Introduction
HIV continues to be an important challenge and a
major global public health issue. According to the last WHO report,
there were approximately 36.7 million individuals living with HIV,
with 1.0 million individuals succumbing to HIV-related causes and
1.8 million newly infected ones at the end of 2016, globally
(1). Despite global policies and
the general advice to treat every HIV patient, only 54% of adults
and 43% of children living with HIV are currently receiving
antiretroviral therapy (1).
The HIV treatment regimen is termed highly active
antiretroviral therapy (HAART) and its main goal is to suppress HIV
replication and reduce viral loads (VLs) below the detectable
level. In fact, it has been proven that suppression of HIV
replication improves life expectancy and quality of life (2,3). The
second aim of HAART is immune recovery. Both effects are essential
for any HIV drug: Failing an acceptable immune recovery is one of
the leading causes for comorbidities in HIV-infected patients,
because of a constant pro-inflammatory status (4,5) that
leads to several chronic diseases including diabetes mellitus,
chronic kidney dysfunction, cardiovascular diseases and cancer
(4–30).
Over 25 drugs and their combinations have been
approved for clinical use, however, the optimal drug regimen has
yet to be identified. HAART is a long-life effective treatment.
Available drugs can achieve undetectable VL and immune restoration
but co-morbidities, adverse effects (AEs), drug interactions and
insurgence of resistance are unsolved problems. Several drugs have
been studied to resolve these problems. Among them there are new
possible components of the ‘entry and fusion inhibitors’ class. All
the drugs of the ‘entry and fusion inhibitors’ class share some
peculiar characteristic that could represent an advance in HIV
control. They are the only drugs that prevent entry of the virus
into the human cells. This mechanism of action may prevent the
infection of new cells during the rounds of ongoing replication
that are present also in patients on HAART and which is thought to
be the cause of refueling of the HIV reservoir (31). These drugs potentially play a role
in the prevention of new infections through the Pre-exposure
prophylaxis (PrEP) and in multidrug-resistant patients.
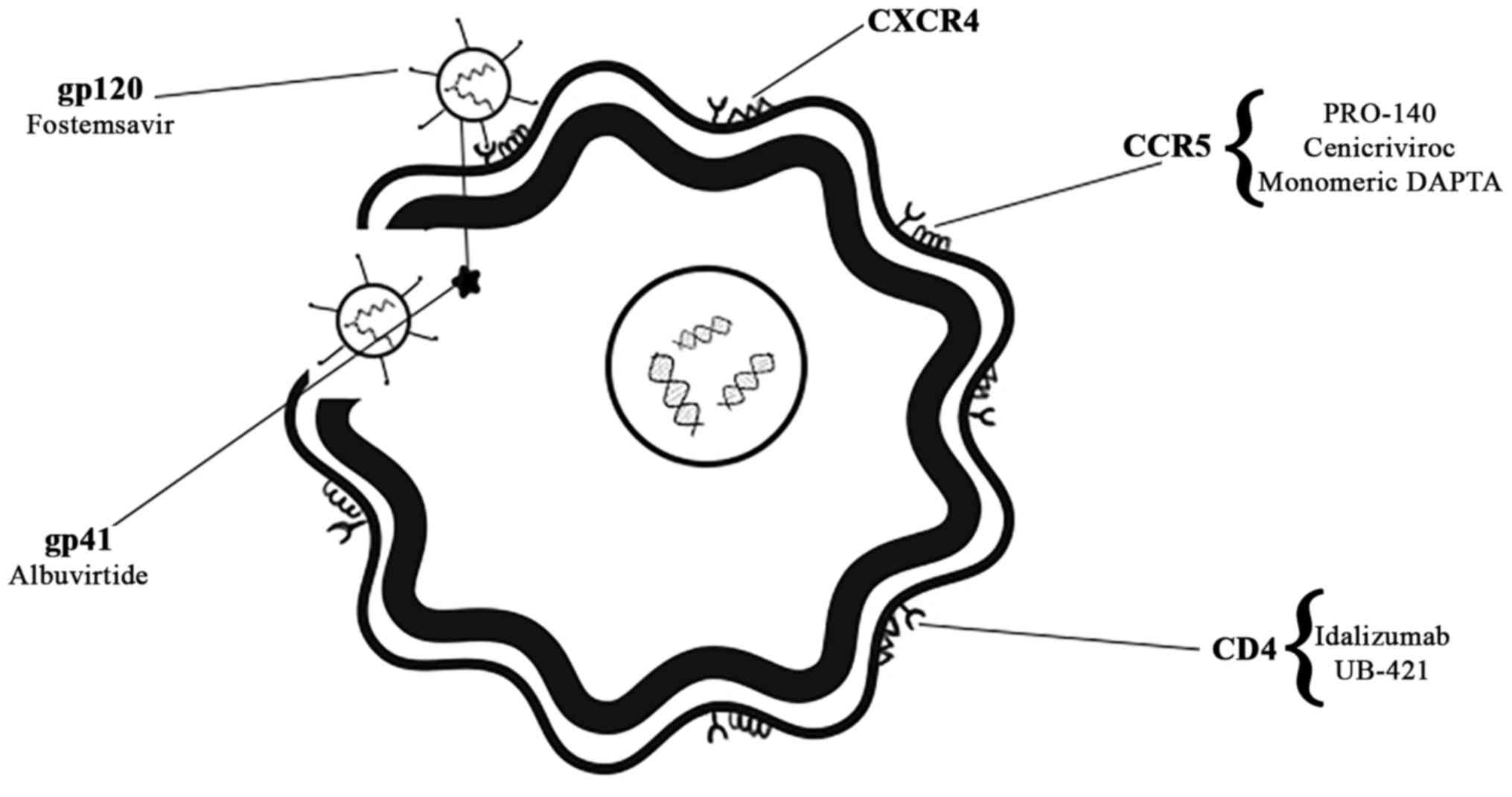
The very first steps of the infectious cycle of HIV
are attachment, fusion and entry of viral particles in the human
cells. During this phase, HIV glycoproteins such as gp120, and
gp41, play a crucial role (32).
