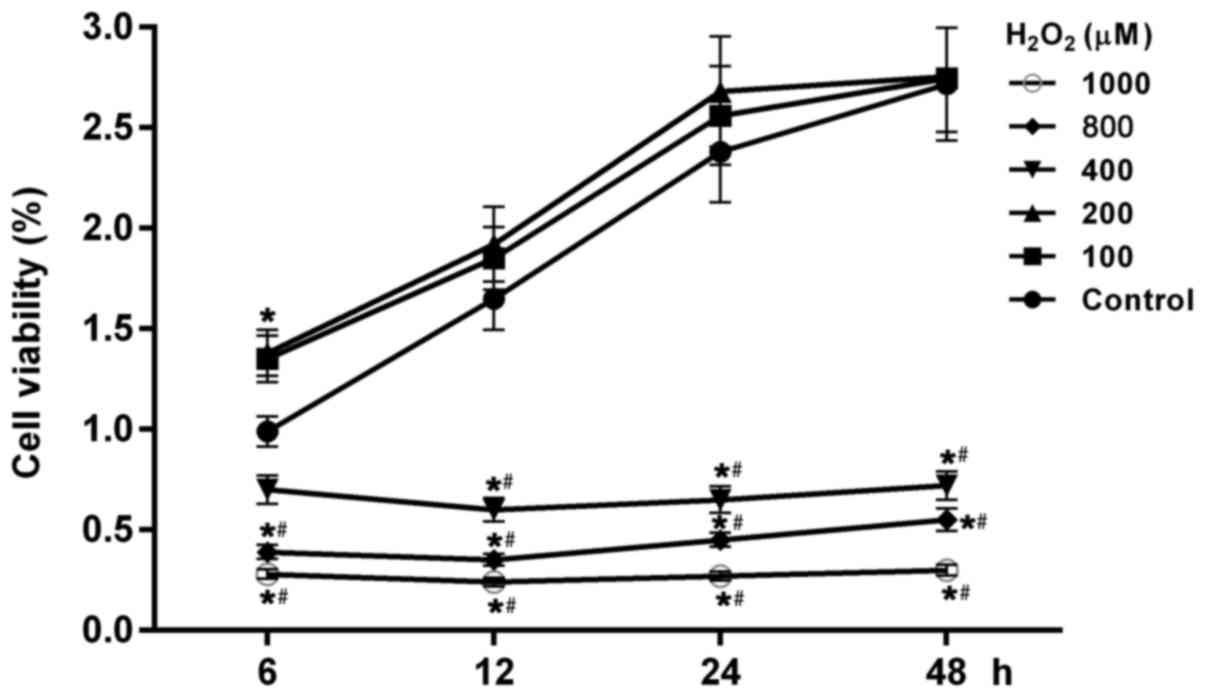
Cell viability is affected by
H2O2 in a dosage- and time-dependent
manner
To test the consequences of increased oxidative
stress on the cell viability of human hepatoblastoma cell line
HepG2, cells were incubated in different concentrations of
H2O2 (0, 100, 200, 400, 800 and 1,000 µmol).
The results demonstrated that cell viability in all groups altered
in a dose- and time-dependent manner (Fig. 1). As well as in the control group,
cell viability in groups with 100 and 200 µmol
H2O2 increased with increased time (Fig. 1). While in the group with the
higher concentrations of H2O2 (400, 800 and
1,000 µmol), cell viability was significant reduced compared with
the control and cells treated with low concentration of
H2O2 (100 and 200 µmol; P<0.05). In groups
with high concentration treatments of H2O2,
cell viability was lowest at 12 h following cell treatment with
H2O2, suggesting that cell growth or cell
apoptosis were affected by high oxidative stress.
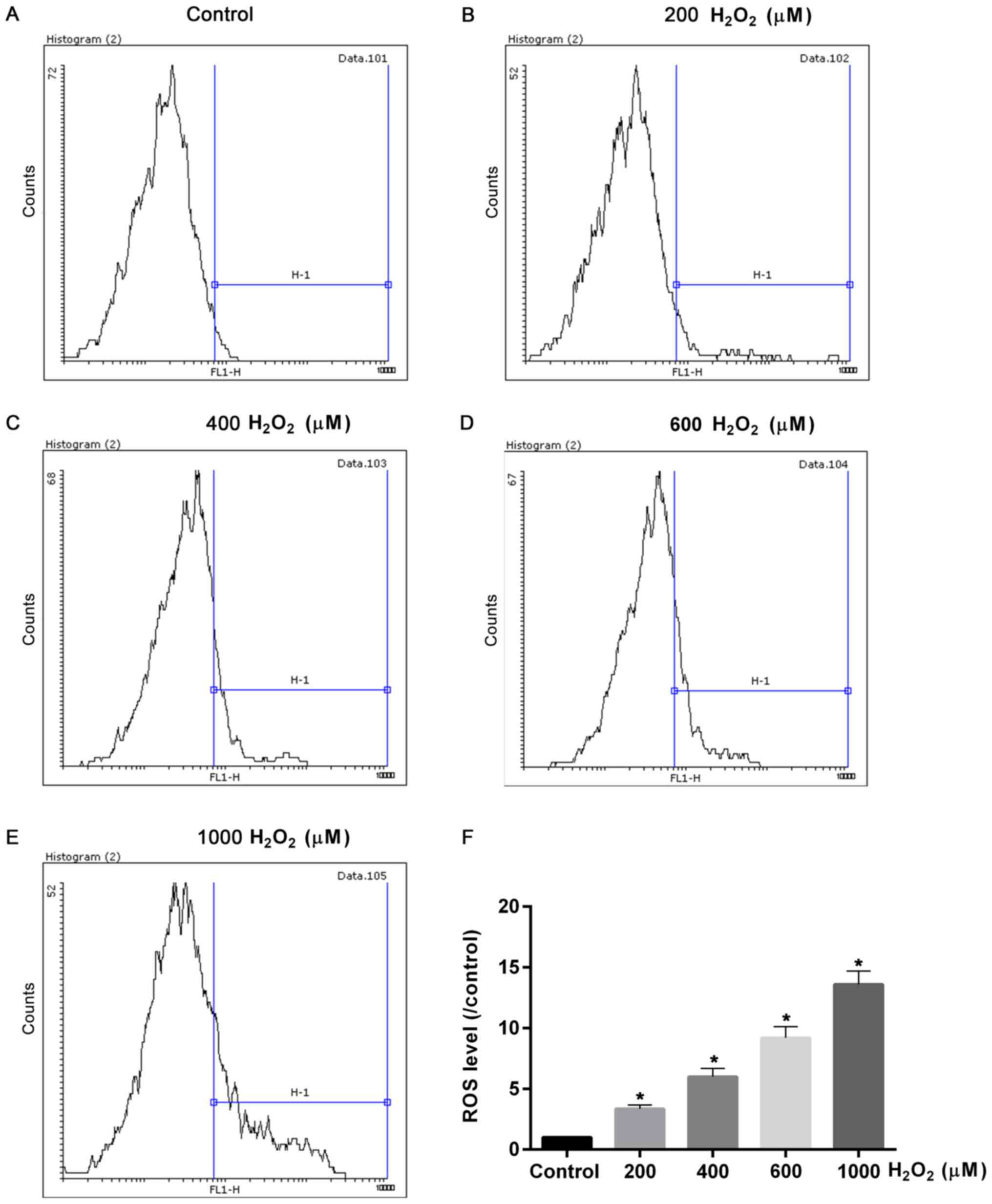
ROS levels in cells treated with
H2O2 are increased in a dosage-dependent
manner
Cell viability was affected by
H2O2 treatments, indicating that cell damage
was induced by oxidative stress. Therefore, the ROS levels induced
by different concentrations of H2O2 were
measured. The flow cytometry assay demonstrated that ROS levels
were significantly elevated with increased treatment concentrations
of H2O2 (P<0.05; Fig. 2). The ROS levels in cells treated
with 600 and 1,000 µmol H2O2 were ~10 and 15
fold of that of control, respectively (Fig. 2). Hence, together with the results
of cell viability test, the cells treated with 600 µmol
H2O2 had moderate injuries; therefore this
dose was chosen for the oxidative damage model and used in
subsequent experiments.
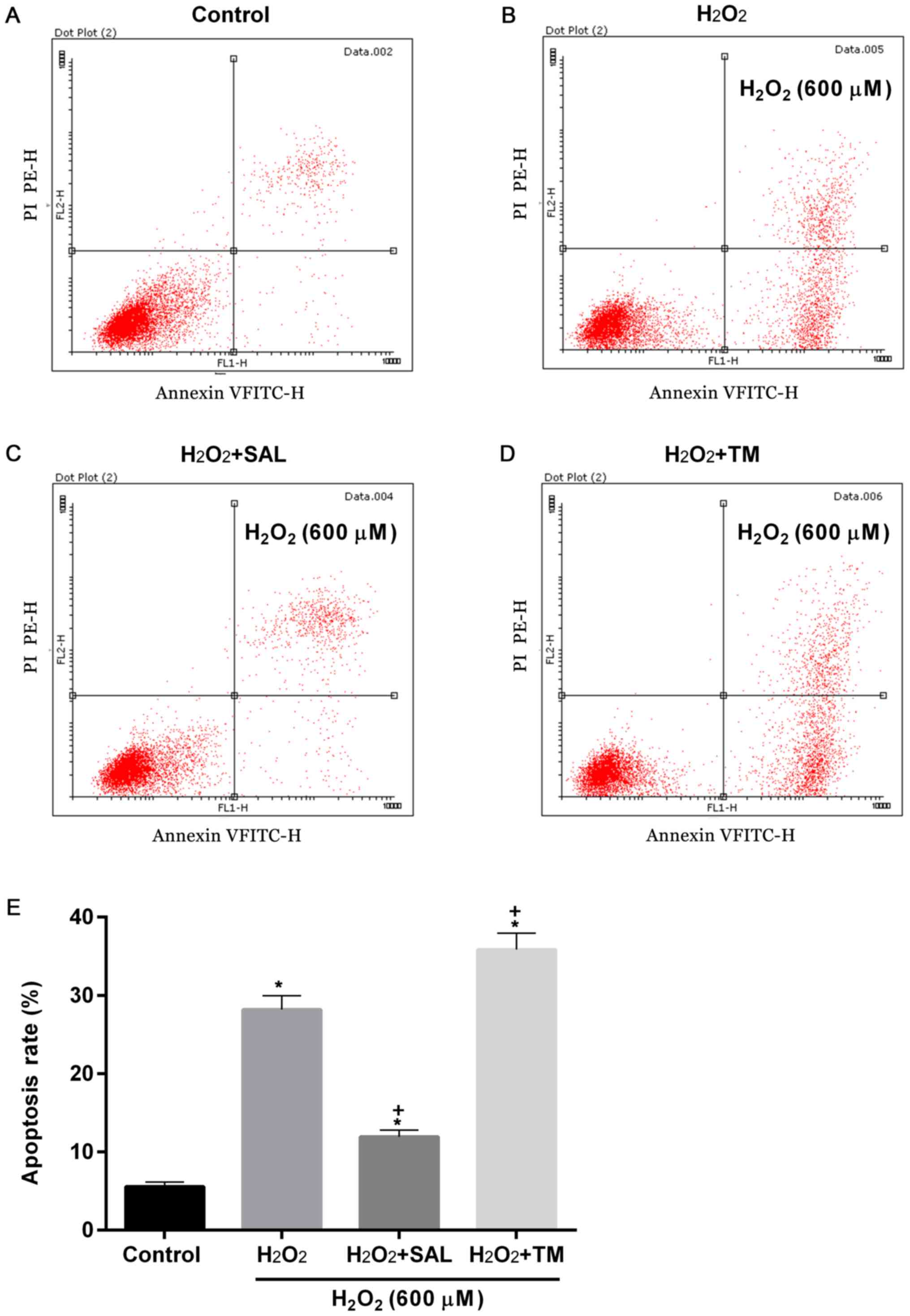
Increased apoptosis and cell autophagy
rates induced by H2O2 are significantly
affected by SAL and TM
To examine apoptosis and cell autophagy rates in
cells with ERS induced by oxidative stress, cells were treated with
H2O2 + ER inhibitor SAL and ERS inducer TM.
Compared with the control, apoptosis rates were significantly
increased in groups treated with H2O2
(P<0.05; Fig. 3). However,
compared with cells treated with H2O2 alone,
the apoptosis rates were decreased and increased in cells treated
with H2O2 + SAL and with
H2O2 + TM, respectively (P<0.05; Fig. 3).
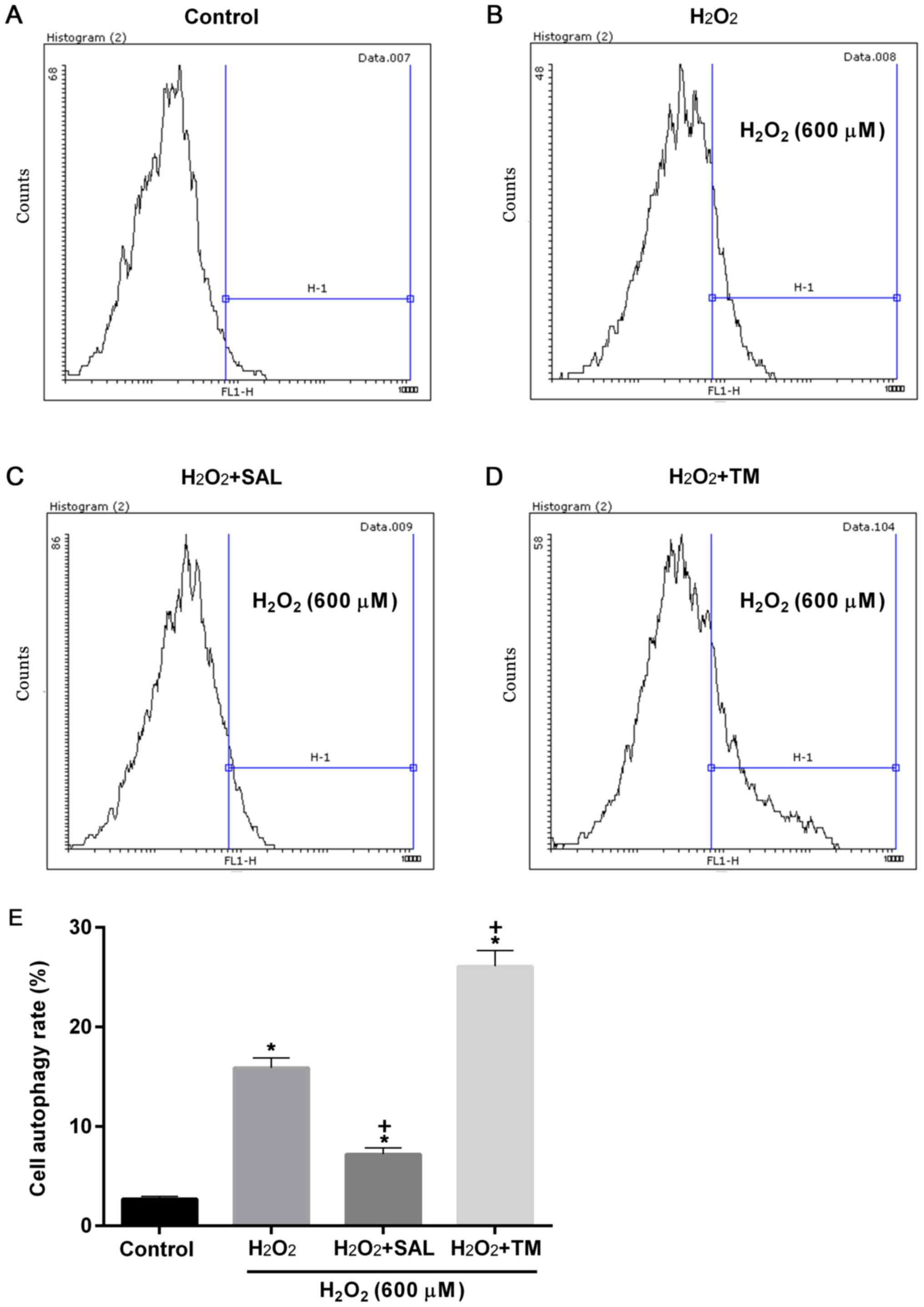
Similarly, cell autophagy rates were significantly
increased in all groups compared with the control (P<0.05;
Fig. 4). Compared with cells
treated with H2O2 alone, cell autophagy rates
were significantly decreased and increased in cells with
H2O2 + SAL and H2O2 +
TM treatments, respectively (P<0.05; Fig. 4).
SAL and TM treatment can reduce and
increase the expression of ERS associated genes, respectively
To further investigate the effects of
H2O2, SAL and TM on ERS, the expression of
ERS-associated genes were determined. As demonstrated in Fig. 5, a western blot assay revealed that
the expression levels of GRP78, IRE1, p50ATF6 and CHOP proteins
were significantly increased following treatment with
H2O2, compared with the control (P<0.05).
The expression level of the aforementioned genes decreased and
increased in cells treated with H2O2 + SAL
and H2O2 + TM, respectively, compared with
the cells treated with H2O2 alone (Fig. 5A and B).
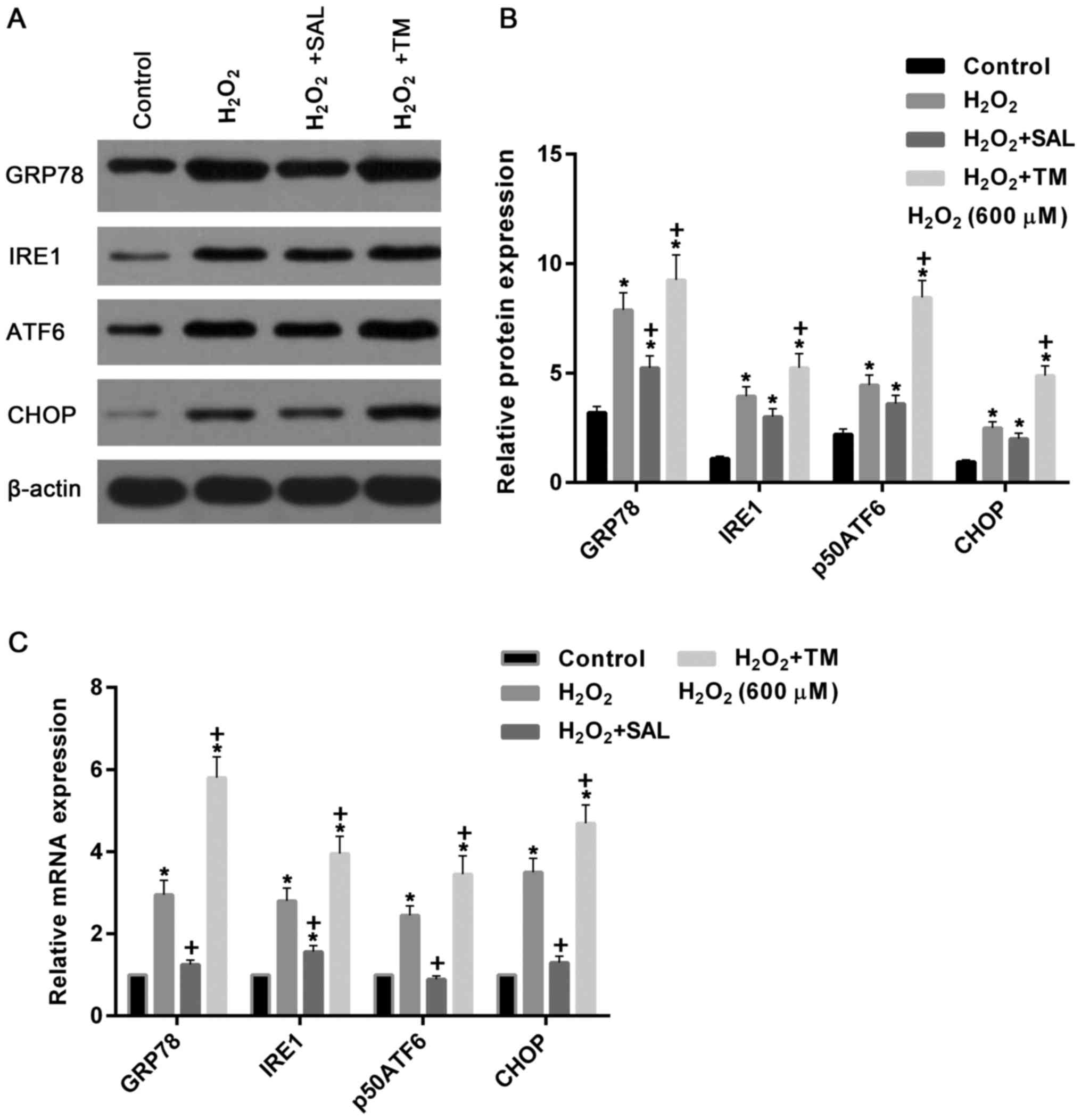 | Figure 5.Expression levels of ERS-associated
proteins were simulated by H2O2 and TM, and
inhibited by SAL. (A) Western blot assay and (B) quantification of
the assay measuring the expression levels of GRP78, IRE1, p50ATF6
and CHOP proteins were increased in cells with
H2O2 treatment. Compared with cells treated
with H2O2 alone, they were reduced and
increased in cells treated with H2O2 + SAL
and H2O2 + TM, respectively. (C) Reverse
transcription-quantitative polymerase chain reaction assay
demonstrated that expression levels of GRP78, IRE1, p50ATF6 and
CHOP mRNA were increased in cells treated with
H2O2 alone and with
H2O2 + TM, compared with the control. These
repression levels were significantly reduced and increased in cells
treated with H2O2 + SAL and
H2O2 + TM, compared with cells treated with
H2O2 alone, respectively. *P<0.05 vs. the
control group. +P<0.05 vs. the
H2O2 group. TM, Tunicamycin; SAL, Salbrinal;
