Introduction
Chronic kidney disease (CKD) is a major disease that
threatens human health (1–4), and renal fibrosis is the final
pathological manifestation of CKD progression (1–4).
Previous reports, and our previous studies, have suggested that
aldosterone can regulate water and salt metabolism, and also serves
a key role in the progression of renal fibrosis (5,6).
However, many studies have only focused on the mechanisms that
promote fibrosis (7–11). Klotho (KL) is predominantly
expressed in the distal tubules of the kidney and is a cofactor for
the fibroblast growth factor 23 receptor (12–14).
Previous studies have suggested that KL reduction is closely
associated with the progression of CKD fibrosis, as kidney KL
expression is reduced in different forms of CKD, including chronic
glomerulonephritis, acute renal tubular injury and diabetic
nephropathy, leading to the promotion of transforming growth factor
β1 and the expression of other fibrosis factors (12–20).
In addition, renal fibrosis was significantly reduced in neonatal
mice that overexpressed KL (16,19).
The above studies suggest that, in accelerating CKD progression,
aldosterone increase is accompanied by the reduction of KL
expression. There may be some association between the changes in
these two factor in CKD.
Histone deacetylase (HDACs) are a class of enzymes
that remove acetyl groups from histone amino acid residues, and
serve an important role in chromatin structural modification and
gene transcription regulation (16,21–26).
Generally, HDACs can inhibit gene transcription activation by
blocking DNA dissociation from histone octamers and tightening the
structure of nucleosomes, which inhibits binding between DNA and
transcription factors or synergistic transcription regulators
(16,21–26).
The current study therefore explored whether high
aldosterone significantly reduces acetylation of the H3K9 site by
activating HDAC1 protein expression in the renal distal convoluted
tubule cells, inducing the loss of KL gene transcriptional
activity and ultimately leading to the exacerbation of renal
fibrosis.
Patients and methods
Selection of patients with CKD
Adult patients were screened if they had a CKD stage
between 1–5, according to the Kidney Disease Outcomes Quality
Initiative (KDOQI) criteria published in January 2012 (27). According to the KDOQI criteria,
patients are classified into five stages based on their estimated
GFR (eGFR). The eGFR was determined by the CKD Epidemiology
Collaboration creatinine equation (28). A total of 115 adults were screened,
of whom three were excluded from the study as they were diagnosed
with acute kidney injury. Finally, 112 patients (77 men and 35
women; 31–88 years, mean age 64.5±12.7 years) were enrolled and
underwent a 6-year follow-up between January 2010 and December 2015
at Huashan Hospital, Fudan University, Shanghai, China.
Blood samples for laboratory measurements were
obtained at the time of enrolment and at 1.5-years follow-up. The
concentration of serum soluble α-KL was determined using an ELISA
kits obtained from Immuno-Biological Laboratories Co., Ltd. (cat.
no. 27998; Fujioka, Japan), according to the manufacturer's
instructions. ∆ aldosterone was calculated using the concentration
of aldosterone at baseline and at 1.5-years follow-up (∆value=value
at the 1.5-year follow-up-value at the baseline).
The present study was approved by the Ethics
Committee of Huashan Hospital, Fudan University (Shanghai, China).
All provided written informed consent for participation and the
study was conducted in accordance with the Declaration of
Helsinki.
Aldosterone-induced CKD mouse model
and serum aldosterone detection
A total of 20 adult C57 male mice at 7–8 weeks of
age and ~20 g weight, were obtained from the Animal Research
Center, Fudan University (Shanghai, China). They were housed at 4
mice per cage and maintained for 14 days in a
temperature-controlled environment under a 12-h light/dark cycle
with ad libitum access to food and water. The mice were
uninephrectomized. Following a 2 week recovery period, an
ALZET® osmotic pump (model no. 2002; DURECT Corporation,
Cupertino, CA, USA) was implanted subcutaneously (29). Subsequently, mice were divided into
three treatment groups: Group 1, vehicle (0.5% ethanol,
subcutaneously) (n=4) as the WT group; Group 2, vehicle
(subcutaneously) + 1% NaCl in drinking water (n=8) as the low
aldosterone group; Group 3, 0.75 µg/h aldosterone (subcutaneously)
+ 1% NaCl in drinking water (n=8) as the high aldosterone group
(29). Mice were treated for 2
weeks. The study was approved by the Ethics Committee of Fudan
University (reference no. 201702069S; Shanghai, China). All
experiments conformed to standards set by the Laboratory Animal
Regulation of the State Scientific and Technological Commission
(Shanghai Lab. Animal Research Center).
The concentration of serum aldosterone was
ascertained using a chemiluminescent immunoassay obtained from
Shenzhen New Industries Biomedical Engineering Co. Ltd. Shenzhen,
China (cat. no. 0525-2007).
