Introduction
At present, cardiovascular diseases (CVDs) remain
the predominant causes of morbidity and mortality in a number of
countries. Notably, atherosclerosis (AS) is a leading cause of CVD
(1). According to a previous
study, the morbidity of coronary heart diseases caused by AS has
increased in the past decade and almost 400 out of every 100,000
people succumb to the disease in Asia per year (2). Furthermore, coronary heart disease
caused by AS is considered the primary cause of non-infectious
disease-associated mortality worldwide (2). Endothelial dysfunction is a typical
early manifestation of atherogenesis, as well as the basic
pathogeny of multiple CVDs, including hypertension, coronary
disease, angina pectoris and cardiac failure (3–5).
Notably, lipid metabolism disorders are among the most important
causes of endothelial cell function impairment and result in a
series of oxidative stress reactions (6).
Nitric oxide (NO) is the vital vasoactive mediator
for protecting vascular endothelial cells and its production is
catalyzed by endothelial nitric oxide synthase (eNOS). Lipid
metabolism disorders can immediately promote the uncoupling of eNOS
and catabolism of NO, thereby generating superoxide anions,
increasing oxidative stress and reducing NO bioavailability
(7,8). Under conditions of oxidative stress,
low-density lipoprotein (LDL) is oxidized to form ox-LDL, which
penetrates and is deposited under the intima, leading to increased
endothelial permeability and impaired endothelial cell function;
endothelial cells ingest increased amounts of lipids and develop
into foam cells which serve a role in proinflammatory and immune
stimulatory effects (9), thereby
promoting the occurrence and development of AS (10). Additionally, ox-LDL can act as a
carrier of oxygen free radicals and continue to induce reactive
oxygen species generation, aggravating atherosclerotic lesions
(11).
Protein kinase B (Akt), which is a type of
serine/threonine kinase involved in the phosphoinositide-3-kinase
(PI3K)-Akt) signaling pathway (12), is considered a key mediator of cell
proliferation, migration, apoptosis, angiogenesis and metabolism
(13). Importantly, Akt can
directly phosphorylate eNOS at a serine phosphorylation site,
resulting in the enhancement of eNOS enzymatic activity and altered
sensitivity of the enzyme to Ca2+ (14). Under the stimulation of sustained
lipid metabolism, oxidative stress or other factors lead to the
inactivation of PI3K and inhibit Akt phosphorylation, thereby
affecting the synthesis of eNOS and aggravating endothelial cell
dysfunction (15).
Rosuvastatin, an inhibitor of
3-hydroxy-3-methylglutaryl-coenzyme A reductase, is the current
paradigm for lipid management that is used for ameliorating
abnormal lipid levels to improve lipid metabolism (16). A study by Qian et al
(17) demonstrated that
rosuvastatin was more effective in lowering LDL-C compared with
atorvastatin and that it decreased plaque volume and vascular
volume in vulnerable coronary artery plaques of AS and stabilized
angina pectoris, which are closely associated with endothelial
dysfunction (18). To the best of
our knowledge, the effects of rosuvastatin on AS have only been
macroscopically investigated in previous studies (16,19)
and the effects of rosuvastatin on HUVEC dysfunction in
atherogenesis remain unclear. In this study, the effects of
rosuvastatin on ox-LDL-induced HUVEC injury and hyposecretion of NO
were investigated. Furthermore, the possible protective mechanism
of rosuvastatin was investigated.
Materials and methods
Chemical reagents
Human umbilical vein endothelial cells (HUVECs) and
endothelial cell culture medium (Ham's F-12K) were purchased from
Procell Life Science and Technology Co., Ltd., (Wuhan, China),
rosuvastatin (purity: >98%) was purchased from Lunan Better
Pharmaceutical Co., Ltd., (Linyi, China) and ox-LDL was purchased
from Yiyuan Biotechnologies Co., Ltd., (Guangzhou, China). MTT
reagent was obtained from Sigma-Aldrich; Merck KGaA (Darmstadt,
Germany), DAPI and the Hypersensitive ECL chemiluminescence Kit and
bicinchoninic (BCA) protein assay kit were supplied by Beyotime
Institute of Biotechnology (Shanghai, China); Nitric Oxide (NO)
assay kit (A013-2), Superoxide Dismutase (SOD) assay kit (WST-1,
A001-3), Catalase (CAT) assay kit (Ultraviolet, A007-2) and
Malondialdehyde (MDA) assay kit (TBA method, A003-1) were purchased
from Nanjing Jiancheng Bioengineering Institute (Nanjing, China).
Anti-eNOS polyclonal antibody (BS3571), anti-phospho-PI3K p85α
(BS4605) and anti-B-cell lymphoma (Bcl)-2 polyclonal antibody
(BS1511) were obtained from Bioworld (Minneapolis, MN, USA).
Anti-phospho-eNOS (S1177) polyclonal antibody (ab195944) was
purchased from Abcam (Cambridge, UK), anti-PI3K p85 monoclonal
antibody (cat. no. 4257), anti-Akt polyclonal antibody (cat. no.
9272), anti-p-Akt (Ser473) polyclonal antibody (cat. no. 4060) and
anti-Bax polyclonal antibody (cat. no. 2772) were from Cell
Signaling Technology, Inc., (Danvers, MA, USA) and horseradish
peroxidase (HRP)-conjugated anti-rabbit immunoglobulin (Ig)G (cat.
no. IH-0011) or anti-mouse IgG (cat. no. IH-0031) were obtained
from Dingguo Changsheng Biotechnology Co., Ltd., (Beijing,
China).
Cell culture
HUVECs were cultured with endothelial cell culture
medium (Ham's F-12K), which contained 10% fetal bovine serum (FBS;
Procell Life Science and Technology Co., Ltd.), 0.05 mg/ml
endothelial cell growth supplement, 0.1 mg/ml heparin and 1%
penicillin/streptomycin at 37°C in an atmosphere containing 5%
CO2.
Cell viability assay
HUVECs in the logarithmic growth phase were
dispersed by trypsinization and seeded into 96-well plates at a
density of 4×104 cells/ml and 200 µl/well overnight.
HUVECs were treated with 0, 0.01, 0.1, 1 or 10 µmol/l rosuvastatin
for 48 h in order to estimate whether this agent induced HUVEC
injury. In addition, HUVECs were pretreated with the indicated
concentrations of rosuvastatin for 24 h and then treated with or
without ox-LDL and incubated for a further 24 h to estimate the
effect of rosuvastatin on ox-LDL induced HUVECs injury.
Subsequently, 20 µl MTT (5 mg/ml in PBS) solution was added into
each well and the samples were incubated for 4 h. A total of 150 µl
dimethyl sulfoxide was added to each well and the plates were
placed on a shaker for 10 min. The absorbance at 570 nm was
measured with a microplate reader (SpectraMax Plus384; Molecular
Devices, LLC, Sunnyvale, CA, USA). The percentage of surviving
cells was calculated as a fraction of the negative control which
were treated with an equal volume of cell culture medium alone.
DAPI staining
HUVECs in the logarithmic growth phase were
dispersed by trypsinization and seeded at a density of
1×105 cells/ml in the coverslips. After treatment, the
coverslips were washed three times with PBS, fixed in 4%
paraformaldehyde at room temperature for 15 min, permeabilized with
0.1% Triton X-100, stained with 5 µg/ml DAPI and shielded from
light at room temperature for 10 min. Finally, the cells were
observed under a fluorescence microscope (Nikon TE-2000U; Nikon
Corporation, Tokyo, Japan).