The envelope protein gp120 binds the CD4 receptor on the host cell
surface, starting a cascade of conformational changes in gp120 that
exposes the chemokine receptor binding domains and allows them to
interact with the target receptor. The main co-receptors used by
HIV-1 for entry into the cell are the chemokine receptors CCR5 and
CXCR4. Tropism, or binding with the co-receptors, is so important
that HIV-1 is classified either exclusively using CCR5 (R5) or
CXCR4 (X4), or using both co-receptors (R5X4 or dual-tropic). Once
gp120 is bound with the CD4 protein and the co-receptor, the
N-terminal fusion peptide gp41 penetrates the cell membrane and the
loop structure, formed by gp120, CD4 and co-receptor, allowing the
fusion of the viral and host membranes. Subsequently, entry of the
viral capsid occurs (33).
All the chemical compounds that act against these
events have been grouped in the ‘entry and fusion inhibitors’
class. They are further sub-classified depending on their target
in: CD4-receptor inhibitors, co-receptor antagonists (CCR5 and
CXCR4), and fusion inhibitors (34).
At present, only two drugs of this class have been
approved: Μaraviroc, a CCR5 antagonist, and enfuvirtide, a gp41
antagonist. Fig. 1 presents the
drugs currently studied and their targets, while Table I focuses on the study phase the
drugs are currently in.
 | Table I.Entry and fusion inhibitors |
Table I.
Entry and fusion inhibitors
| Drug | Target | Study phase | Clinical
studies |
|---|
| Fostemsavir | gp120 | III | BRIGHTE study
(ongoing) |
| Ibalizumab | CD4 (dominio
2) | III | NCT00784147
(complete) |
| Albuvirtide | gp41 | III | TALENT study
(complete, unpublished results) |
| UB-421 | CD4 (dominio
1) | III | NCT03149211
(ongoing) |
| NCT01668043
(complete) |
| PRO-140 | CCR5 | III | NCT02483078
(ongoing) |
| NCT02355184
(ongoing) |
| NCT02990858
(ongoing) |
| NCT02859961
(ongoing) |
| Cenicriviroc | CCR5 e CCR2 | IIb | NCT02128828
(complete) |
| Monomeric
DAPTA | CCR5 | II | NCT00000392
(complete) |
| Combinectin | gp120 e gp41 | Pre-clinical | --- |
| Vedolizumab | α4β7 integrin | Pre-clinical | --- |
In this paper we reviewed the articles, clinical
trials and conference communications with regard to all the drugs
belonging to the class of entry and fusion inhibitors that are at
least in phase I clinical trials.
CCR5 antagonist
PRO-140
PRO-140 is a humanized IgG4 monoclonal antibody
(mAb), that binds to hydrophilic extracellular domains on CCR5 and
competitively inhibits viral entry of HIV-1 (35,36).
It is used only in patients with an R5-type HIV virus (36,37).
At antiviral concentrations, PRO-140 does not prevent CC-chemokine
signaling (35,36).
Notably, in vitro PRO-140 has exhibited
activity against viruses resistant to maraviLaroc, the only CCR5
antagonist approved for HIV treatment [Jacobson et al
(37): CCR5 monoclonal antibody
PRO 140 inhibited HIV-1 resistant to maraviroc, a small molecule
CCR5 antagonist. International AIDS Conference, Mexico City 2008,
abs TUAA0305].
When a virogical failure (VF) occurs, no change in
co-receptor tropism and no significant change in virus
susceptibility to PRO-140 and maraviroc have been reported
(38) [Lalezari et al: PRO
140 single-agent maintenance therapy for HIV-1 infection: A 2-year
update. Conference on retroviruses and opportunistic infections
(CROI), February 13–16, 2017, Seattle, abs 437].
Currently, PRO-140 appears to be a well-tolerated
drug. No drug-related serious adverse effects (SAEs),
discontinuations because of AEs, or any notable patterns of
toxicity have been previously reported. The most common AE was a
mild, transient, and self-resolving injection site reaction that
has occurred in less than 10% of participants (38,39).
In a randomized, double-blind, placebo-controlled
dose-ranging phase 2a study, Jacobson et al reported that
subcutaneous PRO-140 demonstrated potent and prolonged
antiretroviral activity (35).
Authors of that study recruited adults affected by only CCR5-tropic
virus at screening, with a mean age of 42.4 standard deviation (SD)
± 7.09 years. Inclusion criteria were: Plasma HIV-1 RNA ≥5,000
copies/ml, CD4 + T cell ≥300 cells/mm3 (with a nadir
>200 cells/mm3) and no history of AIDS-defining
illnesses. The 44 participants did not receive antiretroviral
therapy for ≥12 weeks and were randomized into one of four
treatment groups: Placebo, 162 mg subcutaneously once a week, 324
mg subcutaneously once a week, and 324 mg subcutaneously twice a
week.
The reduction for the PRO-140 groups was
statistically significant relative to the placebo group. The best
performances were reached in the 324 mg weekly group in which 73%
of subjects had a VL of <400 copies/ml while no placebo subject
had such a VL (P=0.001). The mean maximum reduction in HIV-1 RNA
observed for the 324 mg weekly dose was 1,65 log. Individual viral
nadirs were typically observed on day 22 (range, day 15–29).
Low-titred anti-PRO-140 antibodies (1:32 or less)
were detected in three subjects treated with the 324 mg weekly
dose.
After these proof-of-concept studies, two different
strategies of treatment with PRO-140 were evaluated in phase
IIb/III clinical trials.
The study ‘A randomized, double-blind,
placebo-controlled trial, followed by single-arm treatment of PRO
140 in combination with optimized background therapy (OBT) in
treatment-experienced HIV subjects’ (NCT02483078; https://clinicaltrials.gov/ct2/show/NCT02483078?term =
NCT02483078&rank =1), evaluated the possibility of adding this
new drug to an OBT, chosen on the basis of a subject's resistance
test results and treatment history, in treatment-experienced
patients with a virologic failure. After one week of overlap with
existing ART and PRO-140 (350 mg SC weekly) all the patients
received PRO-140 and an OBT, while in the second arm, PRO-140 is
due to be substituted with a placebo.
The study ‘PRO 140 SC as single agent maintenance
therapy in virally suppressed subjects with CCR5-tropic HIV-1
infection’ (NCT02859961; https://clinicaltrials.gov/ct2/show/NCT02859961?
term = NCT02859961 & rank =1) is also ongoing. It is a
multi-center study designed to evaluate the efficacy, safety, and
tolerability of the strategy of shifting clinically stable patients
receiving suppressive combination antiretroviral therapy to PRO-140
monotherapy (350 mg sc weekly) and maintaining viral suppression
for 48 weeks following study entry. Consenting patients are to be
shifted from a combination antiretroviral regimen to weekly PRO-140
monotherapy for 48 weeks during the treatment phase with the one
week overlap of existing retroviral regimen and PRO-140 at the
beginning of the study treatment and also one week overlap at the
end of the treatment in subjects that do not experience virologic
failure.
This strategy should be innovative and noteworthy
for stable patients, improving their quality of life and their
adherence to treatment. PRO-140 drug interactions are currently
unknown.
Cenicriviroc
Cenicriviroc (CVC) is a small molecule that binds to
a domain of CCR5 and subsequently blocks HIV-1 entry inhibiting
interaction between HIV-1 gp120 and CCR5 (40). CVC may have an anti-inflammatory
effect as it is also a CCR2 antagonist (41).
Cenicriviroc has a plasma half-life of approximately
35 h and may be used once daily (42).
CVC, administered orally and once daily,
demonstrated potent antiviral activity and good tolerance between
HIV-1-infected subjects, that were antiretroviral-experienced, and
naive to CCR5 antagonists (43).
CVC exhibits high levels of resistance in
vitro. Complete resistance to CVC developed after 67 weeks,
with several amino acid changes in both the V3 and other Env
regions (44).
A phase 2b, multicenter, randomized, double-blind,
double-dummy study evaluated, at 24 and 48 weeks, the proportion of
virologic success. It also compared the safety and tolerability of
two different doses of CVC (100 and 200 mg orally) with those of
EFV (42). Those authors used
TDF/FTC as a backbone for all the participants. The three arms
were: CVC 100 mg, 58 patients; CVC 200 mg, 57 patients; and EFV 28
patients.
The proportion of virologic success was similar in
all the treatment arms at week 24 (76, 73 and 71%) and week 48 (68,
64 and 50%). In addition, the rates of virologic non-response were
not significantly different in the treatment groups at week 48 (15,
20 and 11%).
None of the efavirenz-treated patients with VF had
emergent NRTI mutations. However, NRTI mutations (M184I and/or V)
accounted for 75% of the VFs with CVC 100 mg and for 33% of the VFs
with CVC 200 mg. One participant with VF who was in the 200 mg CVC
group had a tropism switch from R5 to X5R5 tropic virus.
The following percentages of AE, related to
treatment, were found (42): 50%
in the 100 mg CVC group, 44% in the 200 mg CVC group, and 71% in
the EFV group. The most frequent were: Nausea (12%), headache
(10%), diarrhea (7%), and abnormal dreams (7%). SAEs occurred in 1
participant in each group, accounting for 2, 2 and 4 of
patients.
CVC does not inhibit CYP-mediated activity and does
not induce CYP3A4 in human hepatocytes.
The interaction between CVC and ritonavir,
darunavir, atazanavir, efavirenz and dolutegravi [Lefebvre et
al, ‘Pharmacokinetics of cenicriviroc when administered with
and without ritonavir, darunavir/ritonavir or
atazanavir/ritonavir’, International Workshop on Clinical
Pharmacology of HIV Therapy, Amsterdam 2013, abs O_09A and O_09B]
has been previously described.
CVC may play a role in treatment-experienced
patients with a multiresistant R5 virus.
Monomeric DAPTA
Monomeric DAPTA (mDAPTA) is a synthetic compound
derived from the gp120 V-2 region of HIV. It is a selective CCR5
co-receptor antagonist that binds this co-receptor and subsequently
inhibits the interaction between HIV-1 gp120 and CCR5 (46–48).
In vitro, Agrawal et al demonstrated
that mDAPTA is 1,000-fold more potent than maraviroc in inhibiting
virus entry (49). In fact, it
completely inhibited the release of virus (X4 and R5) from PBMCs
from all the samples of HIV-uninfected and HIV-infected patients
with VL <50 copies/ml. The authors' disclosure was that mDAPTA
prevents HIV recovery and the production of replication-competent
HIV from CD8-depleted patient PBMCs.
An interesting phase I trial tested an intra-nasal
administration of mDAPTA 3 times a day for up to 32 weeks, given
either alone or in combination with current HAART. The mean VL did
not change in the study period, being 3.71 log copies/ml at
baseline and 3.85 log copies/ml at week 24. By contrast, a complete
suppression of active HIV replication in the circulating monocyte
(CD14) population was observed. Furthermore, the investigators of
that study were unable to isolate infectious virus from any plasma
sample (50). This fact indicates
a disconnection between PCR detection of HIV RNA and extremely
infectious plasma virus in these long-term non-progressor subjects.
Authors of that study observed an immune restoration with a mean
CD4+ cell count increase from 540 to 652 cell/μl.
A phase 2 study is still ongoing (NCT00951743;
https://clinicaltrials.gov/ct2/show/NCT00951743?term =
NCT00951743 & rank =1). This is a 24-week, double-blind,
two-arm study in which mDAPTA is compared to placebo. In total, 40
HIV-infected individuals in HAART and with VL <200 copies ml
were randomized to receive 0.01 mg of mDAPTA intranasally twice a
day or placebo.
In the phase 1 study (50) conducted neither AEs nor nasal
pathologies were reported.
No resistance or changes in tropism during treatment
was registered. No drug interactions are known.
gp120 antagonist
Fostemsavir
Fostemsavir is a pro-drug of temsavir (TMR) a
molecule that prevents viral entry by binding to the viral envelope
gp120 and interfering with virus attachment to the host CD4
receptor (51). TMR binds directly
to the virus and not human cells, and is active against R5, X4 and
R5X4 HIV-1 (51–55).
In a multiple-ascending dose study, the average
plasma half-life of TMR was 3.2–4.5 h (immediate-release
formulation) and 7–14 h (extended-release formulation) (Mascolini,
‘Levels of novel HIV attachment inhibitor with or without
Ritonavir’, 12 th International Workshop on Clinical Pharmacology
of HIV Therapy, Miami 2011).
After 8 days of monotherapy, fostemsavir achieved a
maximum median decrease in HIV-1 RNA from baseline of 1.21 to 1.73
log copies/ml (56).
Suboptimal efficacy of TMR was associated with the
presence of the M426L, S375, M434 and M475 substitutions (54,55).