Hematoxylin and eosin (H&E)
staining
Briefly, fresh kidney tissues were fixed in 4%
paraformaldehyde (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany)
for 30 min at room temperature. Then, the tissue was dehydrated
using graded ethanol (70, 80, 90 and 100%), embedded in paraffin,
sectioned (6-µm thickness), and immersed in xylene for dewaxing.
The sections were stained with H&E (Sigma-Aldrich; Merck KGaA).
The rehydrated sections were stained in hematoxylin solution for 30
min at room temperature and washed in tap water for 5 min until the
sections turned blue. The sections were differentiated in 70%
ethanol containing 1% HCl for 5 seconds and then washed for 5 min
in tap water until blue. The sections were then stained in eosin
solution for 10 min. The sections were cleared with xylene and
sealed with neutral resin (both Sigma-Aldrich; Merck KGaA). The
sections were observed under a light microscope at magnification,
×100-400 and three fields of view were observed.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total kidney RNA was extracted from each group of
mice using TRIzol® (Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA). The RNA was processed using
DNase I (Sigma-Aldrich; Merck KGaA) and reverse transcribed to cDNA
using the ReverTra Ace-α-® kit [Toyobo (Shanghai) Co.,
Ltd., Shanghai, China]. The RNA and primer Oligo dt were heated in
a 70°C heat block for 5 min then immediately chilled in ice water
for ≥5 min, then centrifuged at 15,725 × g for 10 sec in a
microcentrifuge at 4°C before being stored on ice until reverse
transcription mix was added. qPCR was performed using a
SYBR® Green Realtime PCR Master Mix [Toyobo (Shanghai)
Co., Ltd.] on a MasterCycle RealPlex4 real-time PCR
detection system (Eppendorf, Hamburg, Germany). The sequences of
forward and reverse primers of Klotho was 5′GAGTGGCATAGGGGCTACA and
5′GGCTGGTTTTCAGGTAAAGG, respectively. The sequences of forward and
reverse primers of fibronectin 1 was 5′AGTGGAAGTGTGAGCGACAT and
5′GTGAGTCTGCGGTTGGTAAAT, respectively. The sequences of forward and
reverse primers of 18S was 5′CAGCCACCCGAGATTGAGCA and
5′TAGTAGCGACGGGCGGTGTG, respectively. The thermocycling conditions
were as follows: 40 cycles of denaturation at 95°C for 15 sec,
annealing at 58°C for 30 sec and extension at 72°C for 42 sec. The
relative expression levels of the gene were determined by the
2−ΔΔCq method (30).
The mRNA expression levels were normalized to the internal
reference gene, 18S ribosomal RNA.
Immunohistochemical staining
Briefly, fresh kidney tissues were fixed in 4%
paraformaldehyde (Sigma-Aldrich; Merck KGaA) for 30 min at room
temperature. Then, the tissue was dehydrated in an ethanol gradient
(70, 80, 90 and 100%), embedded in paraffin, sectioned (6-µm
thickness) and immersed in xylene for dewaxing. The sections were
blocked using an immunohistochemical blocking solution (Beyotime
Institute of Biotechnology, Haimen, China) at 37°C for 30 min.
After the blocking solution was discarded, the samples were washed
in washing buffer (Beyotime Institute of Biotechnology) three
times, 5 min each at room temperature. The samples were incubated
with antibodies (rabbit anti-mouse HDAC1; cat. no. 34589 and rabbit
anti-mouse H3K9Ac; cat. no. 9927, 1:200; Cell Signaling Technology,
Inc., Danvers, MA, USA) at 37°C for 45 min. The horseradish
peroxidase-conjugated secondary antibody (cat. no. sc-2768; 1:200;
Santa Cruz Biotechnology, Inc., Dallas, TX, USA) was added and the
sections were incubated for 60 min at room temperature. Finally, a
VECTASTAIN Elite ABC kit (cat. no. PK-6100; Vector Laboratories,
Inc., Burlingame, CA, USA) was used for the color reaction.
Meanwhile, PBS (pH 7.4) was used as a negative control in the place
of the first antibody. After the antibody was discarded, the
samples were washed in washing buffer three times, 5 min each at
room temperature. Finally, the samples were mounted in neutral
resin (Sigma-Aldrich; Merck KGaA).
Masson's trichrome staining was used at room
temperature for 10 min to detect collagen fibers in the tissues:
Collagen fibers were stained blue, nuclei are stained dark brown
and background is stained red. The sections were observed under a
light microscope at magnification, ×100-400 and three fields of
view were observed.
Western blot analysis
Total protein was extracted using NP-40 (Nonidet
P-40) lysis buffer over ice and the protein concentration
determined with a BCA Protein Assay kit (cat. no. 23225; Thermo
Fisher Scientific, Inc.). Protein at 30 µg/lane protein was
separated by 12% SDS-PAGE gels and transferred onto polyvinylidene
fluoride membranes. The membranes were blocked using 5% bovine
serum albumin (cat. no. 37520; Thermo Fisher Scientific, Inc.) in
Tris-buffered saline with Tween-20 or with Triton X-100 and then
incubated with primary antibodies overnight at 4°C. The antibodies
of HDAC1 and H3K9Ac were the same as used for immunohistochemical
staining, dilution 1:500 together with rabbit anti-mouse klotho
antibody (cat. no. ab154163; 1:1,000; Abcam, Cambridge, MA, USA).
Subsequently, the membranes were incubated with horseradish
peroxidase (HRP)-conjugated secondary antibodies (goat anti-rabbit
IgG; cat. no. sc-2357; 1:200; Santa Cruz Biotechnology, Inc.;
rabbit anti-mouse β-actin antibody; cat. no. ab8227; 1:2,000;
Abcam) for 1 h in 37°C. The membranes were washed to remove unbound
antibodies and proteins were detected using ECL Immobilon Western
Chemiluminescent HRP Substrate (cat. no. WBKLS0500; EMD Millipore,
Billerica, MA, USA). The protein bands were visualized using a
Kodak XAR-5 film chemiluminescence imaging system (Sigma-Aldrich;
Merck KGaA). The protein levels were normalized to the reference
protein β-actin.
Chromatin immunoprecipitation
(ChIP)
ChIP reactions were performed using the
EZ-ChIP™ kit (EMD Millipore, Billerica, MA, USA),
according to the manufacturer's instructions. Briefly, the mouse
kidney samples were fixed in 1% paraformaldehyde for 30 min at 37°C
and then 125 mM glycine was added for 10 min at room temperature to
terminate the cross-linking. Ultrasonication was used to fragment
the DNA into 200–1,000 bp chromatin fragments. The sonicator was
adjusted to 30% intensity and 2 mm pulse wave, 50 watts of power.
All samples received 3 sonications for 10 sec each, 15 sec apart.
The chromatin fragments were incubated overnight at 4°C with
primary antibodies (Rabbit anti-mouse H3K9Ac; cat. no. 9649; 1:100;
Cell Signaling Technology, Inc.). Then, Pierce™ Protein
A/G Plus Agarose (Thermo Fisher Scientific, Inc.) was added to the
solution to obtain immunoprecipitated chromatin for PCR analysis.
The PCR conditions were as follows: Denaturation at 95°C for 30
sec, annealing at 55°C for 30 sec and extension at 72°C for 30 sec
for 30 cycles. The ChIP PCR KL promoter primers were as
follows: Forward, 5′-CTCAGGATGGAGGCCACAGG-3′ and reverse,
5′-CACAGCAGGGGTCATAGGGA-3′. The amplified product was visualized on
a 1.2% agarose gel. The ChIP PCR product was on the KL
promoter region from −215 to −134 bp.
Statistical analysis
Statistical analyses were performed using GraphPad
Prism software version 5.0 (GraphPad Software, Inc., La Jolla, CA,
USA) or SPSS software version 17.0 (SPSS, Inc., Chicago, IL, USA).
The data are presented as the mean ± standard error. Differences
among multiple groups were evaluated using one-way analysis of
variance and differences between two groups were evaluated using
Student's t test. Correlation between two continuous variables was
determined using Pearson's correlation coefficient. P<0.05 was
considered to indicate a statistically significant difference.
