Introduction
Prostatitis is the most common type of urinary
disease in males <50 years of age (1) and has a prevalence ranging from 2–9%
in the general male population (2). It was reported that ≤50% of males may
suffer from chronic prostatitis at a certain stage of their life
(3). According to the National
Institutes of Health, prostatitis is divided into four categories:
Acute bacterial prostatitis, chronic bacterial prostatitis, chronic
prostatitis (CP)/chronic pelvic pain syndrome and asymptomatic
inflammatory prostatitis (4). As a
common type of prostatitis, chronic non-bacterial prostatitis
(CNBP; also termed CP) is difficult to diagnose; the long-term
therapeutic effects resulting from the routine use of antibiotics
and α-blockers have not been reported (5), which leads to a high recurrence rate
and a low curative rate for CNBP. Furthermore, CNBP may cause male
infertility and sexual dysfunction (6). In addition, continual and
recalcitrant inflammation in the prostate has been reported to be
associated with prostate cancer and benign prostatic hyperplasia
(BPH) (7,8). As CP has a high incidence rate and is
associated with numerous hazards, including male infertility and
sexual dysfunction, the disease presents a great challenge for
clinicians.
Nucleotide oligomerization domain (NOD)-like
receptors (NLRs) belong to the family of pattern recognition
receptors that recognize pathogen-associated molecular patterns
(PAMPs), as well as host-derived danger-associated molecular
patterns (DAMPs). NOD-like receptor family pyrin domain-containing
proteins (NLRPs) serve a critical role in the innate and adaptive
immune responses, and are involved in the development of chronic
inflammatory diseases, including cryopyrin-associated periodic
syndromes, gout, atherosclerosis and type 2 diabetes (9). As the most widely studied
inflammasome, the NLRP3 inflammasome is composed of NOD-like
receptor protein 3 (NLRP3), apoptosis-associated speck-like protein
containing a caspase recruitment domain (ASC) and caspase-1
(10). NLRP3 interacts with ASC to
activate caspase-1, and activated caspase-1 subsequently
upregulates the production of proinflammatory cytokines, including
interleukin (IL)-1β and IL-18, leading to tissue injury (11).
Autophagy is an evolutionarily conserved cellular
process involved in the isolation and degradation of cytosolic
macromolecules, damaged organelles and several pathogens, and
senescence (12,13). Autophagy is maintained at low
levels under physiological conditions; however, it may be induced
by nutrient deprivation, mitochondrial damage and pharmacological
inhibitors of mammalian target of rapamycin (mTOR), such as
rapamycin (14). It was previously
reported that the inhibition of autophagy promoted
antigen-presenting cells to process and secrete IL-1β in an NLRP3-
and Toll/IL-1 receptor-domain-containing adaptor-inducing
interferon-β-dependent manner (15). Downregulation of autophagy induced
by knockdown of autophagy-related 7 (Atg7) or Atg16L1 promoted the
notably enhanced secretion of IL-1β and IL-18 in response to
lipopolysaccharide and other PAMPs (16). These studies suggested that
autophagy may directly or indirectly inhibit IL-1β and IL-18
secretion, and that this process may be associated with the NLRP3
inflammasome. Our previous study demonstrated that the level of
autophagy was suppressed, and the expression levels of cytokine
IL-1β, a molecule located downstream of the NLRP3 inflammasome
pathway, was significantly increased in CNBP rats (17); however, the association between
autophagy and the NLRP3 inflammasome in CNBP is yet to be
investigated.
Therefore, the present study employed rapamycin, a
well-reported inhibitor of mTOR that serves a key role in
autophagy, was used to assess the effects of autophagy on NLRP3
inflammasome-mediated inflammation in the prostate via a rat model
in order to determine the mechanism underlying the pathogenesis of
CNBP.
Materials and methods
Establishment of the CNBP rat
model
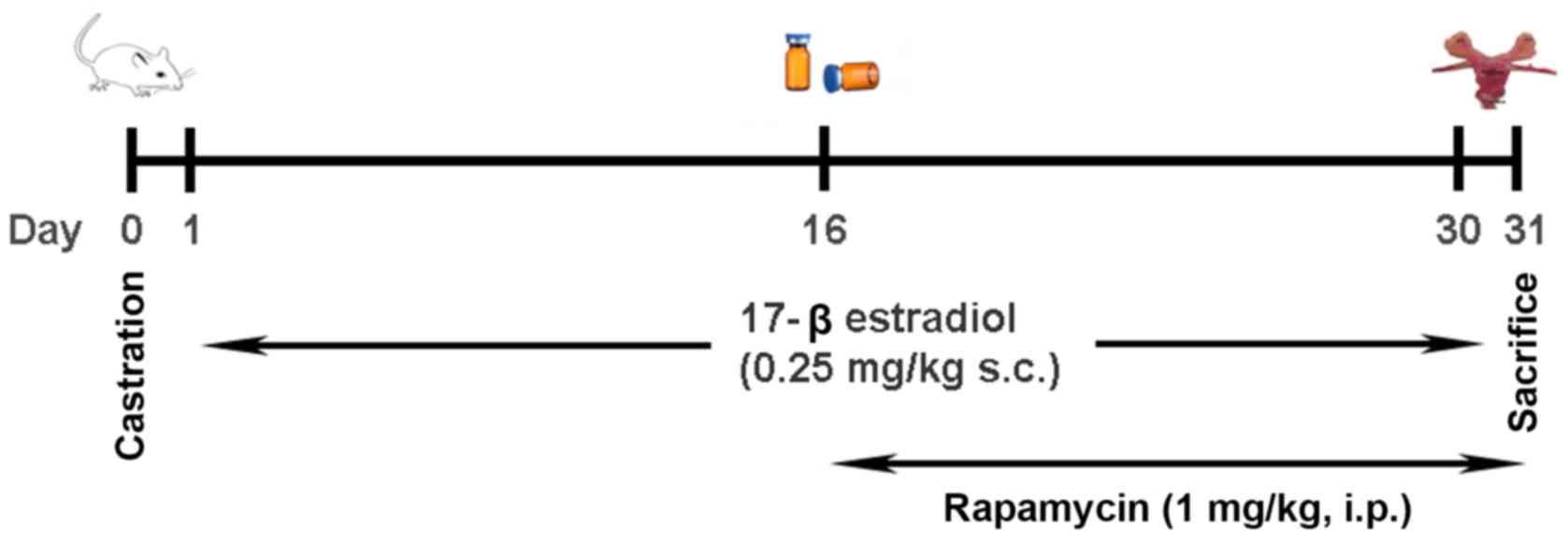
A total of 30 male Sprague-Dawley rats (6 weeks old)
weighing 250±20 g were purchased from the Center of Experimental
Animals of Wuhan University (Wuhan, China). All rats were housed in
a specific pathogen-free facility at a constant ambient temperature
(22±2°C) and humidity (50±10%) under a 12 h light/dark cycle, with
free access to sterile water and regular chow. The present study
was approved by the Ethics Committee for Animal Experiments of
Renmin Hospital of Wuhan University (approval no. WDRM-20170709).
All animal welfare and experimental procedures were performed in
accordance with the guidelines approved by the Care and Use of
Laboratory Animals of Renmin Hospital of Wuhan University. The rats
were randomly divided into three groups (n=10 per group) on the
basis of experimental requirements and anesthetized via inhalation
of 1–3% isoflurane prior to surgery: i) Control group, in which
sham surgery was performed on each of the rats; ii) CNBP group, in
which the rats were castrated and then subcutaneously injected with
17β-estradiol (0.25 mg/kg; Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany) for 30 consecutive days; and iii) rapamycin-treated group,
in which the rats also received a daily intraperitoneal injection
of rapamycin (1 mg/kg; Sigma-Aldrich; Merck KGaA) (18,19)
from the 16th day post-surgery for a total of 15 days, based on the
same model as the CNBP rats. All rats were sacrificed via an
overdose of isoflurane inhalation prior to further analysis. The
experimental outline of the present study is presented in Fig. 1.
Histological examination
The prostates were removed and fixed in 10% neutral
buffered formalin for 24 h at room temperature. All tissue samples
were dehydrated, embedded in paraffin and cut into 4-µm sections.
The sections were then stained with hematoxylin and eosin (H&E)
for 3–5 min at room temperature, and subsequently examined under a
light microscope (magnification, ×200; Olympus Corporation, Tokyo,
Japan), with 10 areas randomly selected from each section to
quantitatively assess the extent of CNBP.
The inflammation scoring criteria used in the
present study were as follows. Extent of inflammatory cell
infiltration: Low, only normal cells or few inflammatory cells (0
points); mild, a small degree of inflammatory cell infiltration (3
points); severe, a high degree of inflammatory cell infiltration (6
points). Gland lumen: Normal, a single columnar or cubic gland
epithelium (0 points); mild, a slightly smaller gland lumen
detected (2 points); severe, markedly smaller or occluded gland
lumen (4 points). Glandular secretion: Normal, a notable degree of
deep-pink staining (0 points); mild, slightly reduced pink staining
(2 points); and severe, decreased luminal secretion or no secretion
with weak or no pink staining (4 points). Fibrous tissue
hyperplasia: Normal, a notably small degree or no fibrous
hyperplasia (0 points); mild, a small degree of fibrous hyperplasia
(3 points); severe, a high degree of fibrous hyperplasia (6
points). The total score was calculated as the sum of the
aforementioned criteria.
Enzyme-linked immunosorbent assay
(ELISA) analysis
The blood samples were collected from the inferior
vena cava and centrifuged at 4°C, 2,000 × g for 10 min. The
separated serum was used for biochemical analysis. The serum levels
of IL-1β (cat. no. EK0393) and IL-18 (cat. no. EK0592) were
measured using a rat ELISA kit purchased from Wuhan Boster
Biological Technology, Ltd. (Wuhan, China). The absorbance at 450
nm was measured on a microplate reader (BioTek Elx9808; BioTek
Instruments, Inc., Winooski, VT, USA).
Immunohistochemistry (IHC)
analysis
Paraffin-embedded prostatic sections were
deparaffinized in xylene for 15 min, rehydrated with descending
alcohol solutions, and heated at 105°C for 10 min in citric acid
buffer (0.01 M, pH 6.0) for antigen retrieval. The slides were
subsequently rinsed with PBS for 10 min, and then transferred to 3%
hydrogen peroxide for 5 min at room temperature to block endogenous
peroxidase activity. After an additional wash with PBS for 5 min,
the sections were blocked with blocking buffer (OriGene
Technologies, Inc., Beijing, China) for 1 h at room temperature
prior to incubation with primary antibodies against NLRP3 (cat. no.
ab214185), ASC (cat. no. ab47092), microtubule-associated protein 1
light chain 3β (LC3B; cat. no. ab48394; all at 1:300 dilution;
Abcam, Cambridge, UK) and caspase-1 (cat. no. ab108362; 1:50;
Abcam) for 60 min at room temperature, and subsequently
biotin-conjugated goat-anti-rabbit immunoglobulin G (IgG) secondary
antibody (cat. no. TA130016; 1:200; OriGene Technologies, Inc.) was
added to each section and incubated at 37°C for 50 min. The SP9000
IHC immunochemistry kit (OriGene Technologies, Inc.) and
3,3′-diaminobenzidine (SK-4100; Vector Laboratories, Inc.,
Burlingame, CA, USA) were used according to the manufacturer's
protocols to detect the immune complexes in the prostate tissues.
Finally, the sections were viewed under a light microscope
(magnification, ×200; Olympus Corporation), and 10 areas were
randomly selected from each slice for further analysis.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Prostate tissues were subjected to RT-qPCR analysis.
Total RNA was extracted from the prostate tissues using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) according to the manufacturer's protocols.
Total RNA (1 µg) was reverse transcribed into cDNA using the
PrimeScript™ RT reagent kit (Takara Bio, Inc., Otsu, Japan)
according to the manufacturer's protocols. qPCR was performed using
the cDNA with the SYBR-Green mix (Takara Bio, Inc.) on an ABI 7500
Real-Time PCR system (Applied Biosystems; Thermo Fisher Scientific,
Inc.). All reactions were conducted in a volume of 20 µl, and the
relative expression of the different genes was normalized against
GAPDH. The sequences of the primers were as follows: Rat NLRP3,
forward 5′-CCAGGGCTCTGTTCATTG-3′, reverse, 5′-CCTTGGCTTTCACTTCG-3′;
rat ASC, forward 5′-AGACATGGGCATACAGGAGC-3′, reverse,
5′-GCAATGAGTGCTTGCCTGTG-3′; rat caspase-1, forward
5′-AAGAAGGTGGCGCATTTCCT-3′, reverse, 5′-GACGTGTACGAGTGGGTGTT-3′;
rat LC3B, forward 5′-GTCGCTAACAAGCAGTGGGA-3′, reverse,
5′-AGGGCTTCTGGGGCTCTAAT-3′; rat Beclin 1, forward
5′-AGCACGCCATGTATAGCAAAGA-3′, reverse, 5′-GGAAGAGGGAAAGGACAGCAT-3′;
rat p62, forward 5′-GCTGCTCTCTTCAGGCTTACAG-3′, reverse,
5′-CCTGCTTCACAGTAGACGAAAG-3′; and rat GAPDH, forward
5′-GGCACAGTCAAGGCTGAGAATG-3′ and reverse,
5′-ATGGTGGTGAAGACGCCAGTA-3′.
The thermocycling conditions were as follows: 95°C
for 30 sec, 40 cycles of denaturation at 95°C for 5 sec, and
extension at 60°C for 40 sec. All samples were repeated three times
and the melting curves of all products were analyzed. The
quantification cycle for each sample were calculated using the
2−ΔΔCq data analysis method (20).
Western blot analysis
Isolated rat prostates were homogenized in a lysis
buffer obtained from Shanghai Biyuntian Bio-Technology Co., Ltd.
(Shanghai, China) with a polytron homogenizer (IKA GmbH,
Königswinter, Germany) on ice. The lysates were subsequently
denatured with SDS loading buffer (5X) at 100°C for 10 min. Protein
samples (40 µg) were equally loaded, which were then separated by
12% SDS-PAGE and subsequently transferred onto polyvinylidene
difluoride membranes (EMD Millipore, Billerica, MA, USA). The
membranes were blocked with 5% non-fat milk dissolved in
Tris-buffered saline with Tween-20 (TBST) for 1 h at room
temperature, and incubated with primary antibodies against GAPDH
(cat. no. ab204481), NLRP3 (cat. no. ab214185), ASC (cat. no.
ab47092), caspase-1 (cat. no. ab108362), Beclin 1 (cat. no.
ab207612), LC3B (cat. no. ab48394), p62 (cat. no. ab91526; all at
1:2,000 dilution; Abcam), mTOR (cat. no. 2983; 1:1,000 dilution;
Cell Signaling Technology, Inc., Danvers, MA, USA) and
phosphorylated mTOR (p-mTOR; phosphorylated on Ser-2448) (cat. no.
2971; 1:1,000 dilution; Cell Signaling Technology, Inc.) overnight
at 4°C. Following three washes in TBST, the membranes were
subsequently incubated with a secondary antibody (cat. no.
C51007-08; 1:10,000 dilution; LI-COR Biosciences, Lincoln, NE, USA)
conjugated to horseradish peroxidase for 1 h at room temperature.
Finally, the membranes were scanned with a two-color infrared
imaging system (Odyssey® Infrared Imaging system; LI-COR
Biosciences). GAPDH (cat. no. ab204481; 1:2,000; Abcam) was used as
the endogenous reference protein. Quantity One 4.6.2 software
(Bio-Rad Laboratories, Inc., Hercules, CA, USA) was used for
densitometry analysis.
Transmission electron microscopy (TEM)
analysis
The isolated prostate tissues were cut into
1-mm3 pieces and fixed in 0.1 M phosphate buffer (pH
7.4) containing 2.5% glutaraldehyde for >3 h at 4°C. Following
three washes with phosphate buffer, the samples were stained with
1% osmium tetroxide for 1 h at 4°C, and then dehydrated with
alcohol and acetone. Subsequently, the tissues were embedded in
Epon 812 resin and polymerized with pure resin at 70°C for 24 h.
The ultrathin sections (~70 nm) were obtained using an
ultramicrotome (RMC Boeckeler Instruments, Inc., Tucson, AZ, USA),
and subsequently stained with 5% uranyl acetate and lead citrate
for 30 min at 37°C. The sections were examined and images were
captured using a transmission electron microscope (Tecnai G2 F30;
FEI Co., Thermo Fisher Scientific, Inc.); five randomly selected
fields from the stained tissue sections were examined
(magnification, ×5,000), and the number of autophagosomes in each
field was counted manually.
Statistical analysis
All experiments were independently repeated three
times. Data are expressed as the mean ± standard deviation.
Statistical analysis was performed with one-way analysis of
variance followed by a Tukey's test using SPSS software version
19.0 (IBM Corp., Armonk, NY, USA). P<0.05 was considered to
indicate a statistically significant difference.
Results
Castration combined with 17β-estradiol
injection causes CNBP in rats
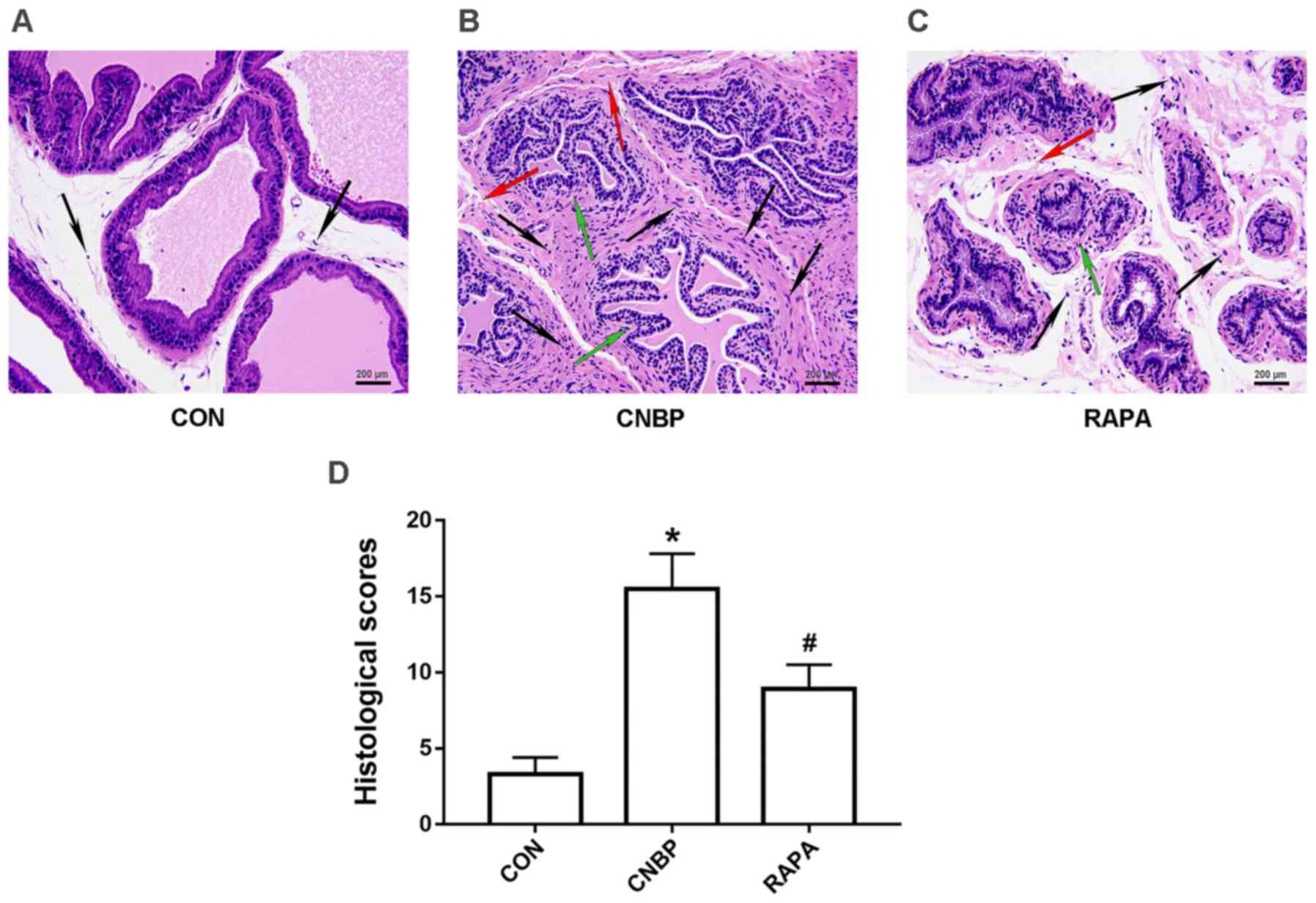
In order to assess prostatic alterations following
CNBP in the present study, H&E staining was performed to assess
the histopathological changes in the prostates. A normal appearance
of the glandular epithelium and stroma was noted in the prostate
tissues of rats in the control group, and the acinus was filled
with eosinophilic secretions; no clear indications of leukocyte
infiltration were observed around the acinus (Fig. 2A). However, glandular epithelial
degeneration, interstitial edema and extensive infiltration of
inflammatory cells were detected in the prostate tissues of rats in
the CNBP group; inflammatory cell infiltration was predominantly
confined to the interstitial area, but was also observed in the
glandular cavity (Fig. 2B). In
addition, when compared with the CNBP group, the number of
inflammatory cells infiltrating around the acinus was notably
reduced in the rapamycin-treated group; the severity of glandular
epithelial degeneration and interstitial edema was also reduced
(Fig. 2C). Subsequent quantitative
analysis supported these findings (P<0.05; Fig. 2D).
Rapamycin inhibits NLRP3 inflammasome
activation in the prostates of the CNBP rats
In order to assess the degree of NLRP3 inflammasome
activation in the prostate tissues of rats, IHC, RT-qPCR and
western blotting were performed to measure the expression levels of
NLRP3, ASC and caspase-1. The results of IHC revealed that
NLRP3-positive staining was rarely observed in the control group,
whereas notable NLRP3 expression was detected in the CNBP group;
however, the number of cells stained positive for NLRP3 was also
decreased in the rapamycin-treated group when compared with the
CNBP group. Similar trends in expression were noted for ASC and
caspase-1 (Fig. 3A). Compared with
the control group, the relative mRNA expression levels of NLRP3,
ASC and caspase-1 were significantly increased in the CNBP group;
however, expression was inhibited upon administration with
rapamycin (P<0.05; Fig. 3B-D).
In addition, the results of western blotting revealed that,
compared with the control group, the protein expression levels of
NLRP3, ASC and caspase-1 were significantly upregulated in the CNBP
group, whereas rapamycin treatment led to a significant inhibition
of NLRP3, ASC and caspase-1 protein expression compared with the
CNBP group (P<0.05; Fig.
3E-H).
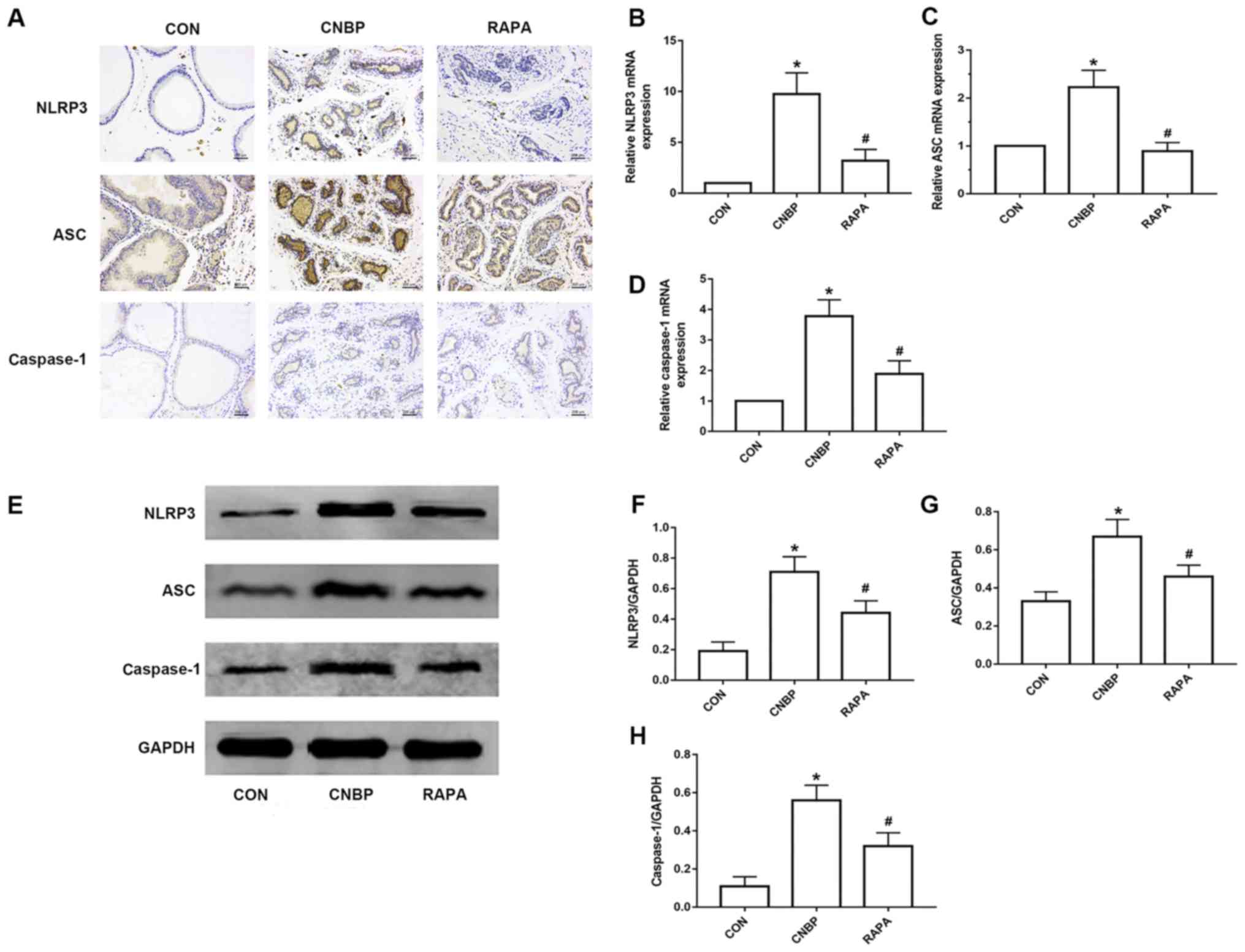 | Figure 3.Rapamycin treatment reduces the
upregulated expression of NLRP3, ASC and caspase-1 in the prostates
of rats. (A) Representative IHC images of NLRP3, ASC and caspase-1
expression in the prostates of rats from the three groups
(magnification, 200×; scale bar, 200 µm). Relative mRNA expression
levels of (B) NLRP3, (C) ASC, and (D) caspase-1 in the prostates of
rats from the different groups as detected by reverse
transcription-quantitative polymerase chain reaction analysis. (E)
Representative images of western blotting of NLRP3, ASC and
caspase-1 in the different groups. Quantitative analysis was used
to assess the levels of (F) NLRP3, (G) ASC and (H) caspase-1 in the
different groups. The values obtained were normalized against
GAPDH. Data are expressed as the mean ± standard deviation.
*P<0.05 vs. the CON group; #P<0.05 vs. the CNBP
group. CON, control; CNBP, chronic non-bacterial prostatitis; RAPA,
rapamycin; IHC, immunohistochemistry; NLPR3, Nod-like receptor
family pyrin domain-containing protein 3; ASC, apoptosis-associated
speck-like protein containing a caspase recruitment domain. |
Rapamycin reduces the expression
levels of IL-1β and IL-18 via suppressing NLRP3 inflammasome
activation
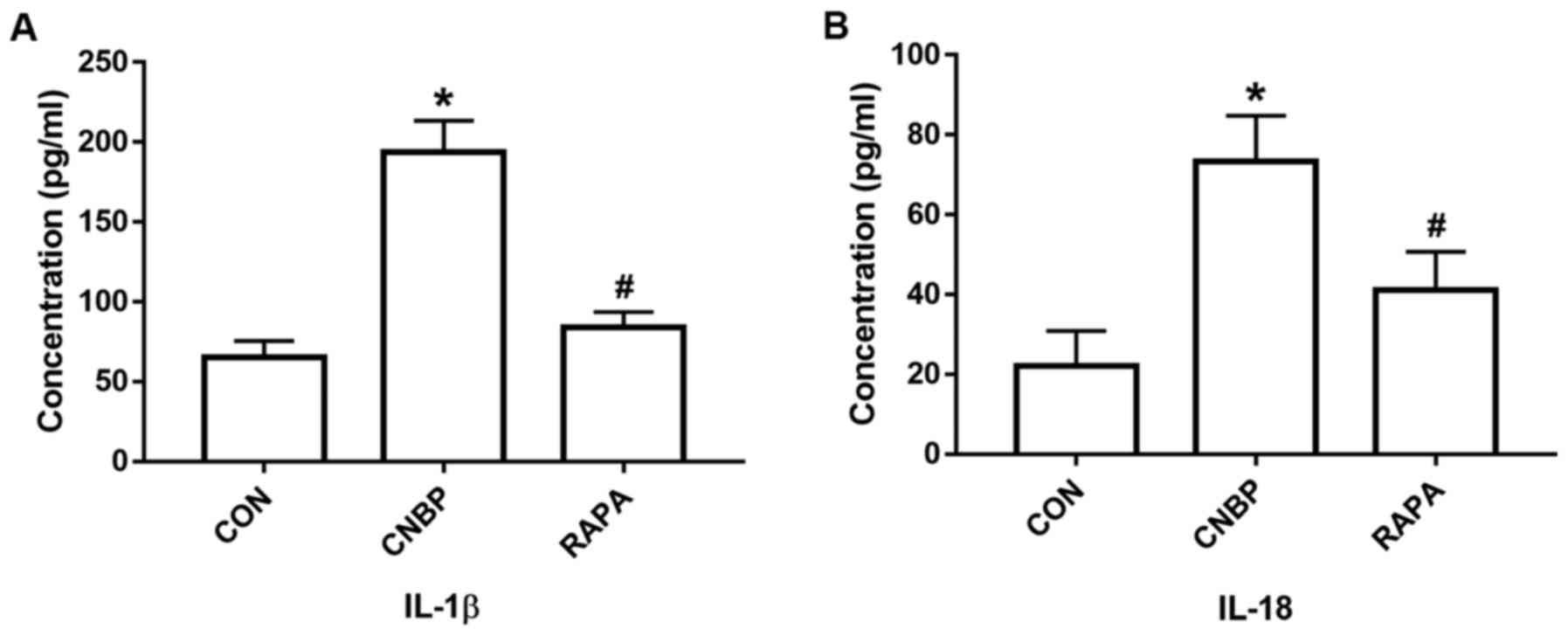
In the present study, following analysis of the
molecules located downstream of the NLRP3 inflammasome pathway, the
concentrations of IL-1β and IL-18 in the serum of the rats from the
different groups were detected using an ELISA kit. The combination
of castration and 17β-estradiol treatment significantly increased
the serum concentrations of IL-1β and IL-18 in the CNBP group
compared with the control group, whereas rapamycin treatment led to
a significant reduction in the levels of IL-1β and IL-18 compared
with the CNBP group (P<0.05; Fig.
4A and B).
Rapamycin induces autophagy by
inhibiting mTOR phosphorylation in CNBP rats
Considering the role of rapamycin in inducing
autophagy, western blotting, RT-qPCR, IHC and TEM analyses were
performed to analyze the level of autophagy in the prostate tissues
of rats from the different groups. As indicated by RT-qPCR
analysis, the relative mRNA expression levels of LC3B and Beclin 1
in the CNBP group were significantly decreased compared with the
control group, whereas rapamycin treatment upregulated the
expression of LC3B and Beclin 1 mRNA compared with the CNBP group
(P<0.05; Fig. 5A and B); the
expression levels of p62 were significantly upregulated in the CNBP
group compared with the control; however, this was reversed in
response to treatment with rapamycin (P<0.05; Fig. 5C). Western blot analysis
demonstrated that, compared with the control group, the protein
expression levels of LC3B-II and Beclin 1 were also significantly
decreased in the CNBP group, but were significantly increased in
the rapamycin-treated group compared with the CNBP group
(P<0.05; Fig. 5D-F).