ERS, endoplasmic reticulum stress; CHOP, C/EBP-homologous protein;
IRE1, inositol-requiring kinase-I; GRP78, glucose-regulated
protein-78; ATF6, activating transcription factor 6. |
Similarly, mRNA expression levels of GRP78, IRE1,
p50ATF6 and CHOP were upregulated in cells treated with
H2O2 alone and with
H2O2 + TM, compared with the control
(P<0.05). However, the mRNA expression levels were significantly
downregulated and upregulated in cells treated with
H2O2 + SAL and H2O2 +
TM, respectively compared with cells treated with
H2O2 alone (P<0.05; Fig. 5C).
TM stimulates the upregulation of
apoptosis-related genes of ER pathway and autophagy
Previous tests revealed that apoptosis and autophagy
were induced following treatment with H2O2
and TM. Consequently, the expression of apoptosis genes and cell
autophagy-associated genes were measured. The results demonstrated
that the protein expression levels of pro caspase-3 and pro
caspase-9 were significantly decreased in the cells treated with
H2O2 alone and with
H2O2 + TM, compared with the control
(P<0.05; Fig. 6). They were
increased and decreased in the cells treated with
H2O2 + SAL and with
H2O2 + TM, respectively, compared with the
cells treated with H2O2 alone (P<0.05;
Fig. 6). By contrast, the protein
levels of caspase-12 were significantly increased in cells treated
with H2O2, compared with the control
(P<0.05). The protein levels were downregulated and upregulated
in cells treated with H2O2 + SAL and with
H2O2 + TM, respectively, compared with the
cells treated with H2O2 alone (Fig. 6A and B).
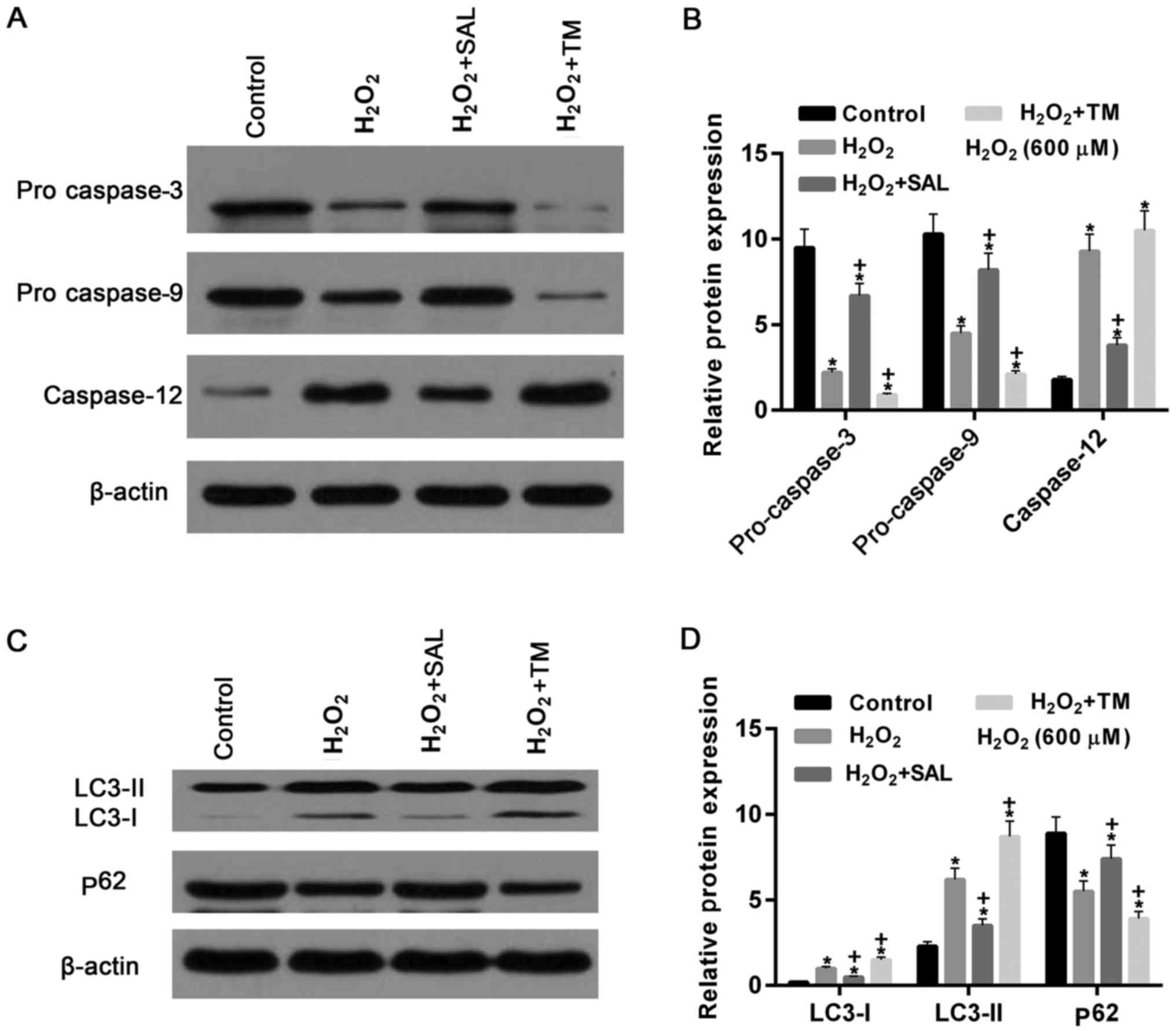 | Figure 6.Apoptosis- and cell
autophagy-associated genes in cells treated with
H2O2 were significantly altered following
co-treatment with SAL and TM. (A) Western blot assay and (B)
quantification of the results demonstrated that the expression of
pro caspase-3 and pro caspase-9 were decreased following cells
treated with H2O2, compared with the control.