Results
High aldosterone levels downregulate
KL and upregulate fibronectin 1 (Fn1) in mice with CKD
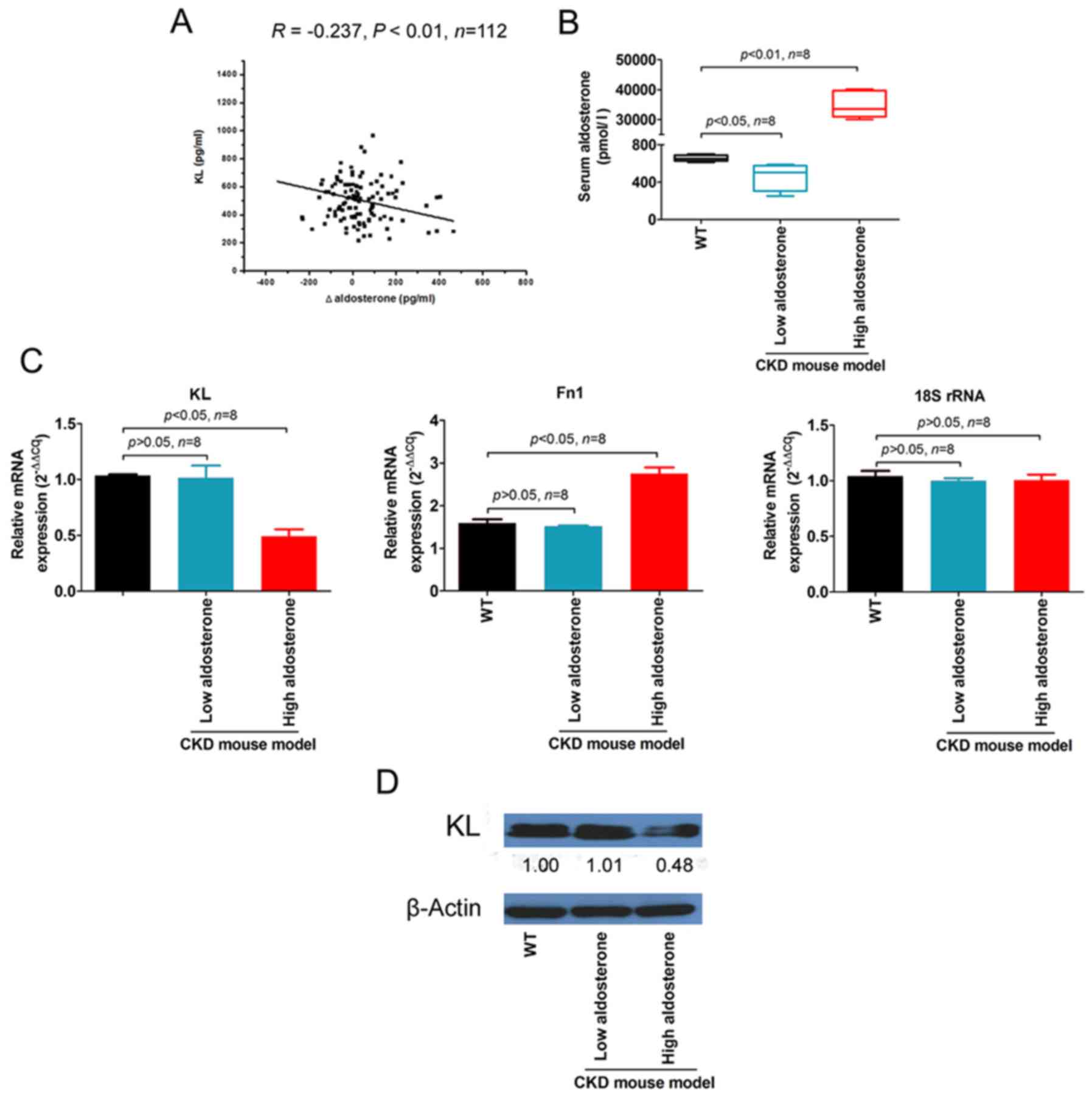
Blood samples were collected from 112 patients with
CKD, and the serum KL and aldosterone concentrations were
determined. Correlation analysis indicated that the serum ∆
aldosterone at 1.5-years follow-up was significantly negatively
correlated with serum KL (R=−0.237, P<0.01; Fig. 1A).
In the aldosterone-induced CKD mouse model,
aldosterone levels in the peripheral blood of mice in the high
aldosterone group (34,702.01±4,307.62 pmol/l) were significantly
higher compared with the low aldosterone (462.19±148.87 pmol/l) and
wild-type (WT) groups (656.11±32.76 pmol/l; Fig. 1B).
qPCR measurement of KL and Fn1
demonstrated that KL mRNA expression levels were
significantly lower, whereas Fn1 expression levels were
significantly higher in the renal tubules of high aldosterone mice
compared with low aldosterone and WT mice (Fig. 1C). The western blot results were
consistent and revealed a significantly lower KL protein expression
in the kidneys of high aldosterone mice compared with low
aldosterone and WT mice (Fig. 1D).
These results suggested that KL expression was inversely
proportional to aldosterone release and renal fibrosis.