Additionally, compared with the control group, the protein
expression levels of p-mTOR in the prostate of the CNBP rats were
significantly increased (P<0.05; Fig. 5D and G), whereas rapamycin
treatment significantly suppressed the upregulated levels of p-mTOR
(P<0.05). In addition, the protein expression levels of mTOR in
the prostate tissues of rats in the three groups did not indicate
any significant differences (Fig. 5H
and I); however, western blot analysis of p62, a marker of
autophagosomal degradation, revealed an expression profile contrary
to that of LC3B-II and Beclin 1 (Fig.
5H and J).
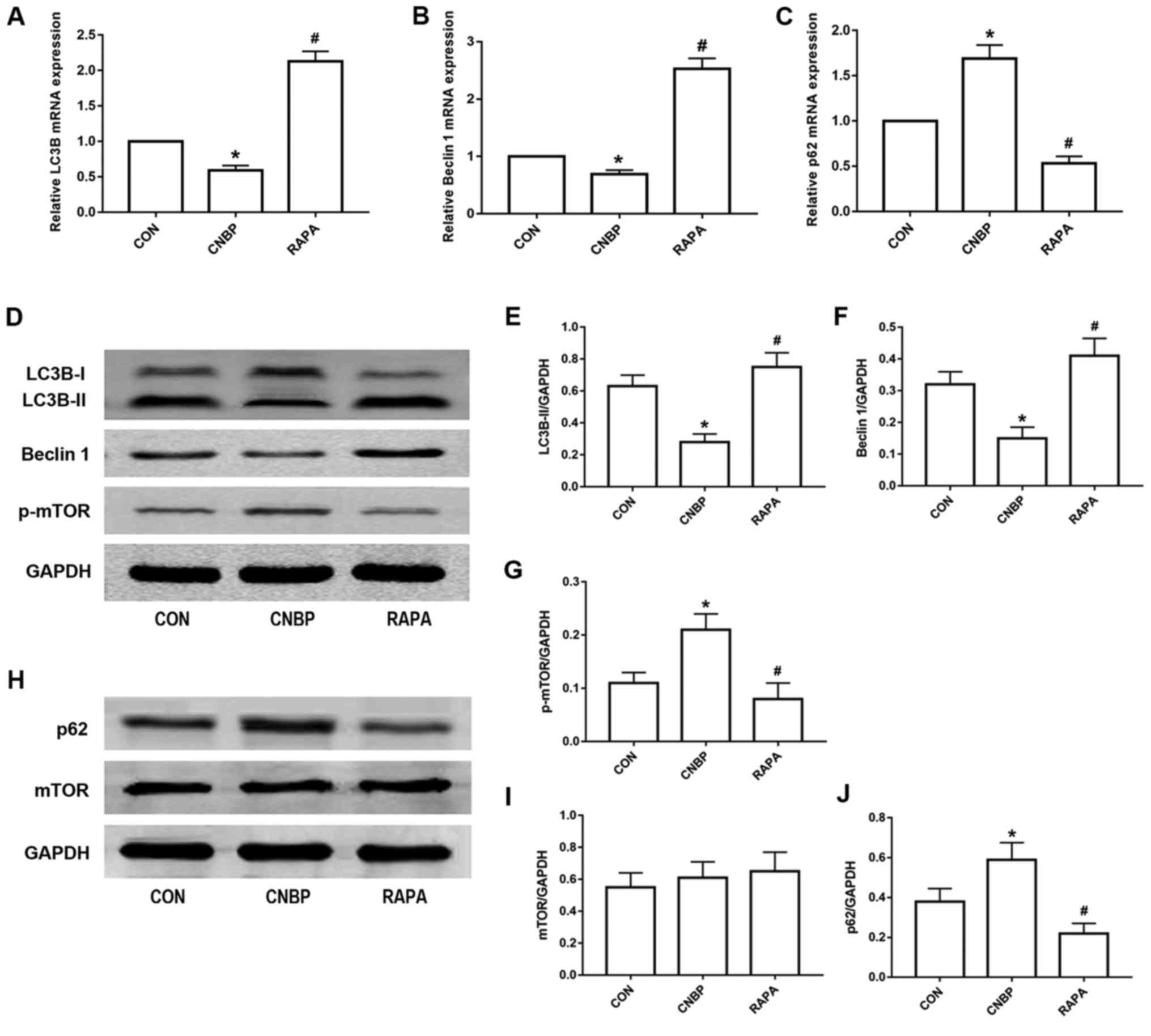 | Figure 5.Rapamycin treatment induces autophagy
in rats via the inhibition of mTOR phosphorylation. The relative
mRNA expression levels of (A) LC3B, (B) Beclin 1, and (C) p62, as
measured by reverse transcription-quantitative polymerase chain
reaction analysis, in the prostates of rats from the three groups.
(D) Representative western blot images of LC3B, Beclin 1 and p-mTOR
from the three groups. Quantitative analysis was used to assess the
levels of (E) LC3B-II, (F) Beclin 1, and (G) p-mTOR in the
different groups. (H) Representative western blot images of p62 and
mTOR from the three groups. Quantitative analysis was used to
assess the expression levels of (I) p62 and (J) mTOR in the
different groups. The values obtained were normalized against
GAPDH. Data are expressed as the mean ± standard deviation.
*P<0.05 vs. the CON group; #P<0.05 vs. the CNBP
group. CON, control; CNBP, chronic non-bacterial prostatitis; RAPA,
rapamycin; mTOR, mammalian target of rapamycin; p-mTOR,
phosphorylated mTOR; LC3B, microtubule-associated protein 1 light
chain 3β. |
Examination of the autophagosomes and LC3B-positive
staining of cells are the techniques considered to be the ‘gold
standard’ to identify autophagy (21). Therefore, TEM analysis was used to
observe cellular autophagy at the ultrastructural level in the
present study. Autophagic vacuoles with a double membrane were
observed in the control group (Fig.
6A), but fewer were detected in the CNBP group (Fig. 6B). Conversely, numerous
autophagosomes were observed in the rapamycin-treated group
(Fig. 6C). Quantitative analysis
of these results also supported these findings (P<0.05; Fig. 6D). Similarly, the results of IHC
revealed that, compared with the control group, the number of cells
positively stained for LC3B were notably decreased in the CNBP
group; however, a marked increase in the number of LC3B-positive
cells was reported in the rapamycin-treated group compared with the
CNBP group (Fig. 6E).
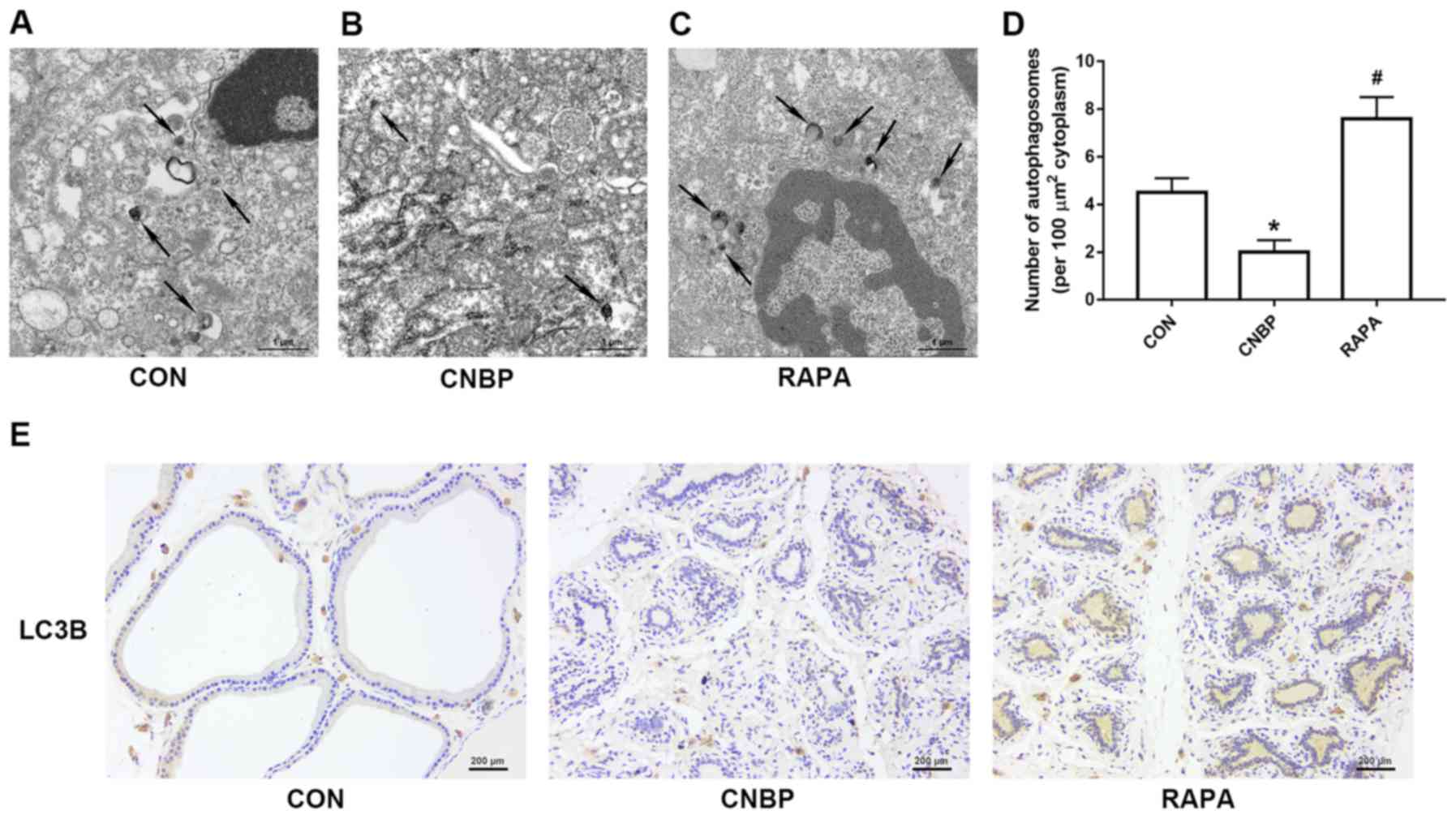 | Figure 6.Ultrastructural alterations due to
autophagy and cells positively stained for LC3B in rats of the
three different groups, as detected by TEM and IHC analyses.