Biochemical assays
HUVECs in the logarithmic growth phase were
dispersed by trypsinization, seeded into 6-well plates at a density
of 1×105 cells/ml (2 ml/well) overnight. After
treatment, the levels of NO in the supernatant, the activity of SOD
and CAT and the content of MDA were estimated using commercial kits
according to the manufacturer's protocol. For the measurement of
SOD activity, HUVECs in 6-well plates were harvested in pre-cooled
PBS using a cell scraper and lysed by ultrasonic decomposition at
300 W for 5 sec, and dissociated for 4 times. Subsequently, 20 µl
cell lysis solution, 20 µl enzyme working liquid, 20 µl enzyme
diluent and 200 µl substrate working liquid were added to each well
of the 96-well plates; the plates were incubated at 37°C for 20 min
and the absorbance at 450 nm was measured with a microplate reader.
The results were calculated and expressed as SOD activity. For the
measurement of CAT activity, 20 µl cell lysis solution was added in
a 1-cm optical path cuvette and rapidly combined with 3 ml
substrate working liquid prior to measuring the absorbance at 240
nm for optical density (OD)1. The absorbance for OD2 was
measured 1 min later. The substrate (H2O2) in
HUVECs can be degraded by CAT. Notably, the
H2O2 concentration was gradually reduced in
the reaction liquid and the corresponding absorbance also gradually
declined. The results were calculated and expressed as CAT
activity. For the measurement of MDA content, 100 µl cell lysis
solution, 100 µl NO. 1 working liquid, 1.5 ml NO. 2 working liquid
and 1.5 ml NO. 3 working liquid were added in test tubes that were
placed in 95°C water baths for 40 min. The tubes were centrifuged
at 4,000 × g for 10 min under room temperature, and the OD values
of the supernatants were measured in 1-cm optical path cuvettes at
532 nm. Notably, the MDA in HUVECs can combine with thiobarbituric
acid through a condensation reaction, resulting in the development
of a red product that has a maximum absorption peak at 532 nm. The
results were calculated and expressed as MDA content. Values were
expressed as the mean ± standard deviation from three independent
experiments.
Western blot analysis
HUVECs in the logarithmic growth phase were treated
with the indicated concentrations of rosuvastatin and incubated
with or without ox-LDL. Subsequently, HUVECs were harvested and
lysed in radioimmunoprecipitation assay buffer (containing moderate
protease inhibitor) for 10 min on ice. The protein concentration
was determined using the BCA protein assay kit. Cell extracts were
centrifuged at 14,000 × g at 4°C and equal amounts of protein
samples (40 µg) were loaded onto 10–12% polyacrylamide-SDS gel.
After electrophoresis, the gel was blotted onto a polyvinylidene
difluoride membrane and blocked with 5% (w/v) non-fat milk for 1 h
at room temperature. The membranes were incubated with rabbit
anti-eNOS polyclonal antibody (1:500), rabbit anti-phospho-eNOS
(phospho S1177) polyclonal antibody (1:500), rabbit
anti-phospho-PI3K p85α polyclonal antibody (1:500), rabbit
anti-PI3K p85 monoclonal antibody (1:1,000), rabbit anti-Akt
polyclonal antibody (1:1,000), rabbit anti-p-Akt (Ser473)
polyclonal antibody (1:1,000), rabbit anti-Bcl-2 polyclonal
antibody (1:500) and rabbit anti-Bax polyclonal antibody (1:1,000)
at 4°C overnight. Primary antibody binding was detected with using
a secondary antibody conjugated to HRP (1:5,000) for 1 h at room
temperature. Bands were visualized using ECL chemiluminescence.
Finally, densitometric analysis of the bands was conducted using
Image J 1.51 (National Institutes of Health, Bethesda, MD,
USA).
Statistical analysis
Statistical analysis was performed using the SPSS
19.0 statistical package (IBM Corp., Armonk, NY, USA). The results
are expressed as the mean ± standard deviation. Statistical
differences among all groups were evaluated using one-way analysis
of variance with Tukey's post hoc test. P<0.05 was considered to
indicate a statistically significant difference.
Results
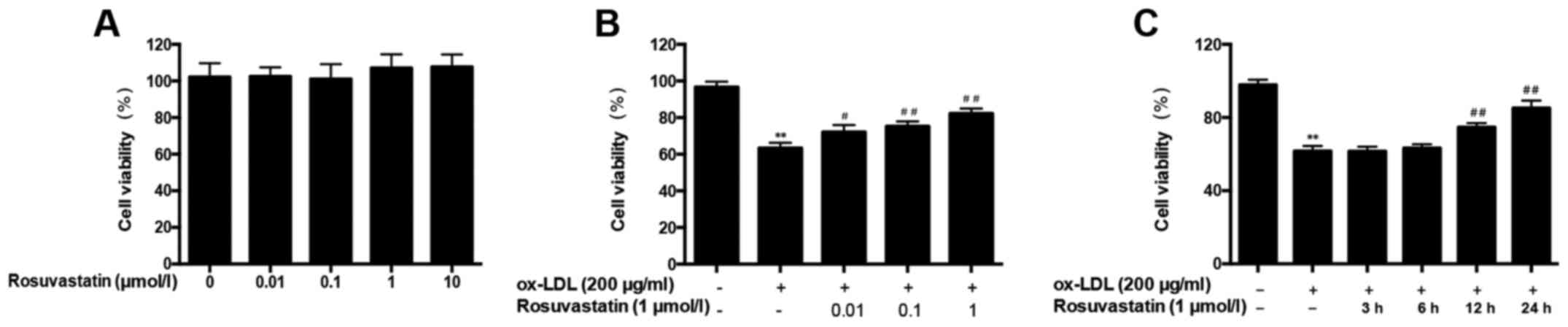
Effects of rosuvastatin on cell
viability
To evaluate the protective effects of rosuvastatin
on HUVECs, cell viability was assessed using an MTT assay. First,
HUVECs were treated with the different rosuvastatin concentrations
for 48 h. However, no concentration of rosuvastatin significantly
altered the HUVEC viability compared with the control group
(Fig. 1A).
HUVECs were treated with 0.01–1 µmol/l rosuvastatin
prior to stimulation with ox-LDL (200 µg/ml) and compared with the
group only stimulated with ox-LDL. It was demonstrated that 0.01
µmol/l rosuvastatin enhanced HUVEC viability significantly
(P<0.05) and 0.1–1 µmol/l rosuvastatin demonstrated a more
significant effect on enhancing the viability of ox-LDL-induced
HUVECs (P<0.01; Fig. 1B).
Furthermore, HUVECs were pretreated with 1 µmol/l rosuvastatin for
different times to assess the time required for rosuvastatin to
effect HUVECs with ox-LDL-induced injury. As presented in Fig. 1C, the viability of HUVECs
pretreated with rosuvastatin for 12 and 24 h was significantly
improved (P<0.01); furthermore, rosuvastatin pretreatment for 24
h exerted the most pronounced effect in terms of increasing cell
viability (85.29±1.54%; P<0.01). These results demonstrated that
rosuvastatin can enhance the viability of HUVECs treated with
ox-LDL.