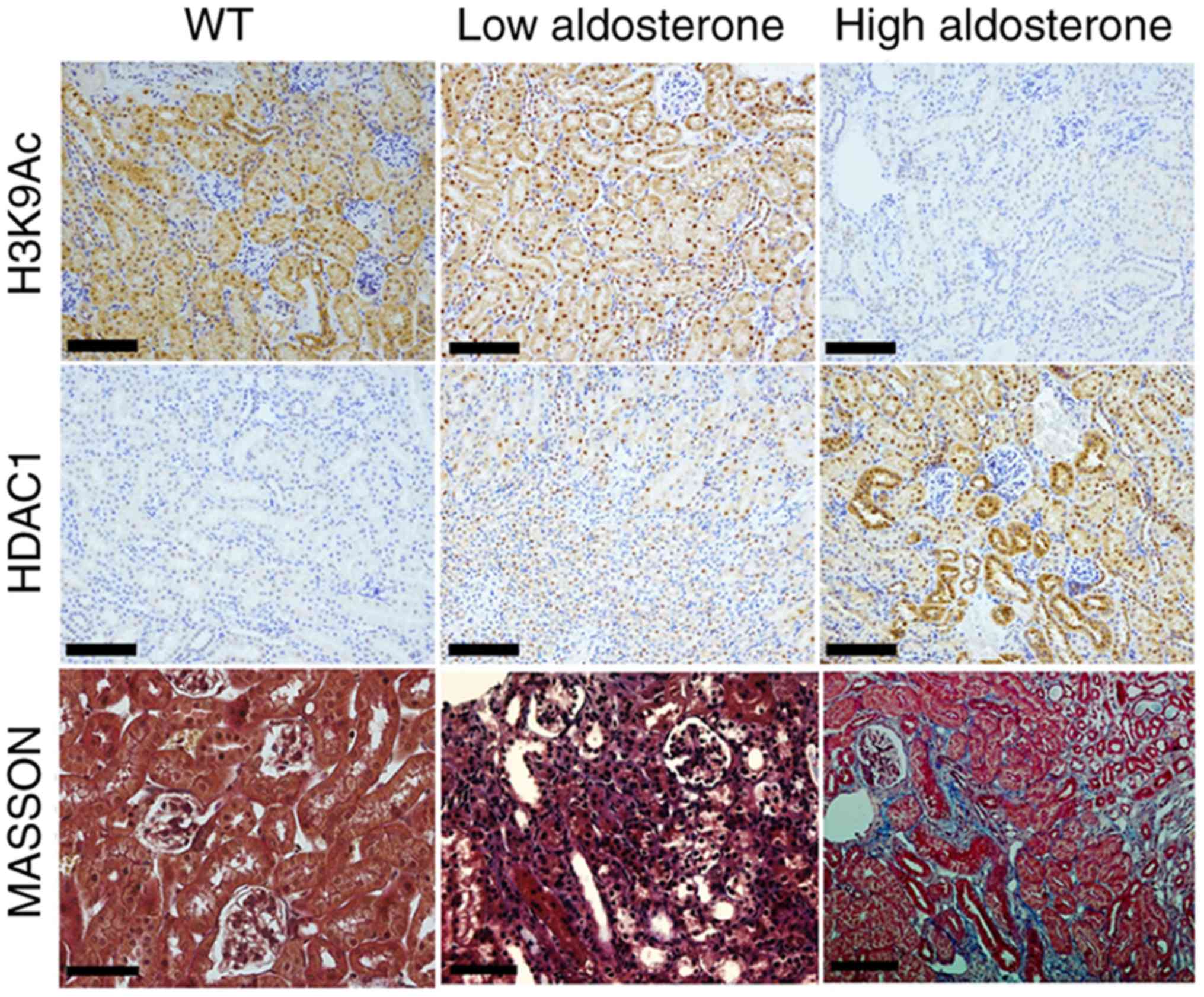
High aldosterone levels induce histone
deacetylation in the distal convoluted tubules of mice with
CKD
Immunohistochemical staining revealed that HDAC1
expression was absent in the distal convoluted tubules of WT mice
and HDAC1 expression was weak in low aldosterone mice. However,
HDAC1 expression was markedly increased in high aldosterone mice.
In addition, H3K9Ac was strongly expressed in the distal convoluted
tubules of WT and low aldosterone mice, whereas H3K9Ac staining was
absent in high aldosterone mice (Fig.
2). Western blotting demonstrated that HDAC1 was highly
expressed and H3K9Ac was rarely modified in the kidneys of high
aldosterone mice (Fig. 3A). The
results indicated that high aldosterone levels induced renal
injury, resulting in high HDAC1 expression in the distal convoluted
tubules and H3K9 deacetylation.