Representative TEM images of autophagosomes in the (A) CON, (B)
CNBP and (C) RAPA groups (magnification, ×5,000; scale bar, 1 µm).
The black arrows indicate autophagosomes with a double membrane in
the prostates of rats. (D) Quantitative analysis of autophagosomes
in the different groups. (E) Representative IHC images of LC3B in
the three groups (magnification, ×200; scale bar, 200 µm). Data are
expressed as the mean ± standard deviation. *P<0.05 vs. the CON
group; #P<0.05 vs. the CNBP group. CON, control;
CNBP, chronic non-bacterial prostatitis; RAPA, rapamycin; TEM,
transmission electron microscopy; IHC, immunohistochemistry; LC3B,
microtubule-associated protein 1 light chain 3β. |
Discussion
CNBP is a very common disease encountered in
urological clinics, and is associated with male infertility, sexual
dysfunction, BPH and prostate cancer (6–8);
however, the etiology of CNBP is yet to be elucidated. To
investigate the underlying mechanisms of CNBP, numerous
experimental animal models that mimic human category III
prostatitis (CP) have been developed by immune manipulation
(22), infection (23), or hormonal treatment (24,25).
Therefore, one possible mechanism that may, at least in part, be
responsible for the pathogenesis of CNBP is sex hormone imbalance.
It has been reported that the increased prevalence of CNBP is
associated with a decrease in the testosterone (T) to estrogen (E2)
ratio in the serum (26), which
was due to the decreased anti-inflammatory effect of T, as well as
an increased proinflammatory function of E2 (26). Therefore, hormone imbalance may
induce prostatic inflammation when the concentration of androgen
decreases and/or the E2 concentration remains unchanged or
increases. Administration of exogenous 17β-estradiol to old (10–13
months) male Wistar rats led to a 3.7-fold increase in the
incidence of CNBP and a 2- to 6-fold increase in the severity of
prostate inflammation (24).
Castration was also reported to have a similar effect (24); combined with estradiol injection,
castration was also used to establish a model of CP in Wistar rats
(27). Additionally, Keith et
al (28) reported that Wistar
rats tend to develop spontaneous prostatitis, which may have a
serious impact on the accuracy of the experimental results
obtained. To the best of our knowledge, spontaneous prostatitis has
not been reported in Sprague-Dawley rats, and providing that this
is the case, Sprague-Dawley rats may be used to build a CNBP model
less prone to these types of experimental error. Thus,
Sprague-Dawley rats, rather than Wistar rats, were selected to
establish the CNBP model in the present study. Although castration
alone is able to induce prostatic inflammation (24), it has been reported that the
combination of castration and estrogen injection induced markedly
severe prostatitis and pathological changes (25). Furthermore, it has been confirmed
that the ratio of T to E2 is negatively correlated with the
majority of inflammatory markers (29), which indicates that combining
castration with E2 injection may provide a more stable approach to
establishing the CNBP model. Therefore, in the present study, the
mechanism underlying the pathogenesis of CNBP was investigated
using a rat model induced by castration and 17β-estradiol
injection. In the present study, prostatic interstitial edema and
infiltration of numerous inflammatory cells around the acinar
cavity were observed in the prostate tissues of CNBP rats upon
H&E staining. Furthermore, varying degrees of tissue damage in
the prostate lumens were also observed in the CNBP rats. These
results indicated that the combination of castration and
17β-estradiol injection may have induced inflammation in the
prostates of rats.
It is well-known that the NLRP3 inflammasome and its
downstream molecules, IL-1β and IL-18, are closely associated with
the pathogenesis of chronic inflammatory diseases, including
cryopyrin-associated periodic syndrome (CAPS), inflammatory bowel
disease (IBD) and type 2 diabetes (30). Jiang et al (31) have demonstrated that the NLRP3
inflammasome is activated in CAPS, and inhibition of the NLRP3
inflammasome exerted significant therapeutic effects against this
condition. The NLRP3 inflammasome, which is composed of NLRP3, ASC
and caspase-1, is usually activated in monocytes and macrophages;
it was originally described as a cytosolic multiprotein structure
of the innate immune system (9).
The stimulation of DAMPs or PAMPs, including lipopolysaccharide and
other molecules derived from cellular damage processes, promotes
the activation of inflammasomes, thereby leading to the activation
of caspase-1; activated caspase-1 ultimately transforms the
endogenous proinflammatory cytokines, IL-1β and IL-18, into their
mature secreted forms (10).
Collectively, activation of the NLRP3 inflammasome may serve an
important role in the maturation and release of inflammatory
cytokines, particularly IL-1β and IL-18. It is well established
that IL-1β and IL-18 are two important cytokines involved in the
inflammatory response, which fulfill critical roles in various
diseases associated with infection, injury, and antigenic
stimulation (32). Vincent and
Mohr (33) reported that IL-1β
increased tissue injury via nuclear factor-κB activation and
leukocyte adhesion. Park et al (34) reported that elevated levels of
IL-18 were associated with severe lupus nephritis in patients with
systemic lupus erythematous. Furthermore, activated NLRP3
inflammasome was detected in patients with IBD and myelodysplastic
syndromes (35,36). Therefore, activation of the NLRP3
inflammasome is a critical step in various inflammatory responses.
In the present study, via IHC, RT-qPCR and western blot analyses,
it was demonstrated that the expression levels of NLRP3, ASC and
caspase-1 in the prostate tissues of CNBP rats were significantly
increased compared with in the control group. Furthermore, the
serum levels of IL-1β and IL-18 detected by ELISA were also
increased in the CNBP group. These results indicated that the NLRP3
inflammasome was activated in the prostate tissues of the CNBP
rats, and the activated NLRP3 inflammasome, together with the
increased expression levels of IL-1β and IL-18, may contribute to
the inflammatory cell infiltration and histological changes
observed in the prostates of CNBP rats.
Autophagy is an important cellular homeostatic
process by which cells degrade damaged or senescent components.
This process involves a variety of autophagy-associated proteins,
of which Beclin 1, LC3 and p62 serve crucial roles (37–39).