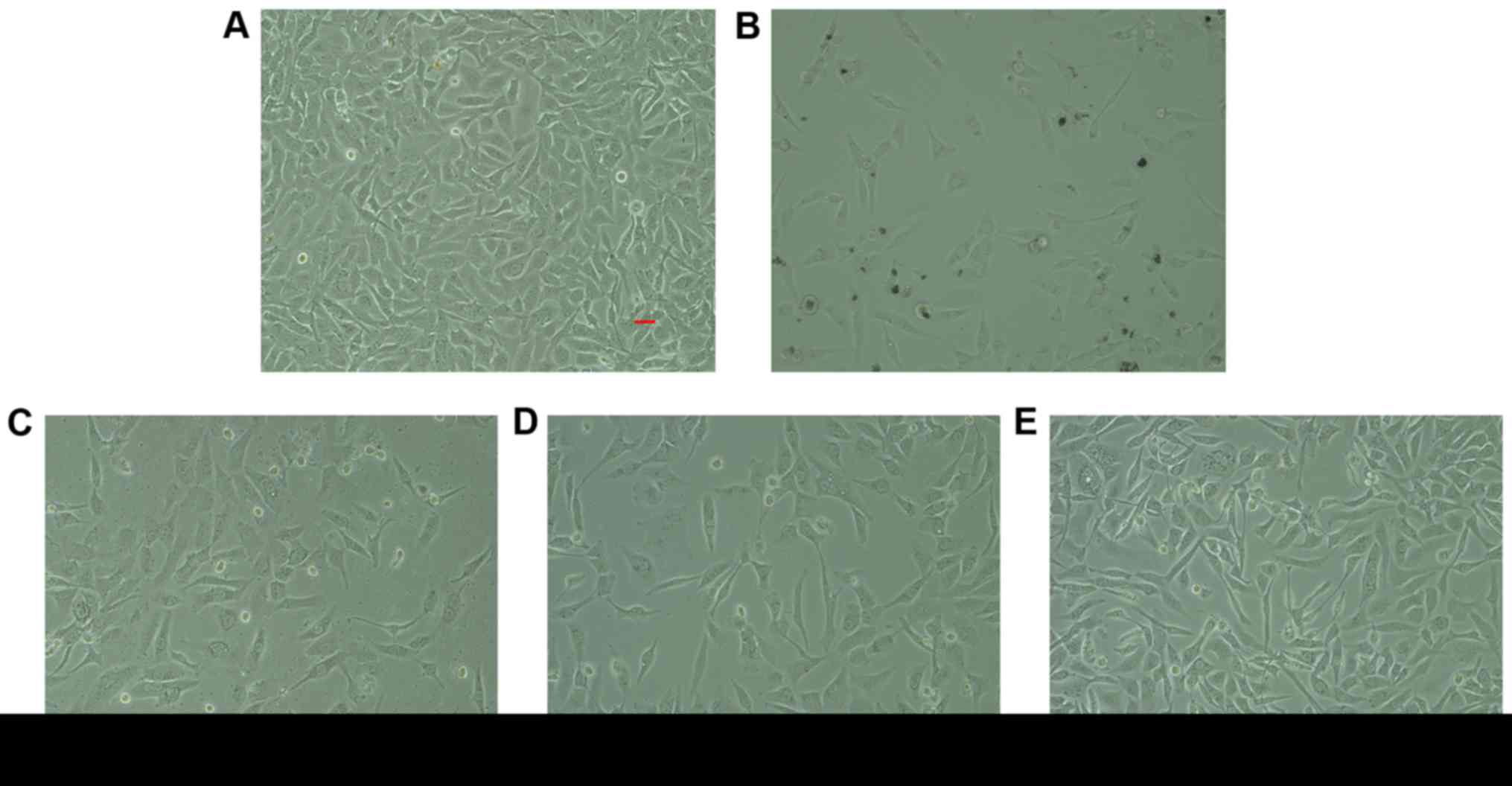
Effects of rosuvastatin on the
morphological alterations of HUVECs
The present study investigated the morphological
changes of HUVECs. Cell growth was almost homogeneous in a
monolayer and the cells appeared to be organized in a
cobblestone-like manner (Fig. 2).
After HUVECs were cultured with ox-LDL, the cell morphology became
irregular, the outline was not clear and nuclear condensation, and
fragmentations were observed under an optical microscope. However,
HUVECs that were pre-incubated with 0.01–1 µmol/l rosuvastatin in
the presence of ox-LDL had started to display signs of
normalization, suggesting that rosuvastatin exerts protective
effects against ox-LDL-induced HUVEC injury.
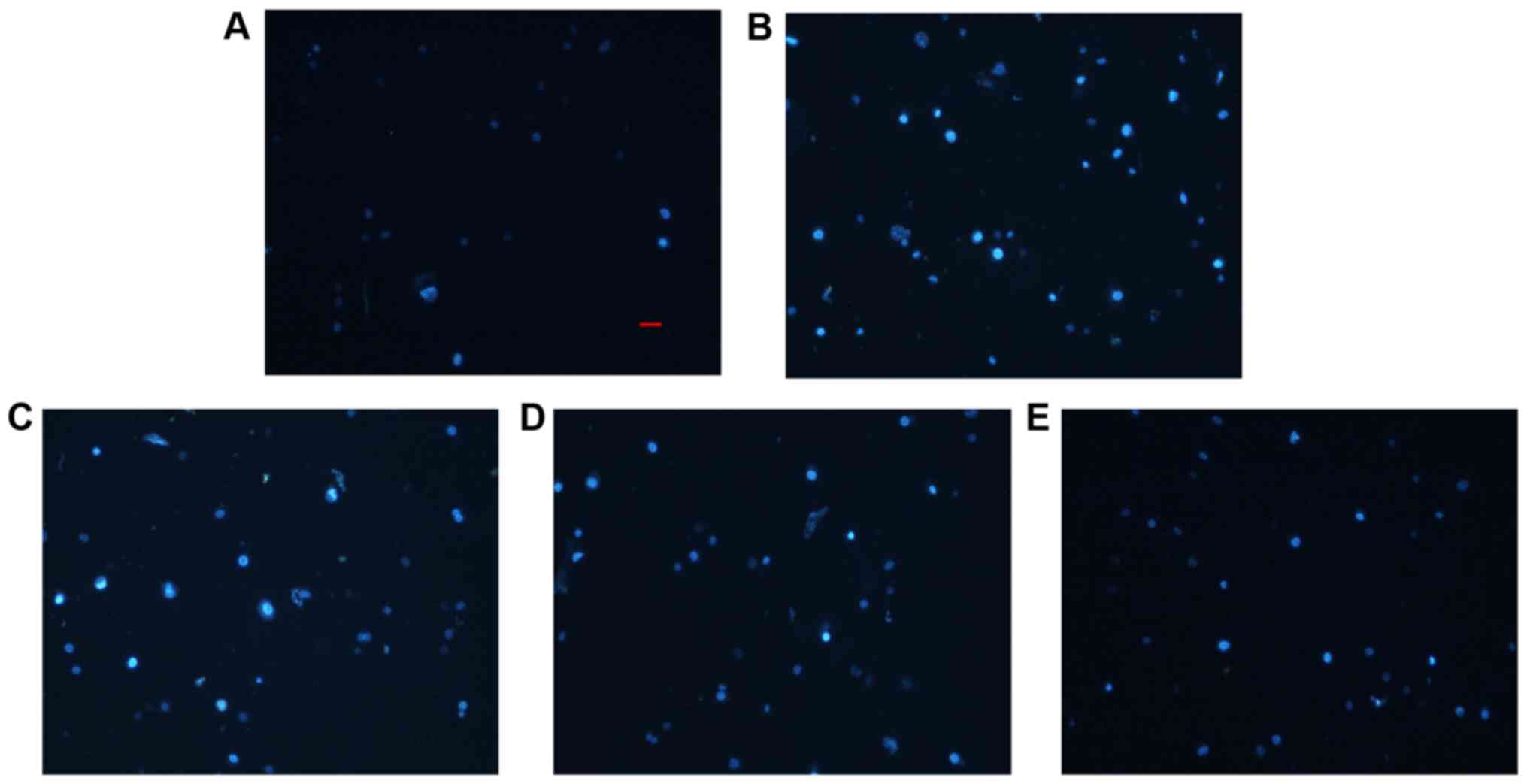
Effect of rosuvastatin on HUVEC
apoptosis induced by ox-LDL
The morphological characteristics of HUVECs in which
apoptosis was induced by ox-LDL were assessed using DAPI staining.
DAPI-positive cells produced a brighter and steady fluorescence. As
presented in Fig. 3, control
HUVECs emitted tiny areas of blue fluorescence. Compared with the
control, ox-LDL induced severe HUVEC functional impairment and
apoptosis, manifesting as intense blue fluorescence that indicated
extensive nuclear injury and fragmentation, which was in accordance
with the MTT results. In contrast, treatment with rosuvastatin
decreased nuclear injury induced by ox-LDL to different degrees.
Notably, the highest rosuvastatin concentration was associated with
less apoptotic fluorescence. The results indicated that
rosuvastatin serves a protective role in ox-LDL-induced apoptosis
in HUVECs.
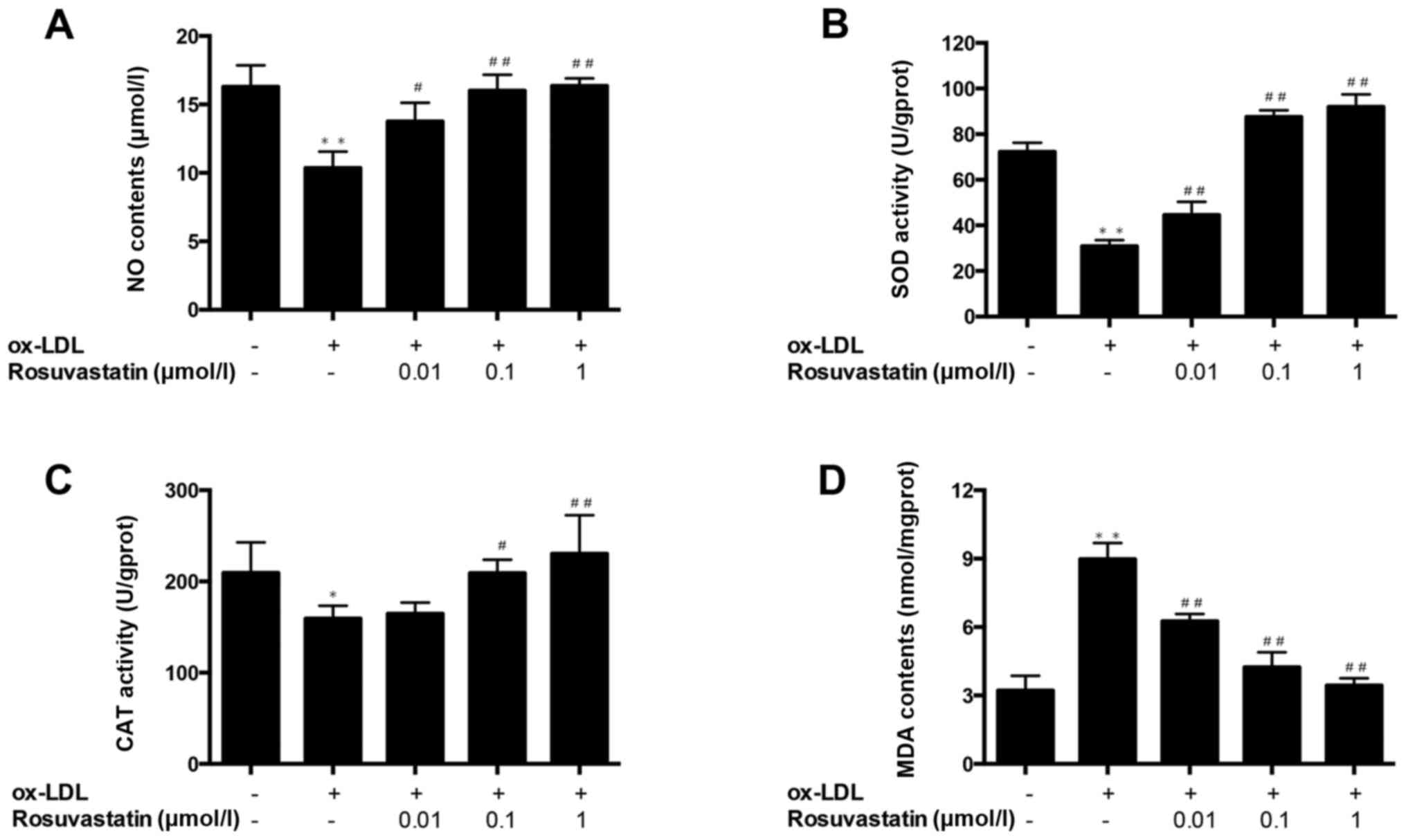
Effect of rosuvastatin on NO levels in
HUVEC injury induced by ox-LDL
To elucidate the implication of NO in ox-LDL-induced
injury of HUVECs, the contents of NO in the cell culture
supernatant were detected. Compared with the control group, the NO
level was significantly decreased in HUVECs with ox-LDL-induced
injury (P<0.01; Fig. 4A),
whereas 0.01 µmol/l rosuvastatin pretreatment significantly
increased NO secretion compared with the ox-LDL-induced injury
group (P<0.05). Notably, the 0.1 and 1 µmol/l rosuvastatin
pretreatment groups demonstrated superior effects (P<0.01).
These results indicated that rosuvastatin may increase the levels
of the endothelial protective factor NO.
Effect of rosuvastatin on oxidative
stress in HUVEC injury induced by ox-LDL
To elucidate the implications of oxidative stress in
HUVEC injury induced by ox-LDL, the intracellular MDA content and
SOD and CAT activities were measured. Compared with the control
group, the activity of SOD and CAT were significantly decreased
(P<0.01 and P<0.05, respectively; Fig. 4B and C) and MDA content was
significantly increased (P<0.01; Fig. 4D) in HUVECs with ox-LDL-induced
injury. While HUVECs pretreated with rosuvastatin (0.01–1 µmol/l)
exhibited a significant improvement of SOD activity and a reduction
of MDA content (P<0.01), 0.1 and 1 µmol/l of rosuvastatin
significantly improved the CAT activity compared with HUVECs with
ox-LDL-induced injury (P<0.05 and P<0.01, respectively). The
results suggested that rosuvastatin has antioxidant properties and
may relieve the oxidative stress of ox-LDL-induced injury in
HUVECs.
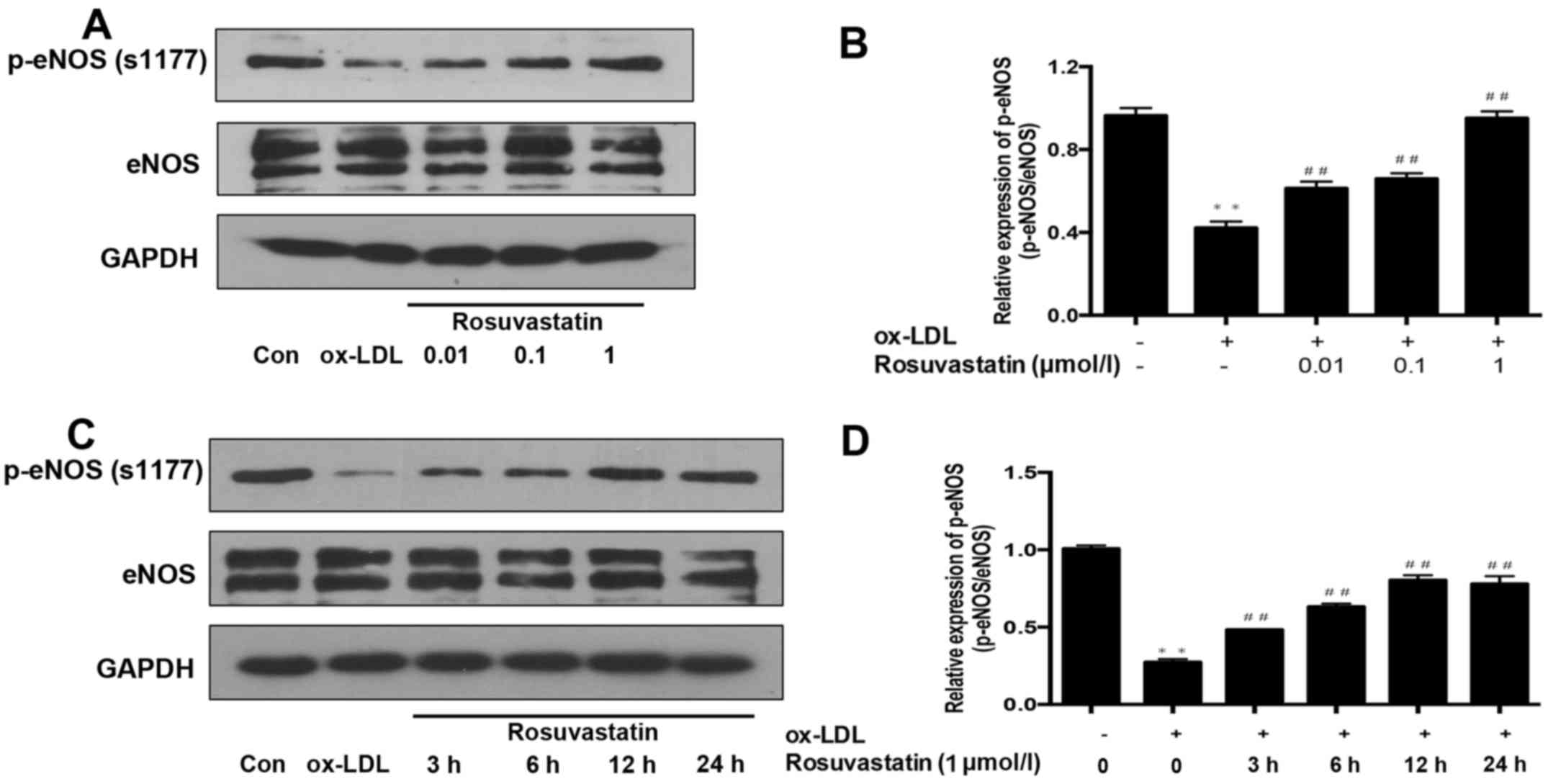
Effect of rosuvastatin on the
phosphorylation of eNOS in HUVEC injury induced by ox-LDL
The phosphorylation of eNOS, which is the regulatory
factor of NO production was measured. As presented in Fig. 5A and B, the phosphorylation of eNOS
in ox-LDL-stimulated HUVECs was significantly decreased compared
with the control group (P<0.01). Furthermore, HUVECs were
pretreated with different concentrations of rosuvastatin and the
phosphorylation levels of eNOS were significantly enhanced
(P<0.01). However, the expression levels of total eNOS were not
notably different between these groups. Subsequently, HUVECs were
pretreated with rosuvastatin 1 µmol/l for different times (3, 6, 12
and 24 h) and then stimulated with ox-LDL to determine the
pretreatment time of rosuvastatin that altered the phosphorylation
of eNOS. The results demonstrated that rosuvastatin pretreatment
for 3 h could significantly affect the phosphorylation of eNOS
(P<0.01) and as the pretreatment time of rosuvastatin increased,
the phosphorylation of eNOS also significantly increased
(P<0.01; Fig. 5C and D). The
results suggested that rosuvastatin exerts a prominent effect on
enhancing eNOS phosphorylation in HUVECs with ox-LDL-induced injury
and pretreatment with rosuvastatin for only 3 h can significantly
promote the phosphorylation of eNOS, which is a shorter time
compared with that required for enhancing HUVEC viability following
ox-LDL stimulation (Fig. 1D).
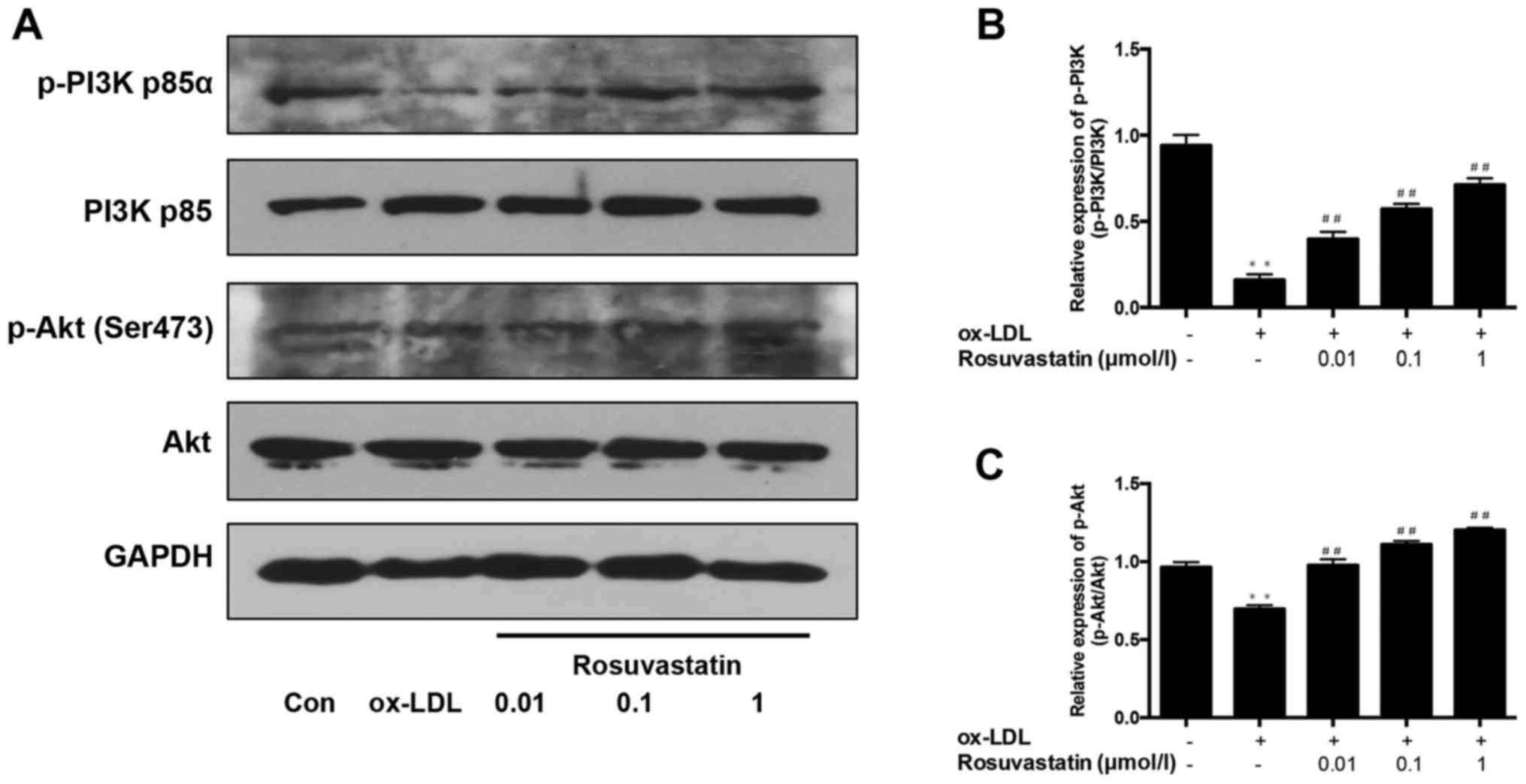
Effects of rosuvastatin on the
phosphorylation of the PI3K/Akt signaling pathway
The PI3K/Akt signaling pathway is considered as a
key mediator of eNOS activity that is implicated in the secretion
of NO in HUVECs. In the present study, the phosphorylation of PI3K
and Akt was determined. As presented in Fig. 6, compared with the control group,
the phosphorylation of PI3K (Fig. 6A
and B) and Akt (Fig. 6A and C)
were significantly decreased in ox-LDL-stimulated HUVECs
(P<0.01), and 0.01–1 µmol/l rosuvastatin significantly enhanced
the phosphorylation of PI3K and Akt to varying degrees (P<0.01).
Furthermore, the effects of rosuvastatin were dose-dependent. The
expression of total PI3K and total Akt did not notably differ
between the control and HUVECs with ox-LDL-induced injury. These
results suggested that rosuvastatin may affect the PI3K/Akt
signaling pathway.
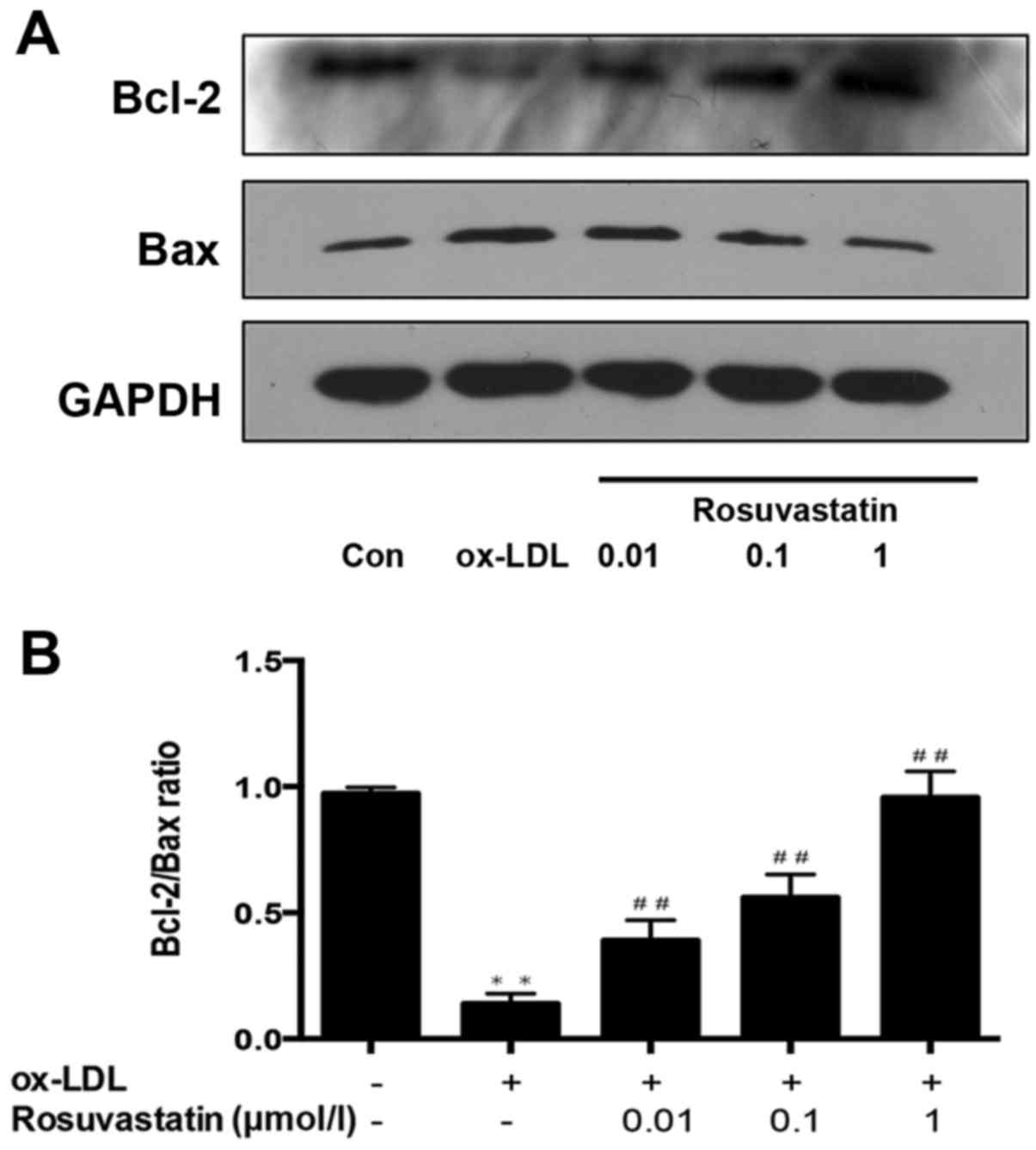
Effect of rosuvastatin on the
expression of Bcl-2/Bax
Bcl-2 and Bax are important hallmarks of apoptosis
that are regulated by the PI3K/Akt signaling pathway. As presented
in Fig. 7, it was demonstrated
that the expression of apoptotic protein (Bax) increased and the
expression of antiapoptotic protein (Bcl-2) was significantly
decreased in HUVECs with ox-LDL-induced injury (P<0.01).
Notably, rosuvastatin enhanced the expression of Bcl-2 and
significantly decreased the expression of Bax (P<0.01) in the
ox-LDL-stimulated HUVEC group. The results suggested that
rosuvastatin may protect HUVECs against ox-LDL-induced apoptosis by
regulating the expression of Bcl-2/Bax.
Discussion
The pathogenesis of AS may be initiated with
systemic inflammation and acute lipid oxidation (20). Under conditions of high blood lipid
levels, the lipids invade and are deposited into subintimal cells,
leading to macrophage infiltration under the vascular intima to
phagocytose lipids, which promotes the formation of atherosclerotic
plaques and thrombi (2). The
endothelial cell is an important barrier of blood vessels, which
can resist the damage caused by inflammatory cell infiltration,
disturbed blood flow and any other external stimulating factors
(21). The excessive LDL modified
by oxidation or enzymes (ox-LDL) disturbs endothelial function,
including disruption of the endothelial barrier, impairment of NO
release followed by ox-LDL penetrating into the intima in the
earliest stages of AS (21). AS
can accelerate the progression of CVDs (6). Notably, chronic lipoprotein
abnormalities induce a decline in kidney function (22), with the exception of hyperlipemia
induced by AS. AS has become a high-risk complication in a number
of other diseases, including diabetes; persistent hyperglycemia
suppresses the phosphorylation levels of Akt and eNOS (23) and disrupts L-arginine-NO
metabolism, which is a key protective factor associated with the
inhibition of apoptosis in HUVECs. It has been demonstrated that
tumor necrosis factor-α-induced apoptosis was inhibited by low
concentrations of NO in a cyclic guanosine
monophosphate-independent manner and the cellular suicide program
was inhibited in HUVECs via S-nitrosylation of members of the
caspase family (24). Furthermore,
NO protects cells from apoptosis by stimulating the production of
vascular endothelial growth factor (25). According to the results above, NO
bioavailability disruption may induce a series of endothelial cell
dysfunctions (26), including
aggravated inflammation (27),
cardiac cell death and acute coronary syndrome (28). Ongoing research has focused on
investigating the mechanisms underlying endothelial cell injury and
improving endothelial cell dysfunction in different diseases.