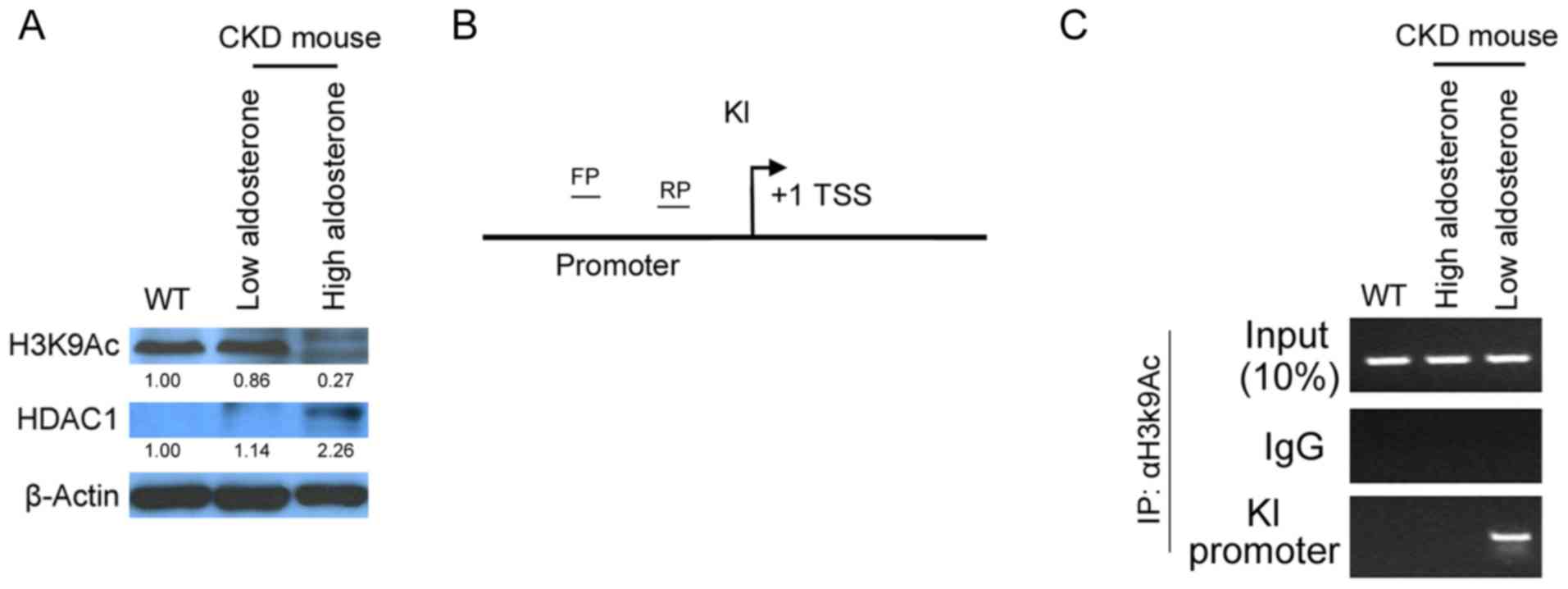 | Figure 3.High aldosterone inhibits KL
gene transcription in the distal convoluted tubules of CKD mice.
(A) Western blot analysis revealed high HDAC1 protein expression
levels and low H3K9Ac expression levels in the distal convoluted
tubules of high aldosterone mice. (B) Schematic diagram
demonstrating ChIP PCR verification of H3K9Ac binding to the
KL gene promoter region, to determine transcriptional
activation of the KL gene. (C) ChIP qPCR revealed presence
of H3K9Ac at the KL promoter in kidney of the low
aldosterone mice. The KL promoter binding product was absent
in high aldosterone mice. ChIP PCR, chromatin immunoprecipitation
polymerase chain reaction; WT, wild-type; CKD, chronic kidney
disease; H3K9Ac, acetylated H3K9; HDAC1, histone deacetylase 1; FP,
forward primer; RP, reverse primer; KL, klotho; TSS; transcription
start site; IgG, immunoglobulin G. |
High aldosterone levels inhibit
transcription of the KL gene in the distal convoluted tubules of
mice with CKD
To determine the transcriptional activation status
between KL and H3K9Ac, ChIP-PCR was used to confirm the binding of
the promoter region of KL gene to H3K9Ac site. The binding
site of the KL promoter region is between 215 and −134 bp
(Fig. 3B). The results showed that
the binding product of KL promoter was obtained in the
H3K9Ac site in the distal convoluted tubules of normal mice and low
aldosterone mice (Fig. 3C).
However, in high aldosterone mice there was rarely a binding
product of KL promoter at the H3K9Ac site. Thus, the results
demonstrated that high aldosterone inhibits the acetylation of the
H3K9 site, leading to its inability to bind to the KL
promoter and ultimately inhibits the transcription of the KL
gene.
Discussion
Several studies have reported that high aldosterone
can induce renal fibrosis (5) and
that KL downregulation promotes CKD fibrosis progression (11). However, the mechanism that leads to
KL downregulation when aldosterone is high remains unclear.
In the present study, KL expression in a high
aldosterone-induced CKD mouse model was investigated. KL expression
was significantly downregulated by high aldosterone at the mRNA and
protein level. The significant differences between the KL
mRNA expression levels of the high aldosterone and control groups
suggested that aldosterone may regulate KL transcriptional
activity to induce KL mRNA and protein expression.
In addition, it was identified that histone
acetylation was different between the high aldosterone and control
groups. The results demonstrated that the H3K9 locus was
deacetylated in the renal tissues of the high aldosterone group.
According to epigenetic theory, histone acetylation is necessary to
promote transcriptional activation of genes and is maintained by
histone acetyltransferases (HAT). Conversely, gene transcription is
inhibited when the specific lysine residue at the end of the
histone undergoes deacetylation. In general, histone acetylation
causes DNA and histone octamer dissociation and nucleosome
structure relaxation, enabling gene transcription activation by the
specific binding between DNA binding sites and various
transcription factors or synergistic transcription regulators.