Compared with the cells treated with H2O2
alone, they were increased and decreased in cells treated with
H2O2 + SAL and with
H2O2 + TM, respectively. However, caspase-12
was significantly increased in cells treated with
H2O2. They were reduced and increased in
cells treated with H2O2 + SAL and with
H2O2 + TM, compared with cells treated with
H2O2 alone, respectively. (C) Western
blotting and (D) quantification of the western blot. The levels of
LC3-I, LC3-II protein were increased in cells treated with
H2O2 alone. Protein levels of LC3-I, LC3-II
were decreased and increased in cells treated with
H2O2 + SAL and with
H2O2 + TM, compared with cells treated with
H2O2 alone, respectively. By contrast, the
expression levels of p62 protein were decreased by
H2O2 treatment compared with the control
group, and upregulated and downregulated in cells treated with
H2O2 + SAL and with
H2O2 + TM, compared to cells treated with
H2O2 alone, respectively. *P<0.05 vs.
control group. +P<0.05 vs. H2O2
group. TM, Tunicamycin; SAL, Salbrinal. |
The expression levels of LC3-I and LC3-II protein
were increased in cells treated with H2O2 and
with H2O2 + TM, compared with the control.
The levels of LC3-I and LC3-II protein were downregulated and
upregulated in cells treated with H2O2 + SAL
and with H2O2 + TM, compared to cells treated
with H2O2 alone, respectively. By contrast,
the levels of p62 protein were significantly downregulated after
cells were treated with H2O2 (P<0.05;
Fig. 6C and D). Compared with
cells treated with H2O2 alone, the expression
levels of p62 protein were increased and reduced, respectively in
cells treated with H2O2 + SAL and with
H2O2 + TM. These results confirmed the
observation of increased autophagy rates detected by flow cytometry
assay, indicating that the formation of autophagosomes was
increased in cells treated with H2O2 or
H2O2 + TM.
Discussion
The present study investigated the role of
ERS-autophagy on the oxidative damage induced by
H2O2 and the mechanism involved. The present
study demonstrated that oxidative damage induced by
H2O2 can be inhibited and enhanced by the ERS
inhibitor SAL and inducer TM, respectively. Furthermore, TM
treatment increased apoptosis and autophagy rates, indicating that
ERS-autophagy is involved the apoptosis of HepG2 cells. In
addition, these processes were inhibited by the ERS inhibitor SAL,
confirming the role of ERS-autophagy on the apoptosis induced by
oxidative stress. Consistent with these observations, TM increased
the expression of ERS-associated genes, including GRP78, IRE1,
p50ATF6 and CHOP, and also the expression of autophagy-associated
genes LC3-I, LC3-II and p62.
Primary and secondary resistance remains a
bottleneck in the clinical treatment of cancer (41). The mechanism of the formation of
drug-resistant tumor cells is not only associated with the
increased expression of resistance and anti-apoptotic genes of
tumor cells, but also the results of the present study suggest that
autophagy, ERS and other signaling pathways are involved in the
formation of drug resistance mechanisms of tumor cells (42,43).
ERS and autophagy are adaptive responses of cells to injury stimuli
(43). ERS-induced autophagy can
maintain homeostasis of the intracellular environment, serving a
certain protective role (43).
When ERS occurs, the UPR can activate autophagy and subsequently
autophagy can reduce the load of the ER by degrading the misfolded
or unfolded protein and suppressing the excessive activation of ERS
(5,43). In addition, degradation products
produced by these processes also provide the raw material for the
synthesis of novel proteins in the cell, the reconstruction of the
cell structure and the formation of ATP (15,17).
However, the over-activation of ERS can increase
cell damage and even cause cell death (19,20,44).
The present study investigated the association between oxidative
stress, the ERS inhibitor SAL and ERS inducer TM to identify the
effect of ERS on oxidative damage induced by
H2O2 in the liver cancer cell line HepG2 and
its mechanism. The results of the present study demonstrated that
oxidative damage induced by H2O2 can be
increased by TM-induced ERS, which was confirmed by the elevations
of apoptosis and autophagy rates. It also demonstrated that ERS was
induced in cells treated with a high concentration of
H2O2 (600 µM), which can be attenuated
in cells treated with the ER inhibitor SAL, resulting in a
reduction of apoptosis and autophagy rates. These results suggested
that ER inducer can enhance oxidative stress damage in HepG2
cells.
The subsequent experiments revealed that the
expression levels of ERS-related genes, such as GRP78, IRE1,
p50ATF6 and CHOP, were increased in cells treated with TM +
H2O2, compared with cells treated with
H2O2 alone. It is well known that in the
absence of ERS, the N-terminus of IRE1, p50ATF6 and PERK binds to
the chaperone Grp78/Bip and they are present in inactive state
(6,11). When ERS occurs, a large number of
unfolded proteins accumulate in the ER cavity, IRE1, p50ATF6 and
PERK are then separated from Grp78/Bip, and Grp78/Bip binds to the
unfolded proteins (11,14,24).
ERS-regulated apoptosis is mediated by activation of the
transcription factor CHOP/GADD153, the apoptotic signaling kinase 1
(ASK1)/c-Jun N-terminal kinase 1 (JNK) kinase pathway and caspase
12 (19). CHOP, also known as
GADD153, is a member of the C/EBP transcription factor family and
is a specific transcription factor for ERS (18,19).
When the ERS occurs, activated PERK, IRE-1 and p50ATF6 can elevate
CHOP expression, and ultimately activate caspase 3 to induce
apoptosis (18,19,45).
Consistent with this mechanism, the results in present study
demonstrated that the elevation of CHOP expression and the
reduction of pro-caspase 3 level were observed when the
upregulation of IRE-1 and p50ATF6 were induced by ERS. In addition,
when IRE-1 is activated, it can bind to tumor necrosis factor
receptor factor 2 (TRAF2) and ASK1 to form IRE1/TRAF2/ASK1 complex,
and further activate JNK, which finally results in apoptosis
(14,46). Furthermore, a number of studies had
revealed that activated calpain kinase can activate caspase 12 when
ERS is activated by an imbalance in intramolecular calcium storage,
whereas activated PERK and IRE-1 can also further activate
caspase-12 by activating TRAF2 protein (14,46,47).
Activated caspase-12 is then transferred to the cytoplasm, causing
the caspase-9 precursors to cleave and activate caspase-9 to
activate caspase-3, leading to apoptosis, whereas this pathway does
not depend on mitochondria (48).
The results revealed that caspase-12 was increased and levels of
pro caspase-3 were reduced, indicating that ERS-induced apoptosis
is involved in CHOP/GADD153, the ASK1/JNK kinase and caspase 12
pathway in HepG2 cells. In addition, the present study demonstrated
that the ERS inducer TM can enhance the apoptosis of HepG2 cells
induced by H2O2 via elevating ERS.
Previously, it has been demonstrated that
starvation, ERS and other factors can induce cell autophagy
(37). Similar to a number of
investigations, the results of the present study demonstrated that
cell autophagy was stimulated by ERS (37,49).
Previous studies have demonstrated that two ubiquitin-like binding
systems are involved in the formation of autophagosomes (37,49).
One is the AtgS/Atg 12 system, including Atg5, Atg 12, Atg7, Atg 10
and Atg 16 (29,49). The other ubiquitin-like binding
system is comprised of LC3/Atg8 and its targeted molecular
phosphatidylethanolamine (PE) (49,50).
With the help of Atg 4, Atg 7 and Atg 3, part of the amino acid
residue at the carboxy terminus of LC3/Atg8 is first cleaved to
reveal the end of the aminoacetic acid, and then LC3/Atg8 and PE
are bound to each other through a dialkylamine bond (49,50).
Following binding, LC3/Atg 8 are transformed from type I, which is
dispersed in the cytoplasm, to type II, which is localized on the
autophagic membrane (49,50). Therefore, an increase in LC3-II
expression is considered to be a marker of autophagy (50,51).
However, in the present study no statistical differences were found
for the ratio of LC3-II and LC-I between different groups (data not
shown). ERS involves the whole process of protein synthesis, it can
also induce the expression of LC3 II and Beclin 1 to promote the
formation of autophagosomes (52).
However, the specific mechanism of ERS in autophagy of tumor cells
is not very clear. Previous studies suggested that ERS induced by
overexpression of mutant proteins can induce the activation of
autophagy, which is mediated in part by the PERK/eIF2a pathway
(53–55). At the molecular level, elevated
levels of autophagy by PERK may be mediated by the upregulation of
autophagy-related gene Atg 12 (45,55).
Therefore, autophagy is a defense mechanism of cells against
misfolded proteins and this process is regulated by UPR.
Researchers also demonstrated that the formation of LC3
autophagosomes is dependent on the IRE-1 pathway, but not by the
PERK pathway (56). Notably,
autophagy activation occurs through IRE-1 kinase activity rather
than endonuclease activity and IRE-1 activates the TRAF2/JNK
pathway to activate autophagy (57,58).
The results demonstrated that LC3 II protein expression was
increased in cells treated with H2O2 alone
and with TM + H2O2, indicating that autophagy
was induced by oxidative stress and TM, and the latter enhanced the
former. Furthermore, activation of autophagy helps cells clear
ubiquitinated proteins by p62 (33). When autophagy occurs, p62 is bound
to the autophagy factor ATG8/LC3 by its ubiquitin-associated region
at the C-terminus interacting with the LC3-interacting region of
LC3 and is transferred to the autophagosome and then is degraded
(33,59). Studies demonstrated that the
misfolding of the proteins produced in the cell may first be
ubiquitinated and then degraded by the p62 autophagy pathway,
decreasing ERS (33,60). The present study also revealed that
in cells treated with H2O2 alone and with TM
+ H2O2, p62 expression was downregulated, and
LC3 II expression was upregulated, compared with the control and
cells treated with SAL + H2O2. Together with
the elevation of autophagy rates in cells treated with
H2O2 alone and with TM +
H2O2, it is hypothesized that autophagy can
be induced by H2O2 and enhanced by ER.
In conclusion, ERS and autophagy can be triggered
by H2O2, which can stimulate apoptosis of the
liver cancer cell line HepG2 when cells are exposed to high
concentrations of H2O2. The results suggested
that ERS inducer TM may be a potential target for treating
oxidative stress damage of tumor cells induced by antitumor drugs
via enhancing ERS and autophagy.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The analyzed data sets generated during the study
are available from the corresponding author on reasonable
request.
Authors' contributions
ZW, HW and CX designed the experiments. ZW and SF
performed the experiments. ZW, HW and SF analyzed the data. ZW, HW
and SF wrote the manuscript. ZW and HW revised the manuscript. All
authors reviewed the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare they have no competing
interests.
References
|
1
|
Benham AM: Protein folding and disulfide
bond formation in the eukaryotic cell: Meeting report based on the
presentations at the European Network Meeting on Protein Folding
and Disulfide Bond Formation 2009 (Elsinore, Denmark). FEBS J.
276:6905–6911. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kato H and Nishitoh H: Stress responses
from the endoplasmic reticulum in cancer. Front Oncol. 5:932015.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Henderson KA: Boric acid localization and
effects on storage calcium release and the endoplasmic reticulum in
prostate cancer cells. Dissertations & Theses-Gradworks.
2009.
|
|
4
|
Schapansky J, Morissette M, Odero G,
Albensi B and Glazner G: Neuregulin beta1 enhances peak
glutamate-induced intracellular calcium levels through endoplasmic
reticulum calcium release in cultured hippocampal neurons. Can J
Physiol Pharmacol. 87:883–891. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mandl J, Mészáros T, Bánhegyi G and Csala
M: Minireview: Endoplasmic reticulum stress: Control in protein,
lipid, and signal homeostasis. Mol Endocrinol. 27:384–393. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rangelaldao R: The unfolded protein
response, inflammation, oscillators, and disease: A systems
biologyapproach. Endo Reticulum Stress Dis. 2:30–52. 2015.
|
|
7
|
Richie DL, Feng X, Hartl L, Aimanianda V,
Krishnan K, Powers-Fletcher MV, Watson DS, Galande AK, White SM,
Willett T, et al: The virulence of the opportunistic fungal
pathogen requires cooperation between the endoplasmic
reticulum-associated degradation pathway (ERAD) and the unfolded
protein response (UPR). Virulence. 2:12–21. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Townsend DM, Manevich Y, He L, Xiong Y,
Bowers RR Jr, Hutchens S and Tew KD: Nitrosative-stress induced
S-glutathionylation of PDI leads to activation of the unfolded
protein response. Cancer Res. 69:7626–7634. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Reddy RK, Mao C, Baumeister P, Austin RC,
Kaufman RJ and Lee AS: Endoplasmic reticulum chaperone protein
GRP78 protects cells from apoptosis induced by topoisomerase
inhibitors: Role of ATP binding site in suppression of caspase-7
activation. J Biol Chem. 278:20915–20924. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bennett HL, Fleming JT, O'Prey J, Ryan KM
and Leung HY: Androgens modulate autophagy and cell death via
regulation of the endoplasmic reticulum chaperone glucose-regulated
protein 78/BiP in prostate cancer cells. Cell Death Dis. 1:e722010.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lu T, Yang W, Wang Z, Hu Z, Zeng X, Yang
C, Wang Y, Zhang Y, Li F, Liu Z, Wang D and Ye Z: Knockdown of
glucose-regulated protein 78/binding immunoglobulin heavy chain
protein expression by asymmetric small interfering RNA induces
apoptosis in prostate cancer cells and attenuates migratory
capability. Mol Med Rep. 11:2492015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Maiuolo J, Bulotta S, Verderio C, Benfante
R and Borgese N: Selective activation of the transcription factor
ATF6 mediates endoplasmic reticulum proliferation triggered by a
membrane protein. Proc Natl Acad Sci USA. 108:7832–1837. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Umebayashi K, Hirata A, Horiuchi H, Ohta A
and Takagi M: Unfolded protein response-induced BiP/Kar2p
production protects cell growth against accumulation of misfolded
protein aggregates in the yeast endoplasmic reticulum. Eur J Cell
Biol. 78:726–738. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gardner BM and Walter P: Unfolded proteins
are Ire1-activating ligands that directly induce the unfolded
protein response. Science. 333:1891–1894. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tang J, Guo YS, Zhang Y, Yu XL, Li L,
Huang W, Li Y, Chen B, Jiang JL and Chen ZN: CD147 induces UPR to
inhibit apoptosis and chemosensitivity by increasing the
transcription of Bip in hepatocellular carcinoma. Cell Death
Differ. 19:1779–1790. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hiss DC and Gabriels GA: Implications of
endoplasmic reticulum stress, the unfolded protein response and
apoptosis for molecular cancer therapy. Part II: Targeting cell
cycle events, caspases, NF-κB and the proteasome. Exp Opin Drug
Discov. 4:907–921. 2009. View Article : Google Scholar
|
|
17
|
Svetlana S, Patricia C, Mnich K, Ayo A,
Pakos-Zebrucka K, Patterson JB, Logue SE and Samali A: Endoplasmic
reticulum stress-mediated induction of SESTRIN 2 potentiates cell
survival. Oncotarget. 7:12254–12266. 2016.PubMed/NCBI
|
|
18
|
Netherton CL, Parsley JC and Wileman T:
African swine fever virus inhibits induction of the stress-induced
proapoptotic transcription factor CHOP/GADD153. J Virol.
78:10825–10828. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Moriya S, Miyazawa K, Kawaguchi T, Che XF
and Tomoda A: Involvement of endoplasmic reticulum stress-mediated
CHOP (GADD153) induction in the cytotoxicity of
2-aminophenoxazine-3-one in cancer cells. Int J Oncol. 39:981–988.
2011.PubMed/NCBI
|
|
20
|
Tan Y, Dourdin N, Wu C, De VT, Elce JS and
Greer PA: Ubiquitous calpains promote caspase-12 and JNK activation
during endoplasmic reticulum stress-induced apoptosis. J Biol Chem.
281:16016–16024. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Niso-Santano M, Bravo-San Pedro JM,
Gómez-Sánchez R, Climent V, Soler G, Fuentes JM and González-Polo
RA: ASK1 overexpression accelerates paraquat-induced autophagy via
endoplasmic reticulum stress. Toxicol Sci. 119:1562011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Nakka VP, Prakash-Babu P and Vemuganti R:
Crosstalk between endoplasmic reticulum stress, oxidative stress,
and autophagy: Potential therapeutic targets for acute CNS
Injuries. Mol Neurobiol. 53:532–544. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Devenish RJ and Klionsky DJ: Autophagy:
Mechanism and physiological relevance ‘brewed’ from yeast studies.
Front Biosci (Schol Ed). 4:1354–1363. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Deegan S, Koryga I, Glynn SA, Gupta S,
Gorman AM and Samali A: A close connection between the PERK and IRE
arms of the UPR and the transcriptional regulation of autophagy.
Biochem Biophys Res Commun. 456:305–311. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Drake KR, Kang M and Kenworthy AK:
Nucleocytoplasmic distribution and dynamics of the autophagosome
marker EGFP-LC3. PLoS One. 5:e98062010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tamura H, Shibata M, Koike M, Sasaki M and
Uchiyama Y: Atg9A, an autophagy-related membrane protein, is
localized in neurons of mouse brain. Neuroscience Research.
68:443–453. 2010. View Article : Google Scholar
|
|
27
|
Jo YK, Kim SC, Park IJ, Park SJ, Jin DH,
Hong SW, Cho DH and Kim JC: Increased expression of ATG10 in
colorectal cancer is associated with lymphovascular invasion and
lymph node metastasis. PLoS One. 7:e527052012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chen ZH, Cao JF, Zhou JS, Liu H, Che LQ,
Mizumura K, Li W, Choi AM and Shen HH: Interaction of caveolin-1
with ATG12-ATG5 system suppresses autophagy in lung epithelial
cells. Am J Physiol Lung Cell Mol Physiol. 306:1016–1025. 2014.
View Article : Google Scholar
|
|
29
|
Hwang S, Maloney NS, Bruinsma MW, Goel G,
Duan E, Zhang L, Shrestha B, Diamond MS, Dani A, Sosnovtsev SV, et
al: Nondegradative role of Atg5-Atg12/Atg16L1 autophagy protein
complex in antiviral activity of interferon gamma. Cell Host
Microbe. 11:397–409. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kung CP, Budina A, Balaburski G,
Bergenstock MK and Murphy M: Autophagy in tumor suppression and
cancer therapy. Critical Reviews in Eukaryotic Gene Exp. 21:71–100.
2011. View Article : Google Scholar
|
|
31
|
Lebovitz CB, Bortnik SB and Gorski SM:
Here, there be dragons: Charting autophagy-related alterations in
human tumors. Clin Cancer Res. 18:1214–1226. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
BenYounès A, Tajeddine N, Tailler M, Malik
SA, Shen S, Métivier D, Kepp O, Vitale I, Maiuri MC and Kroemer G:
A fluorescence-microscopic and cytofluorometric system for
monitoring the turnover of the autophagic substrate p62/SQSTM1.
Autophagy. 7:883–891. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Komatsu M, Kurokawa H, Waguri S, Taguchi
K, Kobayashi A, Ichimura Y, Sou YS, Ueno I, Sakamoto A, Tong KI, et
al: The selective autophagy substrate p62 activates the stress
responsive transcription factor Nrf2 through inactivation of Keap1.
Nat Cell Biol. 12:213–223. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kundu M: ULK1, Mammalian Target of
Rapamycin, and Mitochondria: Linking Nutrient Availability and
Autophagy. Antioxid Redox Signal. 14:1953–1958. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Cheong H and Klionsky DJ: Dual role of
Atg1 in regulation of autophagy-specific PAS assembly in
Saccharomyces cerevisiae. Autophagy. 4:724–726. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Luciani MF, Giusti C, Harms B, Oshima Y,
Kikuchi H, Kubohara Y and Golstein P: Atg1 allows second-signaled
autophagic cell death in Dictyostelium. Autophagy. 7:501–508. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yorimitsu T, Nair U, Yang Z and Klionsky
DJ: Endoplasmic reticulum stress triggers autophagy. J Biol Chem.
281:30299–30304. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Tekirdag KA, Korkmaz G, Ozturk DG, Agami R
and Gozuacik D: MIR181A regulates starvation- and rapamycin-induced
autophagy through targeting of ATG5. Autophagy. 9:374–385. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lopez-Terrada D, Cheung SW, Finegold MJ
and Knowles BB: Hep G2 is a hepatoblastoma-derived cell line. Hum
Pathol. 40:1512–1515. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kenneth J and Livak TD: Analysis of
relative gene expression data using rea l-time quantitative PCR a
nd the 2(-Delta Delta C(T)) method. Method. 25:402–408. 2001.
View Article : Google Scholar
|
|
41
|
Simasi J, Schubert A, Oelkrug C, Gillissen
A and Nieber K: Primary and secondary resistance to tyrosine kinase
inhibitors in lung cancer. Anticancer Res. 34:2841–2850.
2014.PubMed/NCBI
|
|
42
|
Quintás-Cardama A, Kantarjian HM and
Cortes JE: Mechanisms of primary and secondary resistance to
imatinib in chronic myeloid leukemia. Cancer Control. 16:122–131.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Schleicher SM, Moretti L, Varki V and Lu
B: Progress in the unraveling of the endoplasmic reticulum
stress/autophagy pathway and cancer: Implications for future
therapeutic approaches. Drug Resist Updat. 13:79–86. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Li T, Su L, Zhong N, Hao X, Zhong D,
Singhal S and Liu X: Salinomycin induces cell death with autophagy
through activation of endoplasmic reticulum stress in human cancer
cells. Autophagy. 9:1057–1068. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Su CC: Tanshinone IIA could inhibit
pancreatic cancer BxPC-3 cells through increasing PERK, ATF6,
Caspase-12 and CHOP expression to induce apoptosis. J Biom Sci Eng.
8:149–159. 2015. View Article : Google Scholar
|
|
46
|
Liu H, Nishitoh H, Ichijo H and Kyriakis
JM: Activation of apoptosis signal-regulating kinase 1 (ASK1) by
tumor necrosis factor receptor-associated factor 2 requires prior
dissociation of the ASK1 inhibitor thioredoxin. Mol Cell Biol.
20:2198–2208. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Nakagawa T and Yuan J: Cross-talk between
two cysteine protease families activation of caspase-12 by calpain
in apoptosis. J Cell Biol. 150:887–894. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Morishima N, Nakanishi K, Takenouchi H,
Shibata T and Yasuhiko Y: An Endoplasmic Reticulum Stress-specific
Caspase Cascade in Apoptosis cytochrome c-independent activation of
caspase-9 by caspase-12. J Biol Chem. 277:34287–34294. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Bae JY and Park HH: Purification and
characterization of a ubiquitin-like system for autophagosome
formation. J Microbiol Biotechnol. 20:1647–1652. 2010.PubMed/NCBI
|
|
50
|
Satoo K, Noda NN, Kumeta H, Fujioka Y,
Mizushima N, Ohsumi Y and Inagaki F: The structure of Atg4B-LC3
complex reveals the mechanism of LC3 processing and delipidation
during autophagy. EMBO J. 28:1341–1350. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Tanida I, Minematsuikeguchi N, Ueno T and
Kominami E: Lysosomal turnover, but not a cellular level, of
endogenous LC3 is a marker for autophagy. Autophagy. 1:84–91. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Sailaja GS, Praveen B, Bharathi G, Chetty
C, Gogineni VR, Velpula KK, Gondi CS and Rao JS: The secreted
protein acidic and rich in cysteine (SPARC) induces endoplasmic
reticulum stress leading to autophagy-mediated apoptosis in
neuroblastoma. Int J Oncol. 42:188–196. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Jiang Q, Li F, Shi K, Wu P, An J, Yang Y
and Xu C: Involvement of p38 in signal switching from autophagy to
apoptosis via the PERK/eIF2α/ATF4 axis in selenite-treated NB4
cells. Cell Death Dis. 5:e12702014. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Kamada Y: Prime-numbered Atg proteins act
at the primary step in autophagy: Unphosphorylatable Atg13 can
induce autophagy without TOR inactivation. Autophagy. 6:415–416.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Saito A, Ochiai K, Kondo S, Tsumagari K,
Murakami T, Cavener DR and Imaizumi K: Endoplasmic reticulum stress
response mediated by the PERK-eIF2(alpha)-ATF4 pathway is involved
in osteoblast differentiation induced by BMP2. J Biol Chem.
286:4809–4818. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Kimura S, Noda T and Yoshimori T:
Dynein-dependent movement of autophagosomes mediates efficient
encounters with lysosomes. Cell Struct Funct. 33:109–122. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Lee H, Noh JY, Oh Y, Kim Y, Chang JW,
Chung CW, Lee ST, Kim M, Ryu H and Jung YK: IRE1 plays an essential
role in ER stress-mediated aggregation of mutant huntingtin via the
inhibition of autophagy flux. Hum Mol Genet. 21:101–114. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Zhang C, Kawauchi J, Adachi MT, Hashimoto
Y, Oshiro S, Aso T and Kitajima S: Activation of JNK and
Transcriptional Repressor ATF3/LRF1 through the IRE1/TRAF2 pathway
is implicated in human vascular endothelial cell death by
homocysteine. Biochem Biophys Res Commun. 289:718–724. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Shvets E, Fass E, Scherz-Shouval R and
Elazar Z: The N-terminus and Phe52 residue of LC3 recruit
p62/SQSTM1 into autophagosomes. J Cell Sci. 121:2685–2695. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Liu WJ, Ye L, Huang WF, Guo LJ, Xu ZG, Wu
HL, Yang C and Liu HF: p62 links the autophagy pathway and the
ubiqutin-proteasome system upon ubiquitinated protein degradation.
Cell Mol Biol Lett. 21:292016. View Article : Google Scholar : PubMed/NCBI
|