Oxidative stress is the etiology behind arterial
wall alterations. Oxidative stress is due to an imbalance between
the enzymatic activity of antioxidants and free radicals, which
causes the decreased bioavailability of the endothelial protective
factor NO (20). Furthermore,
endothelial cell dysfunction increases the production of oxygen
free radicals derived from NO catabolism, resulting in a vicious
circle of endothelial cell injury (7). Oxidative stress is also implicated in
apoptosis-associated protein expression that results in cell
apoptosis. It has been demonstrated that ox-LDL-induced apoptosis
may be achieved by regulating the expression of Bcl-2 and Bax
protein within human fatty streaks (29). Based on the above-mentioned
findings, clearing the circulating reactive oxygen species or
increasing the antioxidant capacity are considered as key points in
the prevention and therapy of AS. The role of eNOS dysfunction in
AS is well understood. eNOS dysfunction in endothelial cells can
disrupt vascular tone and structure regulation (30). A previous study demonstrated that
increasing the expression and activation of eNOS can promote
neointimal growth, cell migration and re-endothelialization
following arterial balloon catheter injury (31). In endothelial cells, Akt
phosphorylates eNOS directly which promotes cell survival by
nitrosylating the reactive cysteine residue in caspases (13) and critically regulates apoptosis in
endothelial cells including activation of the pro-apoptotic
proteins Bax and caspases 3 and 9 (32). In addition, the activation of Akt
stimulated with activated phosphoinositide-dependent kinase-1
(PDK1) and mammalian target of rapamycin complex 2 by direct
binding and phosphorylation on threonine 308 and serine 473
(13). Notably, PI3K activation is
the key regulator of Akt activation by regulating PDK1. The
expression and activation of eNOS and Akt can be suppressed by PI3K
inhibitors (23,32). The PI3K/Akt signaling pathway
exerts a strong regulatory effect on expression and activation of
eNOS.
The 2013 American College of Cardiology/American
Heart Association cholesterol guidelines recommended using
‘high-intensity’ statin therapy for reducing the risk of
hypercholesterolemia-induced cardiovascular events due to its
beneficial effects on LDL-C reduction (33,34).
Rosuvastatin has multiple advantages compared with other statins,
including stronger regulatory effects on dyslipidemia, a shorter
half-life and high bioavailability (19). In addition to lowering LDL-C, it is
plausible that there are more pronounced benefits of rosuvastatin
therapy in coronary atherosclerotic plaque regression (17). These results called attention to
the effects of rosuvastatin on endothelial cell dysfunction. In the
present study, the influence of ox-LDL on endothelial cell
protection factor eNOS/NO was measured and the effects of
rosuvastatin on ox-LDL induced insufficient expression levels of
eNOS/NO, as well as PI3K/Akt signaling pathway-regulatory factor of
eNOS, aiming to investigate the multiple effects of rosuvastatin on
endothelial cells independent of lipid regulation.
Ox-LDL was used to induce HUVEC injury in
vitro. The present study demonstrated that ox-LDL promoted
extensive HUVEC apoptosis, reduced endothelial cell-derived NO
levels, increased oxidative stress and reduced the activity of SOD
and CAT. Furthermore, the content of MDA was increased compared
with the levels in normal HUVECs. These results were in agreement
with the findings of Ahsan et al (32). Following treatment with
rosuvastatin, cell viability rates were increased, NO secretion in
HUVECs was increased and the high-dose rosuvastatin-treated group
was comparable to the control group; the activity of SOD and CAT
was enhanced, and the MDA content was decreased by rosuvastatin.
The results suggested that rosuvastatin exerted beneficial
antioxidant effects in HUVECs with ox-LDL-induced injury.
Furthermore, ox-LDL-induced eNOS activation was markedly decreased
compared with control HUVECs; however, rosuvastatin treatment
reversed the effects of ox-LDL in a dose-dependent manner. The
present study also evaluated the phosphorylation of PI3K and Akt,
as well as the expression of Bcl-2/Bax in HUVECs with
ox-LDL-induced injury. Notably, phosphorylation of PI3K and Akt was
inhibited and the ratio of Bcl-2/Bax was decreased. However,
rosuvastatin treatment increased the phosphorylation of PI3K and
Akt compared with ox-LDL-stimulated HUVECs. Furthermore, the
upregulation of eNOS by rosuvastatin was regulated via the PI3K/Akt
signaling pathway and the results suggested that rosuvastatin may
protect HUVECs against ox-LDL-induced apoptosis. Furthermore, the
biological functions of vascular eNOS and NO are varied, including
anti-inflammatory, anti-platelet and vasodilatory actions, as well
as endoplasmic reticulum stress (ERs)-derived eNOS dysfunction is a
crucial damage factor of endothelial cell through oxidative stress
and ERs-induced apoptosis serves a key role in the occurrence of AS
(35). Consequently, the authors
intend to investigate the effect of rosuvastatin on ERs and the
relevant apoptosis signal pathways, which are closely associated
with endothelial cell dysfunction in the next step of research.
In conclusion, ox-LDL can induce severe injury and
apoptosis of HUVECs, reduce eNOS phosphorylation and eNOS-derived
NO. However, rosuvastatin treatment can reverse the effects of
ox-LDL, upregulate the phosphorylation of PI3K/Akt, and the
expression of Bcl-2/Bax, indicating that rosuvastatin may attenuate
endothelial dysfunction and apoptosis in AS and in other diseases
accompanied by vascular lesions.
Acknowledgements
The authors would like to thank Dr Wenwen Fu, Mr.
Zeyuan Lu and Mr. Yuchen Wang, of the department of Pharmacology,
College of Pharmacy, Jilin University (Changchun, China), for their
technical assistance.
Funding
The present study was supported by the Science and
Technology development projects of Jilin, China (grant no.
20150101200JC).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
DS and GL conceived and designed the study. JG, HX
and XY performed the experiments. JG and HX wrote the paper. GX and
HC analyzed data for the study, reviewed and edited the manuscript.
All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Samadi S, Bozorgmanesh M, Khalili D,
Momenan A, Sheikholeslami F, Azizi F and Hadaegh F:
Hypertriglyceridemic waist: The point of divergence for prediction
of CVD vs. mortality: Tehran Lipid and Glucose Study. Int J
Cardiol. 165:260–265. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wong MC, Zhang DX and Wang HH: Rapid
emergence of atherosclerosis in Asia: A systematic review of
coronary atherosclerotic heart disease epidemiology and
implications for prevention and control strategies. Curr Opin
Lipidol. 26:257–269. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gkaliagkousi E, Gavriilaki E,
Triantafyllou A and Douma S: Clinical significance of endothelial
dysfunction in essential hypertension. Curr Hypertens Rep.
17:852015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lucia J, Lucia M, Lucia J, Janicko M,
Fedacko J, Novakova B, Chmelarova A, Majernik J and Pella D: Effect
of ivabradine on endothelial function in patients with stable
angina pectoris: Assessment with the Endo-PAT 2000 device. Adv
Ther. 32:962–970. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shaw J and Anderson T: Coronary
endothelial dysfunction in non-obstructive coronary artery disease:
Risk, pathogenesis, diagnosis and therapy. Vasc Med. 21:146–155.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gu X, Yang X, Li Y, Cao J, Li J, Liu X,
Chen J, Shen C, Yu L, Huang J and Gu D: Usefulness of low-density
lipoprotein cholesterol and non-high-density lipoprotein
cholesterol as predictors of cardiovascular disease in Chinese. Am
J Cardiol. 116:1063–1070. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kuhlencordt PJ, Padmapriya P, Rützel S,
Schödel J, Hu K, Schäfer A, Huang PL, Ertl G and Bauersachs J:
Ezetimibe potently reduces vascular inflammation and
arteriosclerosis in eNOS-deficient ApoE ko mice. Atherosclerosis.
202:48–57. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li H, Horke S and Förstermann U: Vascular
oxidative stress, nitric oxide and atherosclerosis.
Atherosclerosis. 237:208–219. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Frostegård J: Immunity, atherosclerosis
and cardiovascular disease. BMC Med. 11:1172013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
van Diepen JA, Berbée JF, Havekes LM and
Rensen PC: Interactions between inflammation and lipid metabolism:
Relevance for efficacy of anti-inflammatory drugs in the treatment
of atherosclerosis. Atherosclerosis. 228:306–315. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mitra S, Khaidakov M, Lu J, Ayyadevara S,
Szwedo J, Wang XW, Chen C, Khaidakov S, Kasula SR, Stone A, et al:
Prior exposure to oxidized low-density lipoprotein limits apoptosis
in subsequent generations of endothelial cells by altering promoter
methylation. Am J Physiol Heart Circ Physiol. 301:H506–H513. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Abeyrathna P and Su Y: The critical role
of Akt in cardiovascular function. Vascul Pharmacol. 74:38–48.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yu H, Littlewood T and Bennett M: Akt
isoforms in vascular disease. Vascul Pharmacol. 71:57–64. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jones SP, Gibson MF, Rimmer DM III, Gibson
TM, Sharp BR and Lefer DJ: Direct vascular and cardioprotective
effects of rosuvastatin, a new HMG-CoA reductase inhibitor. J Am
Coll Cardiol. 40:1172–1178. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang Y, Wang SJ, Han ZH, Li YQ, Xue JH,
Gao DF, Wu XS and Wang CX: PI3K/AKT signaling pathway plays a role
in enhancement of eNOS activity by recombinant human angiotensin
converting enzyme 2 in human umbilical vein endothelial cells. Int
J Clin Exp Pathol. 7:8112–8117. 2014.PubMed/NCBI
|
|
16
|
Aggarwal RK and Showkathali R:
Rosuvastatin calcium in acute coronary syndromes. Expert Opin
Pharmacother. 14:1215–1227. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Qian C, Wei B, Ding J, Wu H, Cai X, Li B
and Wang Y: Meta-analysis comparing the effects of rosuvastatin
versus atorvastatin on regression of coronary atherosclerotic
plaques. Am J Cardiol. 116:1521–1526. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Takayama T, Komatsu S, Ueda Y, Fukushima
S, Hiro T, Hirayama A and Saito S: ALTAIR study group: Comparison
of the effect of rosuvastatin 2.5 mg vs. 20 mg on coronary plaque
determined by angioscopy and intravascular ultrasound in Japanese
with stable angina pectoris (from the Aggressive Lipid-Lowering
Treatment Approach Using Intensive Rosuvastatin for Vulnerable
Coronary Artery Plaque [ALTAIR] Randomized Trial). Am J Cardiol.
117:1206–1212. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kurtoglu E, Balta S, Sincer I, Altas Y,
Atas H, Yilmaz M, Korkmaz H, Erdem K, Akturk E, Demirkol S and Can
C: Comparision of effects of rosuvastatin versus atorvastatin
treatment on plasma levels of asymmetric dimethylarginine in
patients with hyperlipidemia having coronary artery disease.
Angiology. 65:788–793. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Castellon X and Bogdanova V: Chronic
inflammatory diseases and endothelial dysfunction. Aging Dis.
7:81–89. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Byfield FJ, Rothblat GH, Gooch KJ and
Levitan I: OxLDL increases endothelial stiffness, force generation
and network formation. Vascul Pharmacol. 45:e732006. View Article : Google Scholar
|
|
22
|
Ananthakrishnan S and Kaysen GA: Treatment
of hyperlipidemia changes with level of kidney function-rationale.
Adv Chronic Kidney Dis. 23:247–254. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Xing Y, Lai J, Liu X, Zhang N, Ming J, Liu
H and Zhang X: Netrin-1 restores cell injury and impaired
angiogenesis in vascular endothelial cells upon high glucose by
PI3K/AKT-eNOS. J Mol Endocrinol. 58:167–177. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Haendeler J, Weiland U, Zeiher AM and
Dimmeler S: Effects of redox-related congeners of NO on apoptosis
and caspase-3 activity. Nitric Oxide. 1:282–293. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Frank S, Kämpfer H, Wetzler C and
Pfeilschifter J: Nitric oxide drives skin repair: Novel functions
of an established mediator. Kidney Int. 61:882–888. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Vulesevic B, McNeill B, Giacco F, Maeda K,
Blackburn NJ, Brownlee M, Milne RW and Suuronen EJ:
Methylglyoxal-induced endothelial cell loss and inflammation
contribute to the development of diabetic cardiomyopathy. Diabetes.
65:1699–1713. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang Y, Ma KL, Liu J, Wu Y, Hu ZB, Liu L,
Lu J, Zhang XL and Liu BC: Inflammatory stress exacerbates lipid
accumulation and podocyte injuries in diabetic nephropathy. Acta
Diabetol. 52:1045–1056. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Konishi H, Miyauchi K, Shitara J, Endo H,
Wada H, Doi S, Naito R, Tsuboi S, Ogita M, Dohi T, et al: Impact of
lipoprotein(a) on long-term outcomes in patients with diabetes
mellitus who underwent percutaneous coronary intervention. Am J
Cardiol. 118:1781–1785. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yang X, Li Y, Li Y, Ren X, Zhang X, Hu D,
Gao Y, Xing Y and Shang H: Oxidative stress-mediated
atherosclerosis: Mechanisms and therapies. Front Physiol.
8:6002017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Desjardins F and Balligand JL: Nitric
oxide-dependent endothelial function and cardiovascular disease.
Acta Clin Belg. 61:326–334. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Guo J, Breen DM, Pereira TJ, Dalvi PS,
Zhang H, Mori Y, Ghanim H, Tumiati L, Fantus IG, Bendeck MP, et al:
The effect of insulin to decrease neointimal growth after arterial
injury is endothelial nitric oxide synthase-dependent.
Atherosclerosis. 241:111–120. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ahsan A, Han G, Pan J, Liu S, Padhiar AA,
Chu P, Sun Z, Zhang Z, Sun B, Wu J, et al: Phosphocreatine protects
endothelial cells from oxidized low-density lipoprotein-induced
apoptosis by modulating the PI3K/Akt/eNOS pathway. Apoptosis.
20:1563–1576. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Valentino M, Al Danaf J, Panakos A,
Ragupathi L, Duffy D and Whellan D: Impact of the 2013 American
College of Cardiology/American Heart Association cholesterol
guidelines on the prescription of high-intensity statins in
patients hospitalized for acute coronary syndrome or stroke. Am
Heart J. 181:130–136. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
O'Keefe JH, DiNicolantonio JJ and Lavie
CJ: Statins, ezetimibe, and proprotein convertase subtilisin-kexin
Type 9 inhibitors to reduce low-density lipoprotein cholesterol and
cardiovascular events. Am J Cardiol. 119:565–571. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Dong Y, Fernandes C, Liu Y, Wu Y, Wu H,
Brophy ML, Deng L, Song K, Wen A, Wong S, et al: Role of
endoplasmic reticulum stress signalling in diabetic endothelial
dysfunction and atherosclerosis. Diab Vasc Dis Res. 14:14–23. 2017.
View Article : Google Scholar : PubMed/NCBI
|