Beclin 1 is involved in the formation of autophagosomes; p62 serves
an important role in the process of autophagosomal degradation
(37). LC3 fulfills critical roles
in the maturation of autophagosomes and the formation of
autolysosomes (38). LC3 exists in
the form of LC3-I under normal conditions, but may be converted
into LC3-II by covalent association with the lipid
phosphatidylethanolamine on autophagosomal membranes during
maturation (38,39). At present, LC3-II is the only
well-characterized protein that is specifically localized to
autophagic structures throughout the process of phagophore
formation to lysosomal degradation (40). It should be noted that LC3 is
expressed as three isoforms in mammalian cells, LC3A, LC3B and
LC3C; however, only LC3B-II correlates with an increased number of
autophagic vesicles (41). As
LC3-II tends to have a higher affinity to its antibody than LC3-I,
comparisons of LC3-I expression, or the LC3-I/LC3-II ratio may lead
to numerous false-positive or false-negative results (42). Therefore, LC3B, Beclin 1 and p62
have been considered to be specific markers that may be used to
monitor cellular autophagy. The process of autophagy eliminates
senescent organelles and abnormal cytosolic macromolecules under
physiological conditions; however, autophagy may contribute to the
pathogenesis of various diseases (43). For example, autophagy impairment
has been reported to induce the abnormal accumulation of mutant
proteins in Alzheimer's disease (44). Ehrnhoefer et al (45) demonstrated that the dysfunction of
autophagy promoted an accumulation of mutant huntingtin in
Huntington's disease. Recently, increasing evidence has indicated
that autophagy may be regulated by E2; for example, it was revealed
that E2 functions as a negative regulator of autophagy in the
uterus of ovariectomized mice, and the inhibition of autophagy by
E2 was mediated via mTOR signaling (46). Wang et al (47) demonstrated that E2 protected
cardiomyocytes against damage induced by lipopolysaccharide via
autophagy inhibition. Fu et al (48) demonstrated that E2 suppressed
autophagy in osteocyte-like MLO-Y4 cells and that this effect was
reversed by rapamycin. Therefore, based on the aforementioned
studies, the present study proposed that E2 may promote the
progression of CNBP by inhibiting autophagy. The results of the
present study indicated that the level of autophagy, as reflected
by the levels of LC3B (a more common isoform of LC3), Beclin 1 and
autophagosome abundance, were significantly suppressed in the
prostate tissues of rats in the CNBP group compared with the
control group. Conversely, as a specific marker of autophagosomal
degradation (21), p62 exhibited
an expression profile contrary to that of LC3B and Beclin 1.
In addition, accumulating evidence has demonstrated
an association between the NLRP3 inflammasome and autophagy.
Autophagy has been reported to exert numerous effects on regulating
inflammasome activation, including eliminating endogenous signals
that activate the inflammasome, and degrading inflammasome
components. In particular, it has been reported that the
suppression of autophagy promoted NLRP3 inflammasome activation,
and the subsequent release of IL-1β and IL-18 (49). The impairment of autophagy in
macrophages was demonstrated to promote NLRP3 inflammasome
activation and lead to the hypersecretion of IL-1β (15). X-11-5-27, a daidzein derivative,
attenuated activation of the NLRP3 inflammasome by promoting
autophagy in acute monocytic leukemia (50). Therefore, on the basis of these
findings, the present study proposed that the suppression of
autophagy may contribute to the progression of CNBP via activation
of the NLRP3 inflammasome. In the present study, it was reported
that the expression levels of IL-1β and IL-18 in rats of the CNBP
group were significantly increased compared with in the control
group. Similarly, the expression levels of the NLRP3 inflammasome
components, NLRP3, ASC and caspase-1, were also significantly
increased in the CNBP group compared with the control group.
Collectively, these findings indicated that E2 promoted the
progression of chronic inflammation in the prostate tissues by
inhibiting autophagy. This process may be associated with
activation of the NLRP3 inflammasome and its downstream molecules,
including IL-1β and IL-18.
To further investigate the role of autophagy in
CNBP, rapamycin, a well-known specific inducer of autophagy that
negatively regulates mTOR (51),
was used to induce autophagy in rats with CNBP. Firstly, via
western blotting, IHC and RT-qPCR analyses, it was demonstrated
that, compared with the CNBP group, rapamycin treatment
significantly increased the levels of autophagy-associated markers,
including LC3B and Beclin 1 in the prostate tissues of rats;
rapamycin induced autophagy in the prostates of rats by inhibiting
the phosphorylation of mTOR. TEM analysis also demonstrated that
rapamycin significantly promoted the formation of autophagosomes
compared with the CNBP group. In addition, the present study
revealed that rapamycin treatment notably attenuated inflammatory
cell infiltration, glandular epithelial degeneration and
interstitial edema in the prostates of the CNBP rats, and rapamycin
treatment was also reported to significantly suppress the
upregulated expression of NLRP3 inflammasome-associated components,
and reduced the levels of IL-1β and IL-18 in rats of the CNBP group
as detected by IHC, RT-qPCR, western blotting and ELISA analyses.
Collectively, the findings of the present findings indicated that
the combination of castration and 17β-estradiol injection induced a
hormone imbalance-induced, NLRP3 inflammasome-mediated chronic
inflammatory response in the prostates of rats via the inhibition
of autophagy, a phenomenon that could be reversed by rapamycin
treatment. However, there was a limitation of the present study.
The size of prostates from the CNBP rats were notably smaller and
the parenchyma of these prostates were decreased when compared with
the control group.
In conclusion, the present study reported that the
imbalance in sex hormones induced by castration and 17β-estradiol
injection promoted inflammation in the prostates of rats.
Furthermore, it was demonstrated that rapamycin alleviated the
chronic inflammatory response in the prostate tissues of rats via
the induction of autophagy and inhibiting the activation of the
NLRP3 inflammasome, as suggested by alterations in the expression
levels of IL-1β and IL-18. The findings of present study may not
only improve understanding of the mechanism underlying the
pathogenesis of sex hormone imbalance-induced CNBP, but also
provides a potential therapeutic target for clinicians; however,
further investigation is required to determine the molecular
pathways by which autophagy affects the progression of CNBP.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81470923, 81470986,
81770078 and 81770688).
Availability of data and materials
The data and materials used or analyzed during this
study are available from the corresponding author on reasonable
request.
Authors' contributions
XC and JZ contributed to the design of the study,
and critically revised the manuscript for important intellectual
content. JL and YS designed the study, performed the experiments
and collected the experimental data. JL drafted this paper. YC, PL
and FL helped to perform experiments and analyze the experimental
data.
Ethics approval and consent to
participate
The present study was approved by the Ethical
Committee on Animal Experiments at Renmin Hospital of Wuhan
University (approval no. WDRM-20170709).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ellem SJ, Wang H, Poutanen M and
Risbridger GP: Increased endogenous estrogen synthesis leads to the
sequential induction of prostatic inflammation (prostatitis) and
prostatic pre-malignancy. Am J Pathol. 175:1187–1199. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Krieger JN, Lee SW, Jeon J, Cheah PY,
Liong ML and Riley DE: Epidemiology of prostatitis. Int J
Antimicrob Agents. 31 Suppl 1:S85–S90. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Vykhovanets EV, Resnick MI, MacLennan GT
and Gupta S: Experimental rodent models of prostatitis: Limitations
and potential. Prostate Cancer Prostatic Dis. 10:15–29. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Krieger JN, Nyberg L Jr and Nickel JC: NIH
consensus definition and classification of prostatitis. JAMA.
282:236–237. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
McNaughton Collins M, MacDonald R and Wilt
TJ: Diagnosis and treatment of chronic abacterial prostatitis: A
systematic review. Ann Intern Med. 133:367–381. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hao ZY, Li HJ, Wang ZP, Xing JP, Hu WL,
Zhang TF, Zhang XS, Zhou J, Tai S and Liang CZ: The prevalence of
erectile dysfunction and its relation to chronic prostatitis in
Chinese men. J Androl. 32:496–501. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Delongchamps NB, de la Roza G, Chandan V,
Jones R, Sunheimer R, Threatte G, Jumbelic M and Haas GP:
Evaluation of prostatitis in autopsied prostates-is chronic
inflammation more associated with benign prostatic hyperplasia or
cancer? J Urol. 179:1736–1740. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sfanos KS and De Marzo AM: Prostate cancer
and inflammation: The evidence. Histopathology. 60:199–215. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Schroder K and Tschopp J: The
inflammasomes. Cell. 140:821–832. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jo EK, Kim JK, Shin DM and Sasakawa C:
Molecular mechanisms regulating NLRP3 inflammasome activation. Cell
Mol Immunol. 13:148–159. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Strowig T, Henao-Mejia J, Elinav E and
Flavell R: Inflammasomes in health and disease. Nature.
481:278–286. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Deretic V: Autophagy in infection. Curr
Opin Cell Biol. 22:252–262. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
He C and Klionsky DJ: Regulation
mechanisms and signaling pathways of autophagy. Annu Rev Genet.
43:67–93. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kroemer G, Mariño G and Levine B:
Autophagy and the integrated stress response. Mol Cell. 40:280–293.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Harris J, Hartman M, Roche C, Zeng SG,
O'Shea A, Sharp FA, Lambe EM, Creagh EM, Golenbock DT, Tschopp J,
et al: Autophagy controls IL-1beta secretion by targeting
pro-IL-1beta for degradation. J Biol Chem. 286:9587–9597. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Saitoh T, Fujita N, Jang MH, Uematsu S,
Yang BG, Satoh T, Omori H, Noda T, Yamamoto N, Komatsu M, et al:
Loss of the autophagy protein Atg16L1 enhances endotoxin-induced
IL-1beta production. Nature. 456:264–268. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Su Y, Lu J, Chen X, Liang C, Luo P, Qin C
and Zhang J: Rapamycin alleviates hormone imbalance-induced chronic
nonbacterial inflammation in rat prostate through activating
autophagy via the mTOR/ULK1/ATG13 signaling pathway. Inflammation.
41:1384–1395. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu RF, Fu G, Li J, Yang YF, Wang XG, Bai
PD and Chen YD: Roles of autophagy in androgen-induced benign
prostatic hyperplasia in castrated rats. Exp Ther Med.
15:2703–2710. 2018.PubMed/NCBI
|
|
19
|
Meng Y, Pan M, Zheng B, Chen Y, Li W, Yang
Q, Zheng Z, Sun N, Zhang Y and Li X: Autophagy attenuates
angiotensin ii-induced pulmonary fibrosis by inhibiting redox
imbalance-mediated NOD-like receptor family pyrin domain containing
3 inflammasome activation. Antioxid Redox Signal. May 7–2018;(Epub
ahead of print).
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Klionsky DJ, Abeliovich H, Agostinis P,
Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA,
Ballabio A, et al: Guidelines for the use and interpretation of
assays for monitoring autophagy in higher eukaryotes. Autophagy.
4:151–175. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu KJ, Chatta GS, Twardzik DR, Vedvick
TS, True LD, Spies AG and Cheever MA: Identification of rat
prostatic steroid-binding protein as a target antigen of
experimental autoimmune prostatitis: Implications for prostate
cancer therapy. J Immunol. 159:472–480. 1997.PubMed/NCBI
|
|
23
|
Kaplan L, Lee C and Schaeffer AJ: Effect
of castration on experimental bacterial prostatitis in rats.
Prostate. 4:625–630. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Naslund MJ, Strandberg JD and Coffey DS:
The role of androgens and estrogens in the pathogenesis of
experimental nonbacterial prostatitis. J Urol. 140:1049–1053. 1988.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Robinette CL: Sex-hormone-induced
inflammation and fibromuscular proliferation in the rat lateral
prostate. Prostate. 12:271–286. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bernoulli J, Yatkin E, Konkol Y, Talvitie
EM, Santti R and Streng T: Prostatic inflammation and obstructive
voiding in the adult Noble rat: Impact of the testosterone to
estradiol ratio in serum. Prostate. 68:1296–1306. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kamijo T, Sato S and Kitamura T: Effect of
cernitin pollen-extract on experimental nonbacterial prostatitis in
rats. Prostate. 49:122–131. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Keith IM, Jin J, Neal D Jr, Teunissen BD
and Moon TD: Cell relationship in a Wistar rat model of spontaneous
prostatitis. J Urol. 166:323–328. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jia YL, Liu X, Yan JY, Chong LM, Li L, Ma
AC, Zhou L and Sun ZY: The alteration of inflammatory markers and
apoptosis on chronic prostatitis induced by estrogen and androgen.
Int Urol Nephrol. 47:39–46. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ozaki E, Campbell M and Doyle SL:
Targeting the NLRP3 inflammasome in chronic inflammatory diseases:
Current perspectives. J Inflamm Res. 8:15–27. 2015.PubMed/NCBI
|
|
31
|
Jiang H, He H, Chen Y, Huang W, Cheng J,
Ye J, Wang A, Tao J, Wang C, Liu Q, et al: Identification of a
selective and direct NLRP3 inhibitor to treat inflammatory
disorders. J Exp Med. 214:3219–3238. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Broz P and Monack DM: Molecular mechanisms
of inflammasome activation during microbial infections. Immunol
Rev. 243:174–190. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Vincent JA and Mohr S: Inhibition of
caspase-1/interleukin-1beta signaling prevents degeneration of
retinal capillaries in diabetes and galactosemia. Diabetes.
56:224–230. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Park MC, Park YB and Lee SK: Elevated
interleukin-18 levels correlated with disease activity in systemic
lupus erythematosus. Clin Rheumatol. 23:225–229. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kanneganti TD: Inflammatory bowel disease
and the NLRP3 inflammasome. N Engl J Med. 377:694–696. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Basiorka AA, McGraw KL,
Abbas-Aghababazadeh F, McLemore AF, Vincelette ND, Ward GA,
Eksioglu EA, Sallman DA, Ali NA, Padron E, et al: Assessment of ASC
specks as a putative biomarker of pyroptosis in myelodysplastic
syndromes: An observational cohort study. Lancet Haematol.
5:e393–e402. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wirawan E, Lippens S, Vanden Berghe T,
Romagnoli A, Fimia GM, Piacentini M and Vandenabeele P: Beclin1: A
role in membrane dynamics and beyond. Autophagy. 8:6–17. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Levine B and Kroemer G: Autophagy in the
pathogenesis of disease. Cell. 132:27–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Autophagy fights disease through cellular
self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Nakatogawa H, Suzuki K, Kamada Y and
Ohsumi Y: Dynamics and diversity in autophagy mechanisms: Lessons
from yeast. Nat Rev Mol Cell Biol. 10:458–467. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Mizushima N and Yoshimori T: How to
interpret LC3 immunoblotting. Autophagy. 3:542–545. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Barth S, Glick D and Macleod KF:
Autophagy: Assays and artifacts. J Pathol. 221:117–124. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kroemer G: Autophagy: A druggable process
that is deregulated in aging and human disease. J Clin Invest.
125:1–4. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Nilsson P and Saido TC: Dual roles for
autophagy: Degradation and secretion of Alzheimer's disease Aβ
peptide. Bioessays. 36:570–578. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ehrnhoefer DE, Martin DDO, Schmidt ME, Qiu
X, Ladha S, Caron NS, Skotte NH, Nguyen YTN, Vaid K, Southwell AL,
et al: Preventing mutant huntingtin proteolysis and intermittent
fasting promote autophagy in models of Huntington disease. Acta
Neuropathol Commun. 6:162018. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Choi S, Shin H, Song H and Lim HJ:
Suppression of autophagic activation in the mouse uterus by
estrogen and progesterone. J Endocrinol. 221:39–50. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wang F, Xiao J, Shen Y, Yao F and Chen Y:
Estrogen protects cardiomyocytes against lipopolysaccharide by
inhibiting autophagy. Mol Med Rep. 10:1509–1512. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Fu J, Hao L, Tian Y, Liu Y, Gu Y and Wu J:
miR-199a-3p is involved in estrogen-mediated autophagy through the
IGF-1/mTOR pathway in osteocyte-like MLO-Y4 cells. J Cell Physiol.
233:2292–2303. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Harris J, Lang T, Thomas JPW, Sukkar MB,
Nabar NR and Kehrl JH: Autophagy and inflammasomes. Mol Immunol.
86:10–15. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zhou W, Liu X, Cheng K, Zhang X, Lu J and
Hu R: X-11-5-27, a daidzein derivative, inhibits NLRP3 inflammasome
activity via promoting autophagy. Exp Cell Res. 360:320–327. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Song Y, Xue H, Liu TT, Liu JM and Chen D:
Rapamycin plays a neuroprotective effect after spinal cord injury
via anti-inflammatory effects. J Biochem Mol Toxicol. 29:29–34.
2015. View Article : Google Scholar : PubMed/NCBI
|