Deacetylated transferases have the opposite effect. In the nucleus,
histone acetylation and deacetylation are in dynamic balance and
coordinated by HATs and HDACs. HATs transfer the acetyl group of
acetyl coenzyme A to the amino-terminal lysine residue of the
histone, whereas HDAC deacetylates the histone, binds closely to
the negatively charged DNA, yielding a dense structure, which
inhibits gene transcription. Based on the above theory, HDAC1
expression was detected in the current study. HDAC1 expression was
significantly increased in the kidney tissues of high aldosterone
mice. Based on this result, it can be hypothesized that histone
deacetylation in the renal tissue of mice with CKD may inhibit
transcription of the KL gene during high aldosterone levels.
Subsequently, ChIP was used to analyze the acetylation of the H3K9
site associated with the KL gene promoter. The experimental
demonstrated deacetylation of the H3K9 site associated with the
KL gene promoter under high aldosterone.
In conclusion, the present study demonstrated that
high aldosterone increased HDAC1 expression levels in the distal
convoluted tubules in CKD mice, leading to histone H3K9
deacetylation, inhibition of KL gene transcription. As a
previous study demonstrated the role of increasing aldosterone in
renal damage (31), the above
results may explain why klotho decreased in CKD and may help lead
to renal fibrosis.
Acknowledgements
The authors would like to thank Ms. Huizhu Shi
(Department of Nephrology, Huashan Hospital, Fudan University) for
her assistance with collection of patient samples.
Funding
The present study was supported by National Natural
Science Foundation of China (grant no. 81670697).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
JX and LL designed the study and participated in
revising the manuscript. LL and YG followed-up the patient cohort
in the CKD clinic. LL, MY and PC performed the experiments and
statistical analyses. LL drafted the manuscript. All authors read
and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of Huashan Hospital, Fudan University (Shanghai, China).
All provided written informed consent for participation and the
study was conducted in accordance with the Declaration of
Helsinki.
Patient consent for publication
All patients in this cohort agreed their data for
publication.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Allison SJ: Chronic kidney disease: The
effect of age on CKD outcomes. Nat Rev Nephrol. 9:32013. View Article : Google Scholar
|
|
2
|
Hallan SI, Matsushita K, Sang Y, Mahmoodi
BK, Black C, Ishani A, Kleefstra N, Naimark D, Roderick P, Tonelli
M, et al: Age and association of kidney measures with mortality and
end-stage renal disease. JAMA. 308:2349–2360. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kovesdy CP, Alrifai A, Gosmanova EO, Lu
JL, Canada RB, Wall BM, Hung AM, Molnar MZ and Kalantar-Zadeh K:
Age and outcomes associated with BP in patients with incident CKD.
Clin J Am Soc Nephrol. 11:821–831. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tangri N, Komenda P and Rigatto C: Age and
outcomes in CKD. Am J Kidney Dis. 62:225–227. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lai L, Chen J, Hao CM, Lin S and Gu Y:
Aldosterone promotes fibronectin production through a
Smad2-dependent TGF-beta1 pathway in mesangial cells. Biochem
Biophys Res Commun. 348:70–75. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Brown NJ: Contribution of aldosterone to
cardiovascular and renal inflammation and fibrosis. Nat Rev
Nephrol. 9:459–469. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Herrada AA, Contreras FJ, Marini NP,
Amador CA, González PA, Cortés CM, Riedel CA, Carvajal CA, Figueroa
F, Michea LF, et al: Aldosterone promotes autoimmune damage by
enhancing Th17-mediated immunity. J Immunol. 184:191–202. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Huby AC, Antonova G, Groenendyk J,
Gomez-Sanchez CE, Bollag WB, Filosa JA and Belin de Chantemèle EJ:
Adipocyte-derived hormone leptin is a direct regulator of
aldosterone secretion, which promotes endothelial dysfunction and
cardiac fibrosis. Circulation. 132:2134–2145. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mayyas F, Alzoubi KH and Van Wagoner DR:
Impact of aldosterone antagonists on the substrate for atrial
fibrillation: Aldosterone promotes oxidative stress and atrial
structural/electrical remodeling. Int J Cardiol. 168:5135–5142.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
McGraw AP, Bagley J, Chen WS, Galayda C,
Nickerson H, Armani A, Caprio M, Carmeliet P and Jaffe IZ:
Aldosterone increases early atherosclerosis and promotes plaque
inflammation through a placental growth factor-dependent mechanism.
J Am Heart Assoc. 2:e0000182013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang M, Chen J, Lai L, You L, Lin S, Hao
C and Gu Y: Aldosterone promotes fibronectin synthesis in rat
mesangial cells via ERK1/2-stimulated Na-H+ exchanger isoform 1. Am
J Nephrol. 31:75–82. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bleskestad IH, Thorsen IS, Jonsson G,
Skadberg Ø, Bergrem H and Gøransson LG: Soluble Klotho and intact
fibroblast growth factor 23 in long-term kidney transplant
patients. Eur J Endocrinol. 172:343–350. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Haruna Y, Kashihara N, Satoh M, Tomita N,
Namikoshi T, Sasaki T, Fujimori T, Xie P and Kanwar YS:
Amelioration of progressive renal injury by genetic manipulation of
klotho gene. Proc Natl Acad Sci USA. 104:2331–2336. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lu X and Hu MC: Klotho/FGF23 axis in
chronic kidney disease and cardiovascular disease. Kidney Dis
(Basel). 3:15–23. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Drew DA, Katz R, Kritchevsky S, Ix J,
Shlipak M, Gutiérrez OM, Newman A, Hoofnagle A, Fried L, Semba RD
and Sarnak M: Association between soluble Klotho and change in
kidney function: The health aging and body composition study. J Am
Soc Nephrol. 28:1859–1866. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lin W, Li Y, Chen F, Yin S, Liu Z and Cao
W: Klotho preservation via histone deacetylase inhibition
attenuates chronic kidney disease-associated bone injury in mice.
Sci Rep. 7:461952017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Park MY, Herrmann SM, Saad A, Eirin A,
Tang H, Lerman A, Textor SC and Lerman LO: Biomarkers of kidney
injury and klotho in patients with atherosclerotic renovascular
disease. Clin J Am Soc Nephrol. 10:443–451. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ritter CS, Zhang S, Delmez J, Finch JL and
Slatopolsky E: Differential expression and regulation of Klotho by
paricalcitol in the kidney, parathyroid, and aorta of uremic rats.
Kidney Int. 87:1141–1152. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang Q, Liu L, Lin W, Yin S, Duan A, Liu
Z and Cao W: Rhein reverses Klotho repression via promoter
demethylation and protects against kidney and bone injuries in mice
with chronic kidney disease. Kidney Int. 91:144–156. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhou L, Mo H, Miao J, Zhou D, Tan RJ, Hou
FF and Liu Y: Klotho ameliorates kidney injury and fibrosis and
normalizes blood pressure by targeting the Renin-Angiotensin
system. Am J Pathol. 185:3211–3223. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Brocks D, Schmidt CR, Daskalakis M, Jang
HS, Shah NM, Li D, Li J, Zhang B, Hou Y, Laudato S, et al: DNMT and
HDAC inhibitors induce cryptic transcription start sites encoded in
long terminal repeats. Nat Genet. 49:1052–1060. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cantley MD, Zannettino ACW, Bartold PM,
Fairlie DP and Haynes DR: Histone deacetylases (HDAC) in
physiological and pathological bone remodelling. Bone. 95:162–174.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Guerriero JL, Sotayo A, Ponichtera HE,
Castrillon JA, Pourzia AL, Schad S, Johnson SF, Carrasco RD, Lazo
S, Bronson RT, et al: Class IIa HDAC inhibition reduces breast
tumours and metastases through anti-tumour macrophages. Nature.
543:428–432. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hull EE, Montgomery MR and Leyva KJ: HDAC
inhibitors as epigenetic regulators of the immune system: Impacts
on cancer therapy and inflammatory diseases. Biomed Res Int 2016.
87972062016.
|
|
25
|
Kwon DY, Zhao YT, Lamonica JM and Zhou Z:
Locus-specific histone deacetylation using a synthetic
CRISPR-Cas9-based HDAC. Nat Commun. 8:153152017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Thoma C: Kidney cancer: Combination of
HDAC inhibitor with IL-2 promising. Nat Rev Urol. 14:6392017.
View Article : Google Scholar
|
|
27
|
Inker LA, Astor BC, Fox CH, Isakova T,
Lash JP, Peralta CA, Kurella Tamura M and Feldman HI: KDOQI US
commentary on the 2012 KDIGO clinical practice guideline for the
evaluation and management of CKD. Am J Kidney Dis. 63:713–735.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Levey AS and Stevens LA: Estimating GFR
using the CKD Epidemiology Collaboration (CKD-EPI) creatinine
equation: More accurate GFR estimates, lower CKD prevalence
estimates, and better risk predictions. Am J Kidney Dis.
55:622–627. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang B, Ding W, Zhang M, Li H and Gu Y:
Rapamycin attenuates aldosterone-induced tubulointerstitial
inflammation and fibrosis. Cell Physiol Biochem. 35:116–125. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Shavit L, Lifschitz MD and Epstein M:
Aldosterone blockade and the mineralocorticoid receptor in the
management of chronic kidney disease: Current concepts and emerging
treatment paradigms. Kidney Int. 81:955–968. 2012. View Article : Google Scholar : PubMed/NCBI
|