Introduction
Sevoflurane is a volatile anesthetic used to induce
and maintain general anesthesia in surgical settings. It is one of
the most commonly used anesthetic agents that progressively
eclipsed a number of existing anesthetics since its first use as an
anesthetic agent. It owes its popularity to its low airway
irritability, ability to rapidly induce anesthesia and good
pharmacokinetic performance (1–3).
However, growing evidence demonstrates that sevoflurane caused
severe side effects, particularly in infants (4,5).
Indeed, sevoflurane was reported to induce high rates of
postoperative cognitive dysfunction in comparison with other
anesthetic agents (6). These
dysfunctions may be associated with sevoflurane-induced
neurotoxicity, which is documented in animals and humans (7–9). To
date, the molecular mechanisms underlying sevoflurane-induced
neurotoxicity have not been fully elucidated. However, a number of
studies have demonstrated that exposure to sevoflurane induces
alterations in the microRNA (miR) expression profile (10–14).
miRs are small non-coding RNA molecules involved in
the regulation of a number of biological processes. They
principally act as post-transcriptional gene regulators by binding
to complementary sequences of target mRNAs, which leads to mRNA
silencing via the inhibition of translation or destabilization of
the mRNAs (15,16). miRs are expressed in response to
particular conditions including exposure to anesthetic agents such
as sevoflurane (12,17). Thousands of miR species have been
identified, sequenced and characterized; however, their functional
roles have yet to be clarified, as one miR may regulate the
expression of a number of genes, including other gene regulators.
The human miR-302 family members have been demonstrated to be
involved in neuronal cell development and biology (18,19).
The miR-302/367 cluster, including miR-367, miR-302a, miR-302b,
miR-302c and miR-302d, has been reported to serve crucial roles in
a variety of biological processes, including the pluripotency of
human embryonic stem cells (20),
reprogramming and self-renewal (21), tumor growth (22) and the regulation of hypoxia
(23). It was additionally
demonstrated that miR-302 is involved in the regulation of
neurulation by inhibiting the differentiation and expansion of
neural progenitors (24). miR-302
is known to regulate DNA repair within cells (25). In addition, the miR-302/367 cluster
is able to orchestrate processes involved in neural tube formation
(26). Notably, a previous study
have indicated that miR-302 protects cells from cytotoxicity by
activating the protein kinase B/nuclear factor erythroid 2-related
factor 2/Nanog axis, to subsequently improve insulin signaling in
neurons (27). An additional study
has demonstrated that miR-302 antagomir protects cells from
apoptosis in hypoxia/reoxygenation injury (28). Additionally, miR-302/367 has
crucial roles in the regeneration of the hippocampus and other
brain structures following neuronal loss (29) and may restore learning and memory
in patients with Alzheimer's disease (30). Therefore, miR-302 family members
were hypothesized to be involved in sevoflurane-induced
neurotoxicity.
Among the miR-302 family members, miR-302e is the
least studied and its functional role is unknown. The present study
aimed to experimentally document the in vitro neurotoxic
effect of sevoflurane and the role of hsa-miR-302e in the
underlying mechanism.
Materials and methods
Cell culture
Human neurons-hippocampal primary cells (HN-h; cat.
no. 1540) were purchased from ScienCell Research Laboratories, Inc.
(San Diego, CA, USA) and plated in Neuronal Medium (cat. no. 1521),
additionally from ScienCell Research Laboratories, Inc. As
recommended by the manufacturer, cells were cultured at 37°C in an
environment of 95% humidity and 5% CO2 incubator for 48
h.
Transient transfection
HN-h cells (5×104 cells/cm2)
were transfected with 5 nM hsa-miR-302e mimic
(5′-UAAGUGCUUCCAUGCUU-3′; MISSION® microRNA mimic), 20
nM hsa-miR-302e inhibitor (5′-UAAGUGCUUCCAUGCUU-3′) or respective
controls (control mimic oligonucleotide,
5′-UCACAACCUCCUAGAAAGAGUAGA-3′; control inhibitor oligonucleotide,
5′-UUGUACUACACAAAAGUACUG-3′; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) using Lipofectamine® reagent (Thermo
Fisher Scientific, Inc.) according to the manufacturer's protocol.
For oxidation resistance gene 1 (OXR1) overexpression, the OXR1
Lentiviral vector (Human; CMV; pLenti-GIII-CMV) was purchased from
Applied Biological Materials Inc. (Richmond, BC, Canada) and was
employed for transfection following the manufacturer's protocol. A
reverse transcription-quantitative polymerase chain reaction
(RT-qPCR) was used to verify the transfection efficiency 72 h
following transfection.
Exposure to sevoflurane
Sevoflurane exposure experiments was performed as
previously described with minor modifications (31). Briefly, hermetically sealed plastic
chambers were used. The chamber was equipped with an inlet
connector coupled to a sevoflurane vaporizer and an outlet
connector linked to a gas monitor (PM 8060; Drager AG & Co.,
KGaA, Lübeck, Germany), which was necessary for checking the gas
concentration in the chamber. Cell cultures (105
cells/well) were kept in the chambers and following the adjustment
of sevoflurane concentration, each chamber was treated with 0, 5,
10 or 15% sevoflurane in the carrier gas (95% air/5%
CO2) for 15 min. Subsequently, once a target
concentration was attained, the corresponding chamber was sealed
and incubated for 6 h at 37°C with renewal of the gas in the
chamber every 3 h. The gas monitor was used to confirm the target
concentration of sevoflurane at the end of the experiment. For
control cells, the same procedure was applied with substitution of
sevoflurane by 5% CO2 air.
Cell viability
An MTT assay was used to evaluate the cytotoxic
effect of sevoflurane on the viability of HN-h cells. Following the
6 h sevoflurane exposure, cells were subsequently cultured for a
period of 48 h prior to the addition of 20 µl MTT reagent (Cell
Proliferation kit I; Roche Applied Science, Penzberg, Germany; 5
mg/ml). Subsequently, the mixture was incubated for an additional 4
h prior to the removal of culture supernatants and their
replacement in each well with 150 µl DMSO as a solubilizing reagent
for the formazan crystals produced by viable cells. Finally, cell
viability was spectrophotometrically assessed via measuring the
optical density at 570 nm with a spectrophotometer (Spectronic
Genesys-5; Milton Roy; Accudyne Industries, Dallas, TX, USA).
Apoptosis assay
A FITC-Annexin V/7-AAD kit (Beckman Coulter, Inc.,
Brea, CA, USA) was used for apoptosis analysis. Following
incubation, ~1×105 cells were collected and rinsed twice
using PBS prior to Annexin V/7-AAD staining for 15 min at room
temperature. Subsequently, cell apoptosis was analyzed using a flow
cytometer (BD Biosciences, Franklin Lakes, NJ, USA; C6). The
software used for analyzing the data was BD FACSDiva software
v8.0.1 (BD Biosciences). Three independent experiments were
performed.
Determination of lactate dehydrogenase
(LDH) release
The culture medium (105
cells/cm2) was taken from the wells and spun in a
microfuge for 2 min at 4°C and 15,000 × g to remove any detached
debris. The supernatant was collected for the measurement of LDH in
the cell-free medium. The cell pellet and the cells remaining in
the multiwell were lysed in 0.5 ml lysis buffer [0.5% Triton X-100
in 0.1 M potassium phosphate buffer, (pH 7.0)]. LDH activity was
assessed using a colorimetric method by monitoring the absorbance
at 340 nm in the presence of 0.2 mM NADH and 2 mM pyruvate. A total
of 1 unit of LDH activity is defined as the amount of enzyme that
catalyzes the formation of 1 µmol NAD mm−1 under the
assay conditions and the percentage of LDH released was estimated
as the cell-free LDH activity divided by the total LDH
activity.
Reactive oxygen species (ROS) level
detection
To determine the level of ROS produced by the cells,
cells were collected, rinsed three times with PBS and trypsinized
prior to centrifugation at 4°C and 15,000 × g for 10 min. Collected
cells were incubated with diluted dihydroethidium provided with the
Reactive Oxygen Detection kit (cat. no. S0033) purchased from
Beyotime Institute of Biotechnology (Haimen, China) according to
the manufacturer's protocol. Subsequently, the level of ROS was
evaluated by flow cytometry analysis using the software mentioned
above.
Lipid peroxidation assay
To determine the extent of lipid peroxidation, cells
were harvested via centrifugation at 4°C and 15,000 × g for 15 min.
The collected cells were frozen and subsequently thawed. Following
thawing, 1 ml 0.67% thiobarbituric acid (TBA; Wako Pure Chemical
Industries, Ltd., Osaka, Japan) and 0.4 ml 5% trichloroacetic acid
were added to the cell suspensions, followed by heating of the
mixture for 60 min in a boiling water bath. Then, the mixture was
centrifuged at 4°C and 15,000 × g for 15 min and the absorbance
measured at 532 nm with a spectrophotometer (Spectronic Genesys-5;
Milton Roy; Accudyne Industries). The amount of malondialdehyde
(MDA) was deduced from the molar extinction coefficient of the
MDA-TBA complex of 1.56×105 cm−1
M−1.
Measurement of intracellular
Ca2+
In order to measure the intracellular
Ca2+, the Fluo 3-AM reagent (Thermo Fisher Scientific,
Inc.) was used. Cells were cultured with 5 mM glutamate in the
presence or absence of sevoflurane. Following harvesting, cells
were incubated at 37°C for 30 min with Fluo 3-AM. Subsequently,
Ca2+-dependent fluorescence intensity was measured by
flow cytometry and 10,000 cells were acquired and analyzed for each
sample.
RT-qPCR analysis
RNA was isolated using miRNA Isolation kit or
miRNeasy Mini kit (Qiagen GmbH, Hilden, Germany) following the
manufacturer's protocol, and cDNA synthesis was performed with the
TaqMan MicroRNA Reverse Transcription kit (Thermo Fisher
Scientific, Inc.). Subsequently, the qPCR amplification was
achieved using the hsa-miR-302e primers with the CFX96
Touch™ Real-Time PCR Detection System (Bio-Rad
Laboratories, Inc., Hercules, CA, USA) according to the
manufacturer's protocol. The forward and reverse primers sequences
for the hsa-miR-302e were; forward primer, 5′-CGCAGTAAGTGCTTCCA-3′
and reverse primer, 5′-GTCCAGTTTTTTTTTTTTTTTAAGCAT-3′. The primers
for U6 gene were; forward primer, 5′-GTGCTCGCTTCGGCAGCACATATAC-3′
and reverse primer, 5′-AAAAATATGGAACGCTTCACGAATTTG-3′. The primers
for OXR1 were forward primer, 5′-TTCGACCAAACCTAAGTGATCCC-3′ and
reverse primer, 5′-GGGGTGTCTAAACCTGTCATTG-3′. The primers for GAPDH
were forward primer, 5′-ACCCACTCCTCCACCTTTGA-3′ and reverse primer,
5′-CTGTTGCTGTAGCCAAATTCGT-3′ The PCR conditions were: 30 sec at
95°C, 45 amplification cycles at 95°C for 5 sec and 58°C for 34
sec. The expression level hsa-miR-302e was obtained using the
2−∆∆Cq method (32)
with U6 small nuclear RNA as a normalizing control while GAPDH was
used as internal control for OXR1.
Western blot analysis
HN-h cells were collected with trypsin and lysed
with radioimmunoprecipitation assay buffer (Sigma-Aldrich; Merck
KGaA, Darmstadt, Germany) and protein concentration in cell lysates
were measured using the bicinchoninic acid protein assay kit
(Beyotime Institute of Biotechnology). Next, 50 µg of the lysates
were purified by 10% SDS-PAGE and subsequently transferred to
polyvinylidene difluoride membranes. Following blocking at 4°C with
5% bovine serum albumin in Tris-buffered saline with 0.05% Tween-20
(TBST) for 2 h, the membranes were incubated at 4°C overnight with
primary antibodies against OXR1 (cat. no. HPA027375; dilution
1:1,000; Sigma-Aldrich; Merck KGaA), Phospho-CaMKII (Thr286; cat.
no. D21E4; dilution 1:1,000; Cell Signaling Technology, Inc.,
Danvers, MA, USA), CaMKII-α (cat. no. D10C11; dilution 1:1,000;
Cell Signaling Technology, Inc.) and GAPDH (cat. no. ab9485;
dilution 1:1,000; Abcam, Cambridge, UK) overnight at 4°C. Next, the
membranes were washed three times with TBST for 5 min and incubated
with horseradish peroxidase conjugated anti-mouse mouse anti-human
immunoglobulin M antibody (M11; cat. no. sc-66121; 1:1,000; Santa
Cruz Biotechnology, Inc., Dallas, TX, USA) at room temperature for
1 h, followed by 5 min wash with TBST, which was repeated three
times. The membranes were visualized using The Clarity Western
Enhanced Chemiluminescent Substrate (cat. no. 1705060; Bio-Rad
Laboratories, Inc.). Densitometric analysis was achieved using the
Image J software version 1.51k (National Institutes of Health,
Bethesda, MD, USA) for Windows.
Bioinformatics
The online bioinformatics tool Targetscan
(http://www.targetscan.org) was used for
predicting the targeting of OXR1 by hsa-miR-302e.
Luciferase assays
The 3′-untranslate region (UTR) of OXR1 was isolated
and 50 µg was used for cloning into the Renilla luciferase
plasmid pRL-TK vector (Promega Corporation, Madison, WI, USA) in
the downstream region of the Renilla luciferase gene.
Additionally, the mutated 3′-UTR of OXR1 (pRL-TK/OXR1-Mut) was
produced with the QuikChange II Site-Directed Mutagenesis kit
purchased from Agilent Technologies, Inc. (Santa Clara, CA, USA).
For the luciferase assays, HN-h cells were seeded in 24-well plates
at a density of 105 cells/well and co-transfected with
miR-302e mimic (5 nM) and 10 ng of pRL-TK vector (Promega
Corporation) and the Renilla luciferase plasmid pRL-TK
vector (10 ng) [luciferase gene under the control of OXR1 UTR]
using Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.). Cells were collected 48 h following transfection
and lysed prior to the measurement of luciferase activity with the
Dual-Luciferase Reporter Assay System (Promega Corporation).
Renilla luciferase was used for normalization. The
experiments were performed independently in triplicate.
Statistical analysis
GraphPad Prism software (version 6.0; GraphPad
Software, Inc., La Jolla, CA, USA) was used for statistical
analysis. Each data was presented as the mean ± standard deviation.
Statistical differences were evaluated using one-way or two-way
analysis of variance followed by Dunnett's multiple comparisons
test. All experiments were performed at least three times.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Sevoflurane exerts a cytotoxic effect
on HN-h cells
HN-h cells were incubated for 6 h in the presence of
sevoflurane at different concentrations and cell viability was
monitored by MTT assay after 48 h of incubation in culture medium.
The cell viability was decreased in a dose-dependent manner in
presence of sevoflurane. At 5% sevoflurane, cell viability was
significantly decreased to 88.35±2.1% (P<0.01) compared with the
control. In presence of 15% sevoflurane, cell viability was
significantly reduced to 39.85±4% (P<0.0001) compared with the
control cells (Fig. 1A; Table I). To further understand the
mechanism of this decrease in cell viability, the level of
apoptosis was also determined following incubation of cells in the
presence (15% sevoflurane) or absence of sevoflurane. In the
absence of sevoflurane, the apoptosis rate was estimated to be
0.11±0.08 (n=3). The apoptosis rate significantly increased to
9.25±1.5 in cell cultures incubated with 15% sevoflurane (Fig. 1B; P<0.0001 vs. 0%). Markers of
apoptotic pathways were additionally investigated in the cells
cultured in the absence or presence of various concentrations of
sevoflurane (Fig. 1C-G). The
results indicated that sevoflurane significantly increased LDH and
intracellular Ca2+ release in a dose-dependent manner
(P<0.01; Fig. 1C and F). The
level of ROS and MDA were also increased by sevoflurane (Fig. 1D and E). The level of
calcium/calmodulin-dependent protein kinase II (CAMII) did not
significantly alter but the level of phosphorylated CAMII was
significantly increased by sevoflurane exposure (P<0.01;
Fig. 1G).
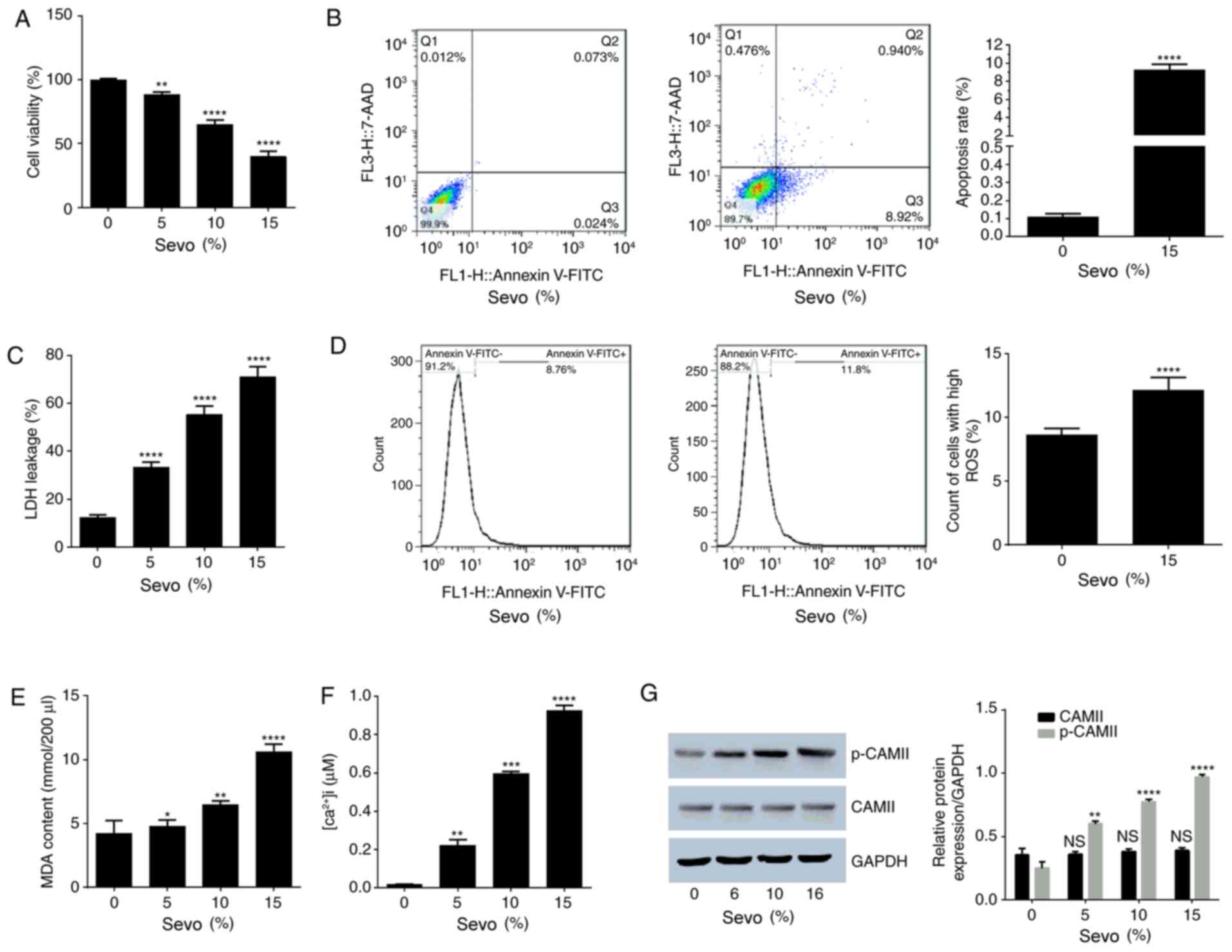 | Figure 1.Sevo exerted cytotoxic effects on
HN-h cells. The effect of 6 h treatment with varying concentrations
of sevo was measured. (A) Cell viability determined using an MTT
assay. (B) Apoptosis rate with flow cytometry results. (C) Level of
LDH leakage. (D) ROS count. (E) Results of lipid peroxidation assay
(MDA content). (F) Concentration of intracellular Ca2+.
(G) Western blotting evaluation of the expression level of CAMII
and phosphorylated CAMII relative to GAPDH. *P<0.05,
**P<0.01, ***P<0.001, ****P<0.0001 vs. control (0% Sevo
group). Sevo, sevoflurane; LDH, lactate dehydrogenase; FITC,
fluorescein isothiocyanate; ROS, reactive oxygen species;
[Ca2+]i, intracellular calcium; MDA, malondialdehyde;
p-, phospho-; CAMII, calcium/calmodulin-dependent protein kinase
II; NS, no significance. |
 | Table I.Sevoflurane-induced cytotoxic
effect. |
Table I.
Sevoflurane-induced cytotoxic
effect.
| Concentration of
sevoflurane, % | Cell viability,
% | Apoptosis rate,
% | LDH leakage, % | [Ca
2+]i, µM | ROS production,
% | MDA, mmol/200
µl | CAMII expression
levela | p-CAMII expression
levela |
|---|
| 0 | 100.000±1.000 | 0.190±0.080 | 12.300±1.500 | 0.018±0.002 | 8.650±0.500 | 4.230±0.910 | 0.360±0.050 | 0.250±0.030 |
| 5 | 88.350±2.100 |
| 33.480±2.100 | 0.222±0.030 |
| 4.790±0.500 | 0.360±0.020 | 0.600±0.031 |
| 10 | 65.140±3.500 |
| 55.300±2.850 | 0.598±0.010 |
| 6.490±0.300 | 0.380±0.010 | 0.770±0.040 |
| 15 | 39.850±4.000 | 25.900±1.500 | 71.200±3.200 | 0.926±0.025 | 12.100±1.000 | 10.610±0.600 | 0.390±0.030 | 0.970±0.020 |
Sevoflurane upregulates hsa-miR-302e
in HN-h cells
RT-qPCR experiments were performed to determine the
level of hsa-miR-302e in cultured cells. The results revealed a
dose-dependent increase in hsa-miR-302e expression following
treatment with sevoflurane (Fig.
2A). The levels of hsa-miR-302e were significantly increased to
2.42±0.60 (P<0.01), 3.98±0.40 (P<0.001) and 8.40±0.32
(P<0.0001) in the 5, 10 and 15% groups, respectively compared
with the control group (1.03±0.20). These results suggested a
potential role of hsa-miR-302e in sevoflurane-induced
cytotoxicity.
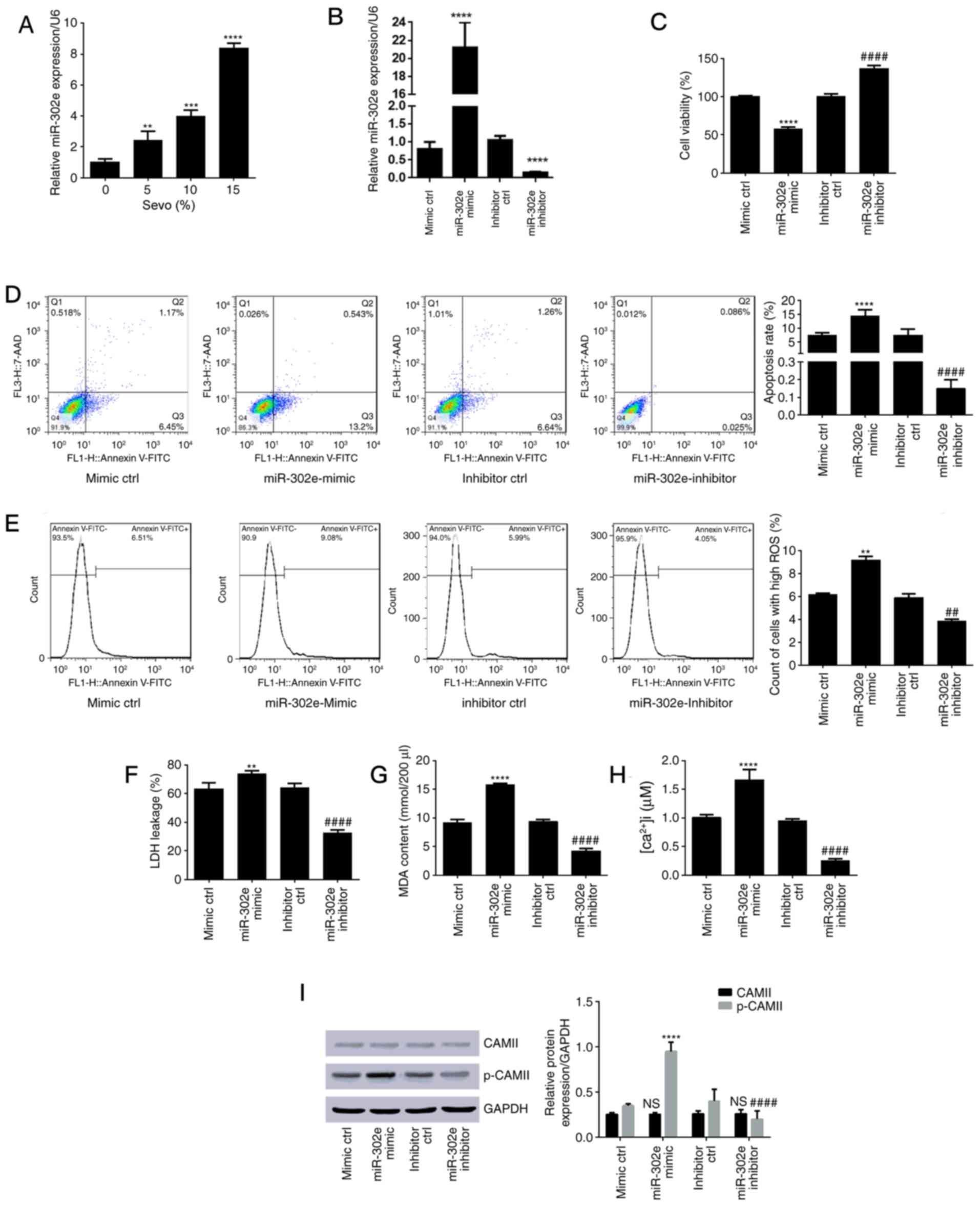 | Figure 2.Sevo upregulated hsa-miR-302e in HN-h
cells and inhibition of hsa-miR-302e alleviated sevo-induced
cytotoxicity. (A) Expression level of hsa-miR-302e following cell
treatment with sevo was measured using the reverse
transcription-quantitative polymerase chain reaction approach.
**P<0.01, ***P<0.001, ****P<0.0001 vs. control (0% Sevo
group). (B) Expression level of hsa-miR-302e following transfection
with mir-302e mimic, inhibitor or corresponding negative controls
was measured using the reverse transcription-quantitative
polymerase chain reaction approach. (C) Cell viability determined
using MTT assay. (D) Apoptosis rate with flow cytometry results.
(E) ROS count. (F) Level of LDH leakage. (G) Results of lipid
peroxidation assay (MDA content). (H) Concentration of
intracellular Ca2+. (I) Expression level of CAMII and
p-CAMII relative to GAPDH. **P<0.01, ***P<0.001,
****P<0.0001 vs. mimic control; ##P<0.01,
####P<0.0001 vs. the inhibitor control. Sevo,
sevoflurane; miR, microRNA; ctrl, control; 7-AAD,
7-aminoactinomycin D; FITC, fluorescein isothiocyanate; ROS,
reactive oxygen species; LDH, lactate dehydrogenase; MDA,
malondialdehyde; [Ca2+]i, intracellular calcium; CAMII,
calcium/calmodulin-dependent protein kinase II; p-, phospho-; NS,
no significance. |
Inhibition of hsa-miR-302e alleviates
sevoflurane-induced cytotoxicity
In order to study the involvement of hsa-miR-302e in
sevoflurane-induced cytotoxicity, cells were transfected with the
hsa-miR-302e mimic or its inhibitor, which efficiently upregulated
or downregulated the expression of hsa-miR-302e in cells,
respectively (Fig. 2B). The cells
were subsequently subjected to 15% sevoflurane and the results are
presented in Fig. 2C-I. The
overexpression of hsa-miR-302e significantly reduced cell viability
from 100 to 58.12±2.10% (P<0.0001; Fig. 2C) and significantly induced cell
apoptosis (Fig. 2D; P<0.0001).
Additionally, the hsa-miR-302e mimic significantly induced ROS
production (Fig. 2E; P<0.01),
LDH release (Fig. 2F; P<0.01),
increased MDA content (Fig. 2G;
P<0.0001), intracellular Ca2+ level (Fig. 2H; P<0.0001) and CAMII
phosphorylation (Fig. 2I;
P<0.0001). In addition, hsa-miR-302e inhibitor induced the
opposite trends and even increased cell viability in comparison
with the controls.
OXR1 is a direct target of
hsa-miR-302e
The online Targetscan tool predicted that OXR1 was
among the candidate targets of hsa-miR-302e (Fig. 3A). Given that oxidative stress
pathways were activated by sevoflurane, western blot analysis was
performed to investigate the effect of sevoflurane on OXR1
expression. The results demonstrated that treatment with
sevoflurane significantly reduced the level of OXR1 protein in a
dose-dependent manner (Fig. 3B;
P<0.01). The expression level of the luciferase reporter gene
fused to OXR1 3′-UTR in control cells and cells harboring the
hsa-miR-302e mimic or inhibitor was presented in Fig. 3C. The hsa-miR-302e mimic
significantly reduced the expression level of the reporter gene
(P<0.0001) whereas the hsa-miR-302e inhibitor led to
significantly upregulated OXR1 expression (P<0.0001). No
significant difference was demonstrated with the mutated 3′-UTR
vector (Fig. 3C). Furthermore,
western blot analysis indicated that hsa-miR-302e overexpression
significantly downregulated OXR1 expression (Fig. 3D; P<0.0001). These results
demonstrated that OXR1 is a direct target of hsa-miR-302e. The
expression of OXR1 was significantly increased following
transfection with the overexpression vector, which suggested a good
transfection efficiency (Fig. 3E;
P<0.0001).
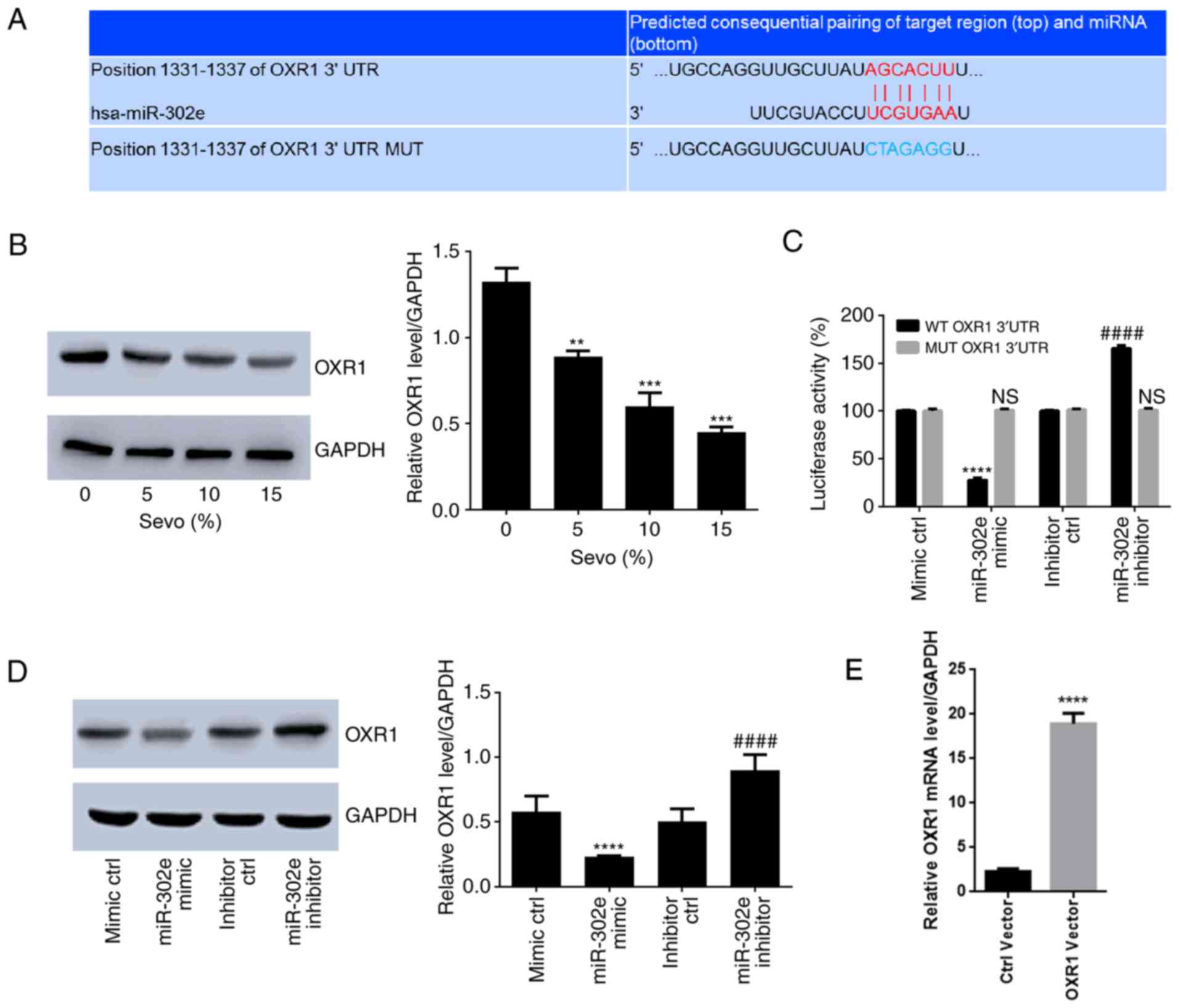 | Figure 3.OXR1 is a specific target of
hsa-miR-302e. (A) Alignment of the hsa-miR-302e sequence with its
target sequence in OXR1 UTR region as predicted by Targetscan.
**P<0.01, ***P<0.001 vs. the control (0% Sevo). (B)
Expression level of OXR1 following treatment with sevo. (C) Results
of luciferase reporter gene assay. (D) Expression level of OXR1 in
cells transfected with hsa-miR-302e mimic or inhibitor. (E)
Expression level of OXR1 in cells transfected with OXR1 expression
vector. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 vs.
the mimic control; ####P<0.0001 vs. the inhibitor
control. OXR1, oxidation resistance gene 1; UTR, untranslated
region; miR, microRNA; MUT, mutant; Sevo, sevoflurane; WT,
wild-type; NS, no significance; ctrl, control. |
Overexpression of OXR1 abolishes
sevoflurane-induced cytotoxicity
OXR1-overexpressing cells were used to study the
role of OXR1 in sevoflurane-induced cytotoxicity. The
OXR1-overexpressing cells were treated with 15% sevoflurane and the
results of its effects are presented in Fig. 4. OXR1-overexpressing cells
demonstrated a significant increase in viability following
sevoflurane exposure compared with the control cells (P<0.0001;
Fig. 4A). In addition, a reduced
apoptotic rate and reduced levels of cytotoxicity markers were
observed in cells overexpressing OXR1 (Fig. 4B-G). These results suggested that
overexpression of OXR1 abrogated sevoflurane-induced
cytotoxicity.
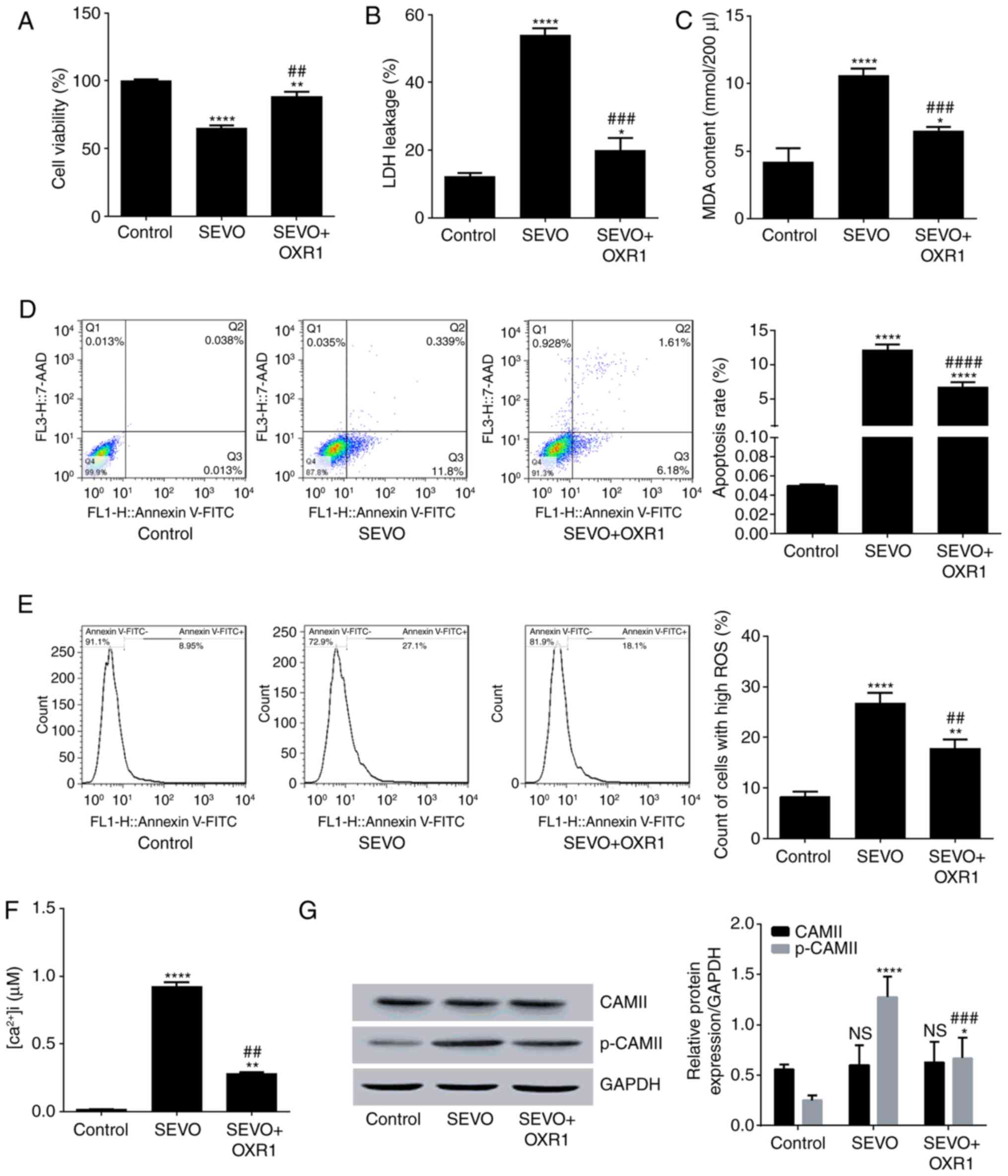 | Figure 4.Overexpression of OXR1 abrogates
Sevo-induced cytotoxicity. (A) Cell viability determined using an
MTT assay. (B) Level of LDH leakage. (C) Results of lipid
peroxidation assay (MDA content). (D) Apoptosis rate with flow
cytometry results. (E) ROS count. (F) Concentration of
intracellular Ca2+. (G) Expression level of CAMII and
p-CAMII relative to GAPDH. *P<0.05, **P<0.01, ****P<0.0001
vs. the control; ##P<0.01, ###P<0.001,
####P<0.0001 vs. the Sevo group. Sevo, sevoflurane;
OXR1, oxidation resistance gene 1; LDH, lactate dehydrogenase; MDA,
malondialdehyde; 7-AAD, 7-aminoactinomycin D; FITC, fluorescein
isothiocyanate; ROS, reactive oxidative species; CAMII,
calcium/calmodulin-dependent protein kinase II; p-, phospho-; NS,
no significance. |
Discussion
Sevoflurane has gained interest due to a number of
advantages over other anesthetics, however its use also has certain
limitations, including neuronal cytotoxicity. This cytotoxicity has
been documented in several studies (7,33).
HN-h cells were used in the present study as an in vitro
model to analyze the cytotoxic effect of sevoflurane on the human
brain. The results confirmed that sevoflurane is toxic to
hippocampal neuronal cells following 6 h of exposure and pointed to
oxidative stress as the principal trigger in sevoflurane-induced
neurotoxicity. The detection of MDA and the release of LDH
demonstrated that the oxidative stress induced by sevoflurane leads
to membrane disruption and necrosis. In addition, apoptotic
molecular markers were also detected demonstrating that the
cytotoxic effect of sevoflurane occurs by activating apoptosis
signaling pathways.
Oxidative stress is one of the most common events
associated with cytotoxic effects in the central nervous system. It
is observed in sclerosis, Alzheimer's and Parkinson's disease
(34). This demonstrates the
necessity to maintain an oxidative equilibrium in neuronal cells.
OXR1 occupies a crucial role among the various molecules that
prevent neuronal cells from oxidation stress-associated death
(35). This eukaryotic, highly
conserved protein does not scavenge ROS, it regulates the molecular
pathways that allow cell protection against oxidative stress and
neurodegenerative diseases including Parkinson's and Alzheimer's
diseases (36). Thus, the
expression level of this protein following cell treatment with
sevoflurane was investigated. Western blotting results demonstrated
that the treatment with sevoflurane significantly reduced the
expression level of OXR1 in HN-h cells.
It is well established that OXR1 is an inducible
protein, which is highly expressed in presence of ROS. Its
expression enhances ROS scavenging, however due to the necessity of
maintaining an equilibrium the protein also has negative regulators
that restore an appropriate level of protein expression when the
redox equilibrium in the cell is established (35,37).
Of these negative-regulators, the 22 nt hsa-miR-302e is involved
OXR1 regulation at the post-transcriptional level. The experiments
using hsa-miR-302e mimic and inhibitors effectively confirmed that
the miR directly targets OXR1 mRNA and downregulates the expression
of this gene by annealing to its 3′UTR sequence as demonstrated in
the reporter gene expression experiment. Notably, it was
demonstrated that sevoflurane treatment increases the expression
level of miR-302. As sevoflurane decreased OXR1 expression
concomitantly with the induction of miR-302e, it was concluded that
sevoflurane induces hsa-miR-302e expression, which in turn
downregulates the expression of OXR1. The downregulation of OXR1
disrupts the equilibrium in the cells and the increased the ROS
levels leading to the oxidation of cellular proteins and lipids.
Oxidative damage results in the activation of different cell death
pathways, including apoptosis and necrosis, which are observed as
sevoflurane-induced cytotoxicity (38).
High expression of hsa-miR-302e was previously
reported to be associated with radio-sensitivity in non-small cell
lung cancer (39). To the best of
our knowledge the present study is the first to specifically
establish experimentally the association of this miR with neuronal
cell resistance to oxidative stress. In miR databases, hsa-miR-302e
is predicted to target >1,000 human genes, including OXR1. This
supports the results of the present study; however, it does not
elucidate the mechanism by which sevoflurane influences the
expression of this miR. Further investigations will be useful in
understanding completely the process by which sevoflurane induces
hsa-miR-302e overexpression. The findings of the present study
provide a novel opportunity for the treatment and management of
diseases associated with oxidative stress in neuronal cells. Deeper
investigation of the hsa-miR-302e/OXR1 pathway could give rise to a
novel generation of treatments for these diseases.
Acknowledgements
Not applicable.
Funding
The present study was supported by a grant from the
Clinical Scientific Research Project of Wuhan Municipal Health
Planning Commission (Wuhan, China; grant no. WX16C06).
Availability of data and materials
The analyzed data sets generated during the study
are available from the corresponding author on reasonable
request.
Authors' contributions
LY, QS, YX, XL and JP designed the study. LY and QS
conducted the experiments and analyzed the data. LY wrote the
manuscript. JP supervised the study. All authors read and approved
the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Freiermuth D, Mets B, Bolliger D,
Reuthebuch O, Doebele T, Scholz M, Gregor M, Haschke M, Seeberger
MD and Fassl J: Sevoflurane and isoflurane-pharmacokinetics,
hemodynamic stability, and cardioprotective effects during
cardiopulmonary bypass. J Cardiothorac Vasc Anesth. 30:1494–1501.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Likhvantsev VV, Landoni G, Levikov DI,
Grebenchikov OA, Skripkin YV and Cherpakov RA: Sevoflurane vs.
total intravenous anesthesia for isolated coronary artery bypass
surgery with cardiopulmonary bypass: A randomized trial. J
Cardiothorac Vasc Anesth. 30:1221–1227. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ružman T, Šimurina T, Gulam D, Ružman N
and Miškulin M: Sevoflurane preserves regional cerebral oxygen
saturation better than propofol: Randomized controlled trial. J
Clin Anesth. 36:110–117. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Walia H, Ruda J and Tobias JD: Sevoflurane
and bradycardia in infants with trisomy 21: A case report and
review of the literature. Int J Pediatr Otorhinolaryngol. 80:5–7.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Conreux F, Best O, Preckel MP, Lhopitault
C, Beydon L, Pouplard F and Granry JC: Effets
électroencéphalographiques du sévoflurane à l'induction chez le
jeune enfant: Étude prospective sur 20 cas. Ann Fr Anesth Réanim.
20:438–445. 2001.(In French). View Article : Google Scholar
|
|
6
|
Geng YJ, Wu QH and Zhang RQ: Effect of
propofol, sevoflurane, and isoflurane on postoperative cognitive
dysfunction following laparoscopic cholecystectomy in elderly
patients: A randomized controlled trial. J Clin Anesth. 38:165–171.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Xiao H, Liu B, Chen Y and Zhang J:
Learning, memory and synaptic plasticity in hippocampus in rats
exposed to sevoflurane. Int J Dev Neurosci. 48:38–49. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hu N, Wang M, Xie K, Wang H, Wang C, Wang
C, Wang C, Li Y, Yu Y and Wang G: Internalization of GluA2 and the
underlying mechanisms of cognitive decline in aged rats following
surgery and prolonged exposure to sevoflurane. Neurotoxicol.
49:94–103. 2015. View Article : Google Scholar
|
|
9
|
Shen X, Liu Y, Xu S, Zhao Q, Guo X, Shen R
and Wang F: Early life exposure to sevoflurane impairs adulthood
spatial memory in the rat. Neurotoxicol. 39:45–56. 2013. View Article : Google Scholar
|
|
10
|
Wu Y, Gu C and Huang X: Sevoflurane
protects against hepatic ischemia/reperfusion injury by modulating
microRNA-200c regulation in mice. Biomed Pharmacother.
84:1126–1136. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sun Y, Li Y, Liu L, Wang Y, Xia Y, Zhang L
and Ji X: Identification of miRNAs involved in the protective
effect of sevoflurane preconditioning against hypoxic injury in
PC12 cells. Cell Mol Neurobiol. 35:1117–1125. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yi W, Li D, Guo Y, Zhang Y, Huang B and Li
X: Sevoflurane inhibits the migration and invasion of glioma cells
by upregulating microRNA-637. Int J Mol Med. 38:1857–1863. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ye J, Zhang Z, Wang Y, Chen C, Xu X, Yu H
and Peng M: Altered hippocampal microRNA expression profiles in
neonatal rats caused by sevoflurane anesthesia: MicroRNA profiling
and bioinformatics target analysis. Exp Ther Med. 12:1299–1310.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fujimoto S, Ishikawa M, Nagano M and
Sakamoto A: Influence of neonatal sevoflurane exposure on nerve
development-related microRNAs and behavior of rats. Biomed Res.
36:347–355. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Friedman R, Farh K, Burge C and Bartel D:
Most mammalian mRNAs are conserved targets of microRNAs. Genome
Res. 19:92–105. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Winter J, Jung S, Keller S, Gregory R and
Diederichs S: Many roads to maturity: MicroRNA biogenesis pathways
and their regulation. Nat Cell Biol. 11:228–234. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang Q, Li G, Li B, Chen Q, Lv D, Liu J,
Ma J, Sun N, Yang L, Fei X and Song Q: Sevoflurane represses the
self-renewal ability by regulating miR-7a,7b/Klf4 signalling
pathway in mouse embryonic stem cells. Cell Prolif. 49:609–617.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhou C, Gu H, Fan R, Wang B and Lou J:
MicroRNA 302/367 cluster effectively facilitates direct
reprogramming from human fibroblasts into functional neurons. Stem
Cells Dev. 24:2746–2755. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang Z, Hong Y, Xiang D, Zhu P, Wu E, Li
W, Mosenson J and Wu WS: MicroRNA-302/367 cluster governs hESC
self-renewal by dually regulating cell cycle and apoptosis
pathways. Stem Cell Reports. 4:645–657. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li HL, Wei JF, Fan LY, Wang SH, Zhu L, Li
TP, Lin G, Sun Y, Sun ZJ, Ding J, et al: miR-302 regulates
pluripotency, teratoma formation and differentiation in stem cells
via an AKT1/OCT4-dependent manner. Cell Death Dis. 7:e20782016.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gao Z, Zhu X and Dou Y: The miR-302/367
cluster: A comprehensive update on its evolution and functions.
Open Biol. 5:1501382015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yang CM, Chiba T, Brill B, Delis N, von
Manstein V, Vafaizadeh V, Oellerich T and Groner B: Expression of
the miR-302/367 cluster in glioblastoma cells suppresses
tumorigenic gene expression patterns and abolishes transformation
related phenotypes. Int J Cancer. 137:2296–2309. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Maadi H, Moshtaghian A, Taha MF, Mowla SJ,
Kazeroonian A, Haass NK and Javeri A: Multimodal tumor suppression
by miR-302 cluster in melanoma and colon cancer. Int J Biochem Cell
Biol. 81:121–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Parchem RJ, Moore N, Fish JL, Parchem JG,
Braga TT, Shenoy A, Oldham MC, Rubenstein JL, Schneider RA and
Blelloch R: miR-302 is required for timing of neural
differentiation, neural tube closure, and embryonic viability. Cell
Rep. 12:760–773. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Suresh B, Kumar AM, Jeong HS, Cho YH,
Ramakrishna S and Kim KS: Regulation of Fanconi anemia protein
FANCD2 monoubiquitination by miR-302. Biochem Biophys Res Commun.
466:180–185. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yang SL, Yang M, Herrlinger S, Liang C,
Lai F and Chen JF: MiR-302/367 regulate neural progenitor
proliferation, differentiation timing, and survival in neurulation.
Dev Biol. 408:140–150. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li HH, Lin SL, Huang CN, Lu FJ, Chiu PY,
Huang WN, Lai TJ and Lin CL: miR-302 attenuates
amyloid-beta-induced neurotoxicity through activation of Akt
signaling. J Alzheimers Dis. 50:1083–1098. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Fang YC and Yeh CH: Inhibition of miR-302
suppresses hypoxia-reoxygenation-induced H9c2 cardiomyocyte death
by regulating Mcl-1 expression. Oxid Med Cell Longev.
2017:79689052017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ghasemi-Kasman M, Baharvand H and Javan M:
Enhanced neurogenesis in degenerated hippocampi following
pretreatment with miR-302/367 expressing lentiviral vector in mice.
Biomed Pharmacother. 96:1222–1229. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ghasemi-Kasman M, Shojaei A, Gol M,
Moghadamnia AA, Baharvand H and Javan M: miR-302/367-induced
neurons reduce behavioral impairment in an experimental model of
Alzheimer's disease. Mol Cell Neurosci. 86:50–57. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhou YF, Wang QX, Zhou HY and Chen G:
Autophagy activation prevents sevoflurane-induced neurotoxicity in
H4 human neuroglioma cells. Acta Pharmacol Sin. 37:580–588. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Altuntas TG, Zager RA and Kharasch ED:
Cytotoxicity of S-conjugates of the sevoflurane degradation product
fluoromethyl-2,2-difluoro-1-(trifluoromethyl) vinyl ether (Compound
A) in a human proximal tubular cell line. Toxicol Appl Pharmacol.
193:55–65. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Manoharan S, Guillemin GJ, Abiramasundari
RS, Essa MM, Akbar M and Akbar MD: The role of reactive oxygen
species in the pathogenesis of Alzheimer's disease, Parkinson's
disease, and Huntington's disease: A mini review. Oxid Med Cell
Longev. 2016:85905782016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Liu KX, Edwards B, Lee S, Finelli MJ,
Davies B, Davies KE and Oliver PL: Neuron-specific antioxidant OXR1
extends survival of a mouse model of amyotrophic lateral sclerosis.
Brain. 138:1167–1181. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Oliver PL, Finelli MJ, Edwards B, Bitoun
E, Butts DL, Becker EB, Cheeseman MT, Davies B and Davies KE: Oxr1
is essential for protection against oxidative stress-induced
neurodegeneration. PLoS Genet. 7:e10023382011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Finelli MJ, Sanchez-Pulido L, Liu KX,
Davies KE and Oliver PL: The evolutionarily conserved
Tre2/Bub2/Cdc16 (TBC), lysin motif (LysM), domain catalytic (TLDc)
domain is neuroprotective against oxidative stress. J Biol Chem.
291:2751–2763. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wu Y, Davies KE and Oliver PL: The
antioxidant protein Oxr1 influences aspects of mitochondrial
morphology. Free Radic Biol Med. 95:255–267. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Chen X, Xu Y, Liao X, Liao R, Zhang L, Niu
K, Li T, Li D, Chen Z, Duan Y and Sun J: Plasma miRNAs in
predicting radiosensitivity in non-small cell lung cancer. Tumour
Biol. 37:11927–11936. 2016. View Article : Google Scholar : PubMed/NCBI
|