No in vitro cross-resistance was observed
with other classes of antiretrovirals (52,53),
with any other entry inhibitors (ibalizumab and enfuvirtide).
In phase 2b studies TMR showed similar efficacy to a
ritonavir-boosted atazanavir (ATV/r) using tenofovir disoproxil
fumarate (TDF) and raltegravir (RAL) as companions (42,57).
Specifically, Thompson et al showed that, through week 48,
the proportion of fostemsavir subjects with a VL <50 copies/ml
was 77–95% versus 88% for ATV/r subjects (42). A difference occurred in virologic
response rates according to the baseline VL. The virologic response
was 74–100% with a baseline VL <100,000 copies/ml VL versus
60–91% in subjects with ≥100,000 copies/ml. Across fostemsavir
arms, median CD4 T cell count increases from baseline were 145–186
cells/μl and similar to the ATV/r arm (142
cells/μl).
The Brighte study (NCT02362503; https://clinicaltrials.gov/ct2/show/NCT02362503?term =
NCT02362503 & rank =1), a phase 3 study, is ongoing and aims to
evaluate whether fostemsavir is an optimal option in heavily
treated experienced adults with limited therapeutic options (≤2
classes of active antiretrovirals remaining). After several phase 2
studies the dosage selected for this clinical trial was 600 mg
tablets orally twice daily. All the patients also received an
OBT.
Grade 2 to 4 treatment-related AEs occurred in
8.5–18% of patients, the most common of which were nausea,
diarrhea, headache, vomiting, fatigue, and asthenia (42,58).
SAEs occurred in 0–2% of patients (42,58).
gp41 antagonists
Albuvirtide
Albuvirtide (ABT) is a peptide derived from gp41
that inhibits the formation of a six-helix bundle structure in
gp41, which is necessary for fusion of the viral and cellular
membranes (59).
The first attempt of combination therapy using ABT
and available drugs led to an open-label, randomized, parallel
phase 2 trial (60). Naïve
patients with a VL >5,000 copies/ml and a CD4+ cell count
>350 cells/μl, were randomized to receive an intravenous
infusion of 160 or 320 mg weekly and lopinavir/ritonavir (LPV/r)
400/100 mg twice daily. At the 47 th day the mean VL decrease was
1.91 log and 2.20 log, while the CD4+ cell count change was −5
cells/μl and 52 cells μl for the 160 and 320 mg
groups, respectively.
The TALENT study, a phase 3 trial, is ongoing
(NCT02369965; http://clinicaltrials.gov/ct2/show/NCT02369965).
Participants are children and adults (aged 16–60 years) with HIV
who have had treatment failure with a standard first-line ART
regimen containing NRTIs or NNRTIs. VL of ≥1,000 copies/ml at
baseline was observed. All the patients received LPV/r and were
randomized to receive ABT (320 mg i.v. weekly) or tenofovir and
lamivudine. An interim analysis (61), showed that, at week 48, 80.4% of
the patients in ABT arm had VL <50 copies/ml versus 66% of the
patients in the other arm. The CD4+ cell count was similar for both
arms.
The most common AEs reported were: Diarrhea (7.5%),
headache (2.2%), and dizziness (2.2%) (61). SAEs occurred in 5.6% of
participants in a phase 3 trial. No injection site reaction has
been reported. ABT does not affect serum cretinine or eGFR levels
but high cholesterol (12.9%) and high triglycerides (32.3%) were
reported as laboratory abnormalities [Xie (61) International congress of drug
therapy in HIV infection, Glasgow 2016].
In Glasgow, Xie (61) presented an interim analysis of the
TALENT study showing that mutations at amino acid positions 36, 40,
126 and 144 could represent mechanism of resistance for ABT.
In patients treated with LPV/r and ABT, a decrease
of LPV/r exposure was identified. However, this decrease did not
lead to a change in the usual doses of these drugs (62). No other drug interactions are known
at present.
An NDA was accepted by China FDA in July 2016 for
this drug that could become the first long-acting anti-HIV new
drug.
CD4 antagonists
Ibalizumab
Ibalizumab is a humanized monoclonal antibody (mAb)
that acts by binding to the interface between domains 1 and 2 of
the CD4 receptor (63). The
post-binding conformational effects caused by this interaction
prevent viral entry and fusion (64). Unlike other monoclonal antibodies
that, targeting domain 1 of CD4, have been found to be
immunosuppressive (65,66), ibalizumab has no immunosuppressive
effects (67,68), because its binding site to CD4
receptors is distant from the binding site of MHC II molecules
(63).
Ibalizumab has an average half-life of 3–3.5 days
when administered subcutaneously (69), and can be administered once a week.
Additionally, an intramuscular administration is being evaluated
(Lin et al: ‘Intramuscular ibalizumab: Pharmacokinetics,
safety, and efficacy vs iv administration’ CROI, Seattle 2017, abs
438). Owing to the limited data, optimal dosage and route of
administration remain to be verified even if the best results have
been obtained with an intravenous administration of 25 mg/kg every
two weeks (70). The first
evidence of a good antiviral effect of ibalizumab in humans was
reported among 30 HIV-positive patients in ART with virologic
failure and a VL of >5,000 copies/ml (70). Those subjects that received 25
mg/kg of ibalizumab had a peak mean reduction of 1.11 log on day 21
after a single dose. The increased peak of CD4+ cells was on day 1
suggesting that an increase may have been due to the redistribution
of CD4 cells from lymphoid tissue rather than an immune
restoration. Khanlou et al confronted ibalizumab and an
optimized background regimen (OBR) versus an OBR alone in
multi-resistant HIV patients (Khanlou et al: ‘Safety,
efficacy and pharmacokinetics of ibalizumab in
treatment-experienced HIV-1 infected patients: Α phase 2b study.’
51 st ICAAC Chicago 2011, abs H2-794b). In this phase 2b trial
ibalizumab was administered intravenously in two doses: 800 mg
every 2 weeks or 2,000 mg every 4 weeks. In the ibalizumab arms,
the VLs showed a significant decrease (1.5–1.6 log copies/ml) while
the mean increase in the CD4 T cell count after 24 weeks was of
37–40 cells/μl. Norris et al, demonstrated that, in
treatment-experienced patients with a multiresistant virus,
ibalizumab added to OBR had a better performance than the OBR
alone. In ibalizumab plus OBR arm the subjects obtained a peak mean
reduction of 0.95–1.16 log versus 0.2 log of the placebo plus OBR
group [Norris, ‘TNX-355 in combination with optimized background
regimen (OBR) exhibits greater antiviral activity than OBR alone in
HIV treatment experienced patients’ Interscience Conference on
Antimicrobial Agents and Chemotherapy, Washington 2005, abs LB-26].
In another phase I trial (69),
ibalizumab was tested as a monotherapy for 9 weeks in subjects with
a VL >5,000 copies/ml and a CD4+ cell count between
100–500/μl. Jacobson et al, chose three different
schedules: 10 mg/kg once a week, 10 mg/kg loading dose followed by
6 mg/kg every 2 weeks, and 25 mg/kg every 2 weeks (69). Almost all the subjects exhibited a
decrease of VL between 0.5 and 1.7 log copies/ml with a peak within
the first week. The VLs returned to baseline values by the end of
treatment. In all the study arms, after an initial increase in CD4+
cell counts, all the subjects returned to the baseline CD4+ cell
count, reinforcing the idea that a redistribution of CD4 cells from
lymphoid tissue is the real cause of this rapid peak, as suggested
by Kuritzkes et al (70).
Moreover, reduced susceptibility to ibalizumab
relative to baseline appeared in 93% of the subjects after the
treatment. This finding discouraged the use of ibalizumab in
monotherapy.
The preliminary results of a phase III trial
(NCT02475629) were presented (Lewis, ‘Long-acting ibalizumab in
patients with multi-drug resistant hiv-1: A 24-week study,’ CROI,
Seattle 2017, abs 449LB). Those authors tested ibalizumab in
treatment-experienced patients infected with multi-drug resistant
HIV-1. Patients must have been treated with HAART for at least 6
months and be failing with a VL >1,000 copies/ml and a mean CD4+
cell counts of 150 cells/μl. After two weeks of monotherapy
the patients received ibalizumab and an OBR. At week 24, 43% of
participants had a VL <50 copies/ml and 50% <200 copies/ml.
The authors disclosed that this schedule had a good tolerability
and high efficacy, even in patients that have failed with >10
ARV agents. Only 1 drug-related AE (IRIS) led to treatment
discontinuation.
Ibalizumab is being evaluated also for PreP in
healthy volunteers at risk for HIV according to clinical history
and sexual behaviors (NCT01292174).
Secondary resistance has been described after one
single omission in a patient that has accidentally received placebo
instead of ibalizumab (71) and
when this drug was used in monotherapy (69). However, this resistance has no
cross-reaction to other entry and fusion inhibitors such as
maraviroc and enfuvirtide (69,72).
Mild AEs have been reported in <15% of the cases,
the most frequent were: rash, headache, nausea and diarrhea
(Khanlou et al, ‘Safety, efficacy and pharmacokinetics of
ibalizumab in treatment-experienced HIV-1 infected patients: A
phase 2b study’. 51 st ICAAC 2011, abs H2-794b). Norris, ‘TNX-355
in combination with optimized background regimen (OBR) exhibits
greater antiviral activity than OBR alone in HIV treatment
experienced patients.’ Interscience conference on antimicrobial
agents and chemotherapy, Washington 2005, abs LB 2–26). Grade 3–4
AEs have been described in 2–4% of subjects in ibalizumab arm
versus 5% in placebo arm [Norris, ‘TNX-355 in combination with
optimized background regimen (OBR) exhibits greater antiviral
activity than OBR alone in HIV treatment experienced patients’
Interscience conference on antimicrobial agents and chemotherapy,
Washington 2005, abs LB 2–26]. No drug-related deaths or
discontinuation occurred during clinical trials.
UB-421
UB-421 is a humanized IgG1 monoclonal antibody (mAb)
that competitively binds to domain 1 of CD4 receptors and inhibits
HIV-1 entry into cells. In vitro, UB-421 has demonstrated
activity ability against both X4 and R5-tropic virus (Wang, ‘A
phase 2 open-label trial of antibody UB-421 monotherapy as a
substitute for HAART|’. CROI, Seattle 2017, abs 450 LB).
In a phase IIa trial (NCT01668043), with naïve
HIV-infected adults in Taiwan the investigators obtained an average
VL reduction of 2.27 and 2.45 log copies/ml after 8-week
monotherapy on 10 mg/kg/weekly or 25 mg/kg/biweekly,
respectively.
In another phase II open label study (Wang, ‘A phase
2 open-label trial of antibody UB-421 monotherapy as a substitute
for HAART|’. CROI, Seattle 2017, abs 450 LB) was administered
UB-421 as a monotherapy as a replacement of HAART in 29
HIV-1-infected adults with virologic suppression. All the subjects
were assigned to Cohort 1 (10 mg/kg weekly of UB-421 for 8 weeks)
or to Cohort 2 (25 mg/kg bi-weekly for 16 weeks). The authors
reported that 27 out of the 29 patients completed the monotherapy
period (8 and 16 weeks) with no virologic rebound (VR) defined as a
VL >400 copies/ml in two consecutive visits. At the end of
treatment 22 patients resumed the previous HAART successfully but 5
refused the HAART treatment. Among them the VR was detected as
35–62 days after the last UB-421 dose. At the end of the study CD4+
cell count remained stable and CD8+ increased. Interestingly, they
observed a mean 2.24-fold reduction in 10 out of 11 patients that
had a proviral DNA >100 copies/106 PBMC at baseline.
The most common drug-related AE was mild to moderate skin rash
(48.3% of subjects), and no death or drug-related SAE occurred.
UB-421 is being evaluated in a phase III trial
(NCT03149211) as substitution therapy for HAART in adults who are
virologically suppressed on a stable ART regimen.
No drug resistance was reported after 8 weeks of
monotherapy with UB-421 (73).
Discussion
Despite the revolution achieved in the field of HIV
treatment in the last three decades, there are still some important
issues that require attention.
First, with HAART HIV changed from a lethal disease
to a chronic one but a cure remains elusive. Furthermore, many
patients are diagnosed at late stage with a low immune recovery
(23). Second, since it is a
chronic disease that needs life-long treatment, HIV is burdened
with a lot of co-morbidities due to the virus and treatment thereof
(17). The safe profile showed by
entry inhibitors could represent advancement in terms of
drug-related toxicities in comparison with actual HAART.
Third, it has been proven that even if we could
obtain a stable virologic suppression, most of the co-morbidities
that come from immune activation cannot be defeated if we are
unable to combat the HIV reservoir (20,21).
Some of these new drugs have shown partial efficacy against the HIV
reservoir, as described above.
Finally, it is imperative to confront the issue of
prevention. Even if condoms and sexual education remain the
milestones of prevention, in recent years the idea of PrEP has
gained ground. Furthermore, in this field the entry inhibitors
could be additional weapons against the spread of HIV
infection.
Entry and fusion inhibitors are a new class of
antiretroviral drugs that could play a role in particular settings.
Being a relatively new class with no or less cross-reactions and
few mechanisms of resistance demonstrated thus far, they could be
used as a rescue therapy in experienced patients with MDR viruses.
Fostemsavir, Ibalizumab, Albuvirtide and Cenicriviroc are being
tested in clinical trials as ‘third drugs’ in patients with problem
of drug resistance.
Another possible use is a monotherapy with an entry
inhibitor (UB-421, PRO-140 and monomeric DAPTA), in stable
undetectable patients. This substitution therapy for stable
patients could be safer especially for older patients with
co-morbidities. In fact, the entry inhibitors showed a very good
safety profile in their first clinical trials, probably also
because they act outside the human cells and target the HIV
proteins having a low toxicity. Furthermore, it has been suggested
by the VISCONTI study that in selected patients, after a period
with an effective therapy that could also reduce the HIV reservoir,
we could stop therapy obtaining a good control of the HIV infection
(74). All those patients have
extremely low HIV DNA levels, and some entry inhibitor showed an
effect in reducing HIV DNA in the first clinical trials.
The most important limitation in clinical practice
is likely to be viral tropism. All those entry inhibitors that bind
one of the two main co-receptors would not be used in every
clinical setting. This is the case of CCR5 antagonists.
In conclusion, entry and fusion inhibitors are very
promising drugs that could reach some features required in new HIV
treatments. They showed good safety and efficacy and seem to be
optimal in long-term treated patients for the lack of significant
drug-drug interactions and drug resistances.
Obviously, all of them have to demonstrate this good
quality in future clinical trials. Currently the possible uses are:
PrEP, rescue therapy for MDR HIV, de-escalation for stable patients
and being the ‘third drug’ in conventional HAART.
Acknowledgements
Not applicable.
Funding
Not applicable.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors’ contributions
EVR and GN conceived and designed the subject. EVR,
MC and MRP retrieved concerned literatures and wrote the article.
FC, AF, GV, FdA and IP reviewed and edited the article. MdR, BC, GP
and GN revised the manuscript. All authors have read and approved
the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
World Health Organization: HIV/AIDS fact
sheet. 2018 12–February. 2018http://www.who.int/en/news-room/fact-sheets/detail/hiv-aids
|
|
2
|
McMahon JH, Elliott JH, Bertagnolio S,
Kubiak R and Jordan MR: Viral suppression after 12 months of
antiretroviral therapy in low- and middle-income countries: A
systematic review. Bull World Health Organ. 91:377–385. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Thompson MA, Aberg JA, Hoy JF, Telenti A,
Benson C, Cahn P, Eron JJ, Günthard HF, Hammer SM, Reiss P, et al:
Antiretroviral treatment of adult HIV infection: 2012
recommendations of the International Antiviral Society-USA panel.
JAMA. 308:387–402. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Castronuovo D, Cacopardo B, Pinzone MR, Di
Rosa M, Martellotta F, Schioppa O, Moreno S and Nunnari G: Bone
disease in the setting of HIV infection: Update and review of the
literature. Eur Rev Med Pharmacol Sci. 17:2413–2419.
2013.PubMed/NCBI
|
|
5
|
Scarpino M, Pinzone MR, Di Rosa M, Madeddu
G, Focà E, Martellotta F, Schioppa O, Ceccarelli G, Celesia BM,
d'Ettorre G, et al: Kidney disease in HIV-infected patients. Eur
Rev Med Pharmacol Sci. 17:2660–2667. 2013.PubMed/NCBI
|
|
6
|
Pomerantz RJ and Nunnari G: HIV and GB
virus C-can two viruses be better than one? N Engl J Med.
350:963–965. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nunnari G, Coco C, Pinzone MR, Pavone P,
Beretta M, Di Rosa M, Schnell M, Giorgio C and Cacopando B: The
role of micronutrients in the diet of HIV-1-infected individuals.
Front Biosci (Elite Ed). 4:2442–2456. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pinzone MR, Berretta M, Cacopardo B and
Nunnari G: Epstein-barr virus- and Kaposi sarcoma-associated
herpesvirus-related malignancies in the setting of human
immunodeficiency virus infection. Semin Oncol. 42:258–271. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pinzone MR, Di Rosa M, Celesia BM,
Condorelli F, Malaguarnera M, Madeddu G, Martellotta F, Castronuovo
D, Gussio M, Coco C, et al: LPS and HIV gp120 modulate
monocyte/macrophage CYP27B1 and CYP24A1
expression leading to vitamin D consumption and hypovitaminosis D
in HIV-infected individuals. Eur Rev Med Pharmacol Sci.
17:1938–1950. 2013.PubMed/NCBI
|
|
10
|
Pinzone MR, Cacopardo B, Condorelli F, Di
Rosa M and Nunnari G: Sirtuin-1 and HIV-1: An overview. Curr Drug
Targets. 14:648–652. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Capetti A, Landonio S, Meraviglia P, Di
Biagio A, Lo Caputo S, Sterrantino G, Ammassari A, Menzaghi B,
Franzetti M, De Socio GV, et al: 96 Week follow-up of HIV-infected
patients in rescue with raltegravir plus optimized backbone
regimens: A multicentre Italian experience. PLoS One. 7:e392222012.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Visalli G, Paiardini M, Chirico C, Cervasi
B, Currò M, Ferlazzo N, Bertuccio MP, Favaloro A, Pellicanò G,
Sturniolo G, et al: Intracellular accumulation of cell cycle
regulatory proteins and nucleolin re-localization are associated
with pre-lethal ultrastructural lesions in circulating T
lymphocytes: The HIV-induced cell cycle dysregulation revisited.
Cell Cycle. 9:2130–2140. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Celesia BM, Nigro L, Pinzone MR, Coco C,
La Rosa R, Bisicchia F, Mavilla S, Gussio M, Pellicanò G, Milioni
V, et al: High prevalence of undiagnosed anxiety symptoms among
HIV-positive individuals on cART: A cross-sectional study. Eur Rev
Med Pharmacol Sci. 17:2040–2046. 2013.PubMed/NCBI
|
|
14
|
Visalli G, Bertuccio MP, Currò M,
Pellicanò G, Sturniolo G, Carnevali A, Spataro P, Ientile R,
Picerno I, Cavallari V, et al: Bioenergetics of T cell activation
and death in HIV type 1 infection. AIDS Res Hum Retroviruses.
28:1110–1118. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Trovato M, Ruggeri RM, Sciacchitano S,
Vicchio TM, Picerno I, Pellicanò G, Valenti A and Visalli G: Serum
interleukin-6 levels are increased in HIV-infected patients that
develop autoimmune disease during long-term follow-up.
Immunobiology. 223:264–268. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
D'Aleo F, Ceccarelli M, Venanzi Rullo E,
Facciolà A, Di Rosa M, Pinzone MR, Condorelli F, Visalli G, Picerno
I, Berretta M, et al: Hepatitis C-related hepatocellular carcinoma:
Diagnostic and therapeutic management in HIV-patients. Eur Rev Med
Pharmacol Sci. 21:5859–5867. 2017.PubMed/NCBI
|
|
17
|
Squillace N, Ricci E, Quirino T, Gori A,
Bandera A, Carenzi L, De Socio GV, Orofino G, Martinelli C, Madeddu
G, et al CISAI Study Group, : Safety and tolerability of
Elvitegravir/Cobicistat/Emtricitabine/Tenofovir Disoproxil fumarate
in a real life setting: Data from surveillance cohort long-term
toxicity antiretrovirals/antivirals (SCOLTA) project. PLoS One.
12:e01792542017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Facciolà A, Venanzi Rullo E, Ceccarelli M,
D'Aleo F, Di Rosa M, Pinzone MR, Condorelli F, Visalli G, Picerno
I, Fisichella R, et al: Kaposi's sarcoma in HIV-infected patients
in the era of new antiretrovirals. Eur Rev Med Pharmacol Sci.
21:5868–5869. 2017.PubMed/NCBI
|
|
19
|
Zanet E, Berretta M, Benedetto FD,
Talamini R, Ballarin R, Nunnari G, Berretta S, Ridolfo A, Lleshi A,
Zanghì A, et al: Pancreatic cancer in HIV-positive patients: A
clinical case-control study. Pancreas. 41:1331–1335. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Nunnari G, Sullivan J, Xu Y, Nyirjesy P,
Kulkosky J, Cavert W, Frank I and Pomerantz RJ: HIV type 1
cervicovaginal reservoirs in the era of HAART. AIDS Res Hum
Retroviruses. 21:714–718. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nunnari G, Leto D, Sullivan J, Xu Y,
Mehlman KE, Kulkosky J and Pomerantz RJ: Seminal reservoirs during
an HIV type 1 eradication trial. AIDS Res Hum Retroviruses.
21:768–775. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Martellotta F, Berretta M, Cacopardo B,
Fisichella R, Schioppa O, Zanghì A, Spartà D, Cappellani A,
Talamini R, Izzi I, et al: Clinical presentation and outcome of
squamous cell carcinoma of the anus in HIV-infected patients in the
HAART-era: A GICAT experience. Eur Rev Med Pharmacol Sci.
16:1283–1291. 2012.PubMed/NCBI
|
|
23
|
Celesia BM, Castronuovo D, Pinzone MR,
Bellissimo F, Mughini MT, Lupo G, Scarpino MR, Gussio M, Palermo F,
Cosentino S, et al: Late presentation of HIV infection: Predictors
of delayed diagnosis and survival in Eastern Sicily. Eur Rev Med
Pharmacol Sci. 17:2218–2224. 2013.PubMed/NCBI
|
|
24
|
Bearz A, Vaccher E, Martellotta F, Spina
M, Talamini R, Lleshi A, Cacopardo B, Nunnari G, Berretta M,
Tirelli U, et al: Lung cancer in HIV positive patients: Τhe GICAT
experience. Eur Rev Med Pharmacol Sci. 18:500–508. 2014.PubMed/NCBI
|
|
25
|
Capetti A, Meraviglia P, Landonio S,
Sterrantino G, Di Biagio A, Lo Caputo S, Ammassari A, Menzaghi B,
De Socio GV, Franzetti M, et al: Four years data of
raltegravir-based salvage therapy in HIV-1-infected,
treatment-experienced patients: The SALIR-E study. Int J Antimicrob
Agents. 43:189–194. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Nuvoli S, Caruana G, Babudieri S, Solinas
P, Pellicanò G, Piras B, Fiore V, Bagella P, Calia GM, Yue M, et
al: Body fat changes in HIV patients on highly active
antiretroviral therapy (HAART): A longitudinal DEXA study. Eur Rev
Med Pharmacol Sci. 22:1852–1859. 2018.PubMed/NCBI
|
|
27
|
Ceccarelli M, Condorelli F, Venanzi Rullo
E and Pellicanò GF: Editorial - Improving access and adherence to
screening tests for cancers: A new, though old, challenge in the
HIV epidemics. World Cancer Res J. 5:e10302018.
|
|
28
|
Ceccarelli M, Rullo EV, Facciolà A,
Madeddu G, Cacopardo B, Taibi R, D'Aleo F, Pinzone MR, Picerno I,
di Rosa M, et al: Head and neck squamous cell carcinoma and its
correlation with human papillomavirus in people living with HIV: A
systematic review. Oncotarget. 9:17171–17180. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
D'Aleo F, Cama BAV, Paolucci IA, et al:
New and old assumptions on lung cancer in People Living with HIV.
World Cancer Res J. 5:e10362018.
|
|
30
|
Visalli G, Facciolà A, D'Aleo F, Pinzone
MR, Condorelli F, Nunnari G, Pellicano GF, Ceccarelli M and Venanzi
Rullo E: HPV and urinary bladder carcinoma: A review of the
literature. World Cancer Res J. 5:e10382018.
|
|
31
|
Pace MJ, Agosto L, Graf EH and O'Doherty
U: HIV reservoirs and latency models. Virology. 411:344–354. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wyatt R and Sodroski J: The HIV-1 envelope
glycoproteins: Fusogens, antigens, and immunogens. Science.
280:1884–1888. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Briz V, Poveda E and Soriano V: HIV entry
inhibitors: Mechanisms of action and resistance pathways. J
Antimicrob Chemother. 57:619–627. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ray S, Fatima Z and Saxena A: Drugs for
AIDS. Mini Rev Med Chem. 10:147–161. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Jacobson JM, Saag MS, Thompson MA, Fischl
MA, Liporace R, Reichman RC, Redfield RR, Fichtenbaum CJ, Zingman
BS, Patel MC, et al: Antiviral activity of single-dose PRO 140, a
CCR5 monoclonal antibody, in HIV-infected adults. J Infect Dis.
198:1345–1352. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
36
|
Trkola A, Ketas TJ, Nagashima KA, Zhao L,
Cilliers T, Morris L, Moore JP, Maddon PJ and Olson WC: Potent,
broad-spectrum inhibition of human immunodeficiency virus type 1 by
the CCR5 monoclonal antibody PRO 140. J Virol. 75:579–588. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Jacobson JM, Thompson MA, Lalezari JP,
Saag MS, Zingman BS, D'Ambrosio P, Stambler N, Rotshteyn Y,
Marozsan AJ, Maddon PJ, et al: Anti-HIV-1 activity of weekly or
biweekly treatment with subcutaneous PRO 140, a CCR5 monoclonal
antibody. J Infect Dis. 201:1481–1487. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
CytoDyn Inc: An extension of protocol PRO
140_CD01 TS Study. ClinicalTrials.gov. Identifier: NCT02355184.
First posted. February 4–2015, https://clinicaltrials.gov/ct2/show/NCT02355184
|
|
39
|
CytoDyn Inc: A randomized, double-blind,
placebo-controlled trial, followed by single-arm treatment of PRO
140 in combination w/optimized background therapy in
treatment-experienced HIV subjects (PRO 140). ClinicalTrials.gov.
Identifier: NCT02483078. First posted. June 26–2015, https://clinicaltrials.gov/ct2/show/NCT02483078
|
|
40
|
Baba M, Takashima K, Miyake H, Kanzaki N,
Teshima K, Wang X, Shiraishi M and Iizawa Y: TAK-652 inhibits
CCR5-mediated human immunodeficiency virus type 1 infection in
vitro and has favorable pharmacokinetics in humans. Antimicrob
Agents Chemother. 49:4584–4591. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Marier J-F, Trinh M, Pheng LH, Palleja SM
and Martin DE: Pharmacokinetics and pharmacodynamics of TBR-652, a
novel CCR5 antagonist, in HIV-1-infected, antiretroviral
treatment-experienced, CCR5 antagonist-naïve patients. Antimicrob
Agents Chemother. 55:2768–2774. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Thompson M, Saag M, DeJesus E, Gathe J,
Lalezari J, Landay AL, Cade J, Enejosa J, Lefebvre E and Feinberg
J: A 48-week randomized phase 2b study evaluating cenicriviroc
versus efavirenz in treatment-naive HIV-infected adults with C-C
chemokine receptor type 5-tropic virus. AIDS. 30:869–878. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Lalezari J, Gathe J, Brinson C, Thompson
M, Cohen C, Dejesus E, Galindez J, Ernst JA, Martin DE and Palleja
SM: Safety, efficacy, and pharmacokinetics of TBR-652, a CCR5/CCR2
antagonist, in HIV-1-infected, treatment-experienced, CCR5
antagonist-naive subjects. J Acquir Immune Defic Syndr. 57:118–125.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Baba M, Miyake H, Wang X, Okamoto M and
Takashima K: Isolation and characterization of human
immunodeficiency virus type 1 resistant to the small-molecule CCR5
antagonist TAK-652. Antimicrob Agents Chemother. 51:707–715. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Visseaux B, Charpentier C, Collin G,
Bertine M, Peytavin G, Damond F, Matheron S, Lefebvre E,
Brun-Vézinet F and Descamps D; ANRS CO5 HIV-2 Cohort, :
Cenicriviroc, a novel CCR5 (R5) and CCR2 antagonist, shows in vitro
activity against R5 Tropic HIV-2 Clinical Isolates. PLoS One.
10:e01349042015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Polianova MT, Ruscetti FW, Pert CB and
Ruff MR: Chemokine receptor-5 (CCR5) is a receptor for the HIV
entry inhibitor peptide T (DAPTA). Antiviral Res. 67:83–92. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Pollicita M, Ruff MR, Pert CB, Polianova
MT, Schols D, Ranazzi A, Perno CF and Aquaro S: Profound anti-HIV-1
activity of DAPTA in monocytes/macrophages and inhibition of
CCR5-mediated apoptosis in neuronal cells. Antivir Chem Chemother.
18:285–295. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Goodkin K, Vitiello B, Lyman WD, Asthana
D, Atkinson JH, Heseltine PN, Molina R, Zheng W, Khamis I, Wilkie
FL, et al: Cerebrospinal and peripheral human immunodeficiency
virus type 1 load in a multisite, randomized, double-blind,
placebo-controlled trial of D-Ala1-peptide T-amide for
HIV-1-associated cognitive-motor impairment. J Neurovirol.
12:178–189. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Agrawal L, Ducoudret O, Baichoo N,
Laznicka M, Ruff M and Pert C: mDAPTA, a potent CCR5 receptor
blocker, prevents viral recovery from CD8-depleted patient PBMCs
with VL< 50 background. International AIDS Society. http://www.abstract-archive.org/Abstract/Share/4495
|
|
50
|
Polianova MT, Ruscetti FW, Pert CB,
Tractenberg RE, Leoung G, Strang S and Ruff MR: Antiviral and
immunological benefits in HIV patients receiving intranasal peptide
T (DAPTA). Peptides. 24:1093–1098. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Langley DR, Kimura SR, Sivaprakasam P,
Zhou N, Dicker I, McAuliffe B, Wang T, Kadow JF, Meanwell NA and
Krystal M: Homology models of the HIV-1 attachment inhibitor
BMS-626529 bound to gp120 suggest a unique mechanism of action.
Proteins. 83:331–350. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Nowicka-Sans B, Gong Y-F, McAuliffe B,
Dicker I, Ho HT, Zhou N, Eggers B, Lin PF, Ray N, Wind-Rotolo M, et
al: In vitro antiviral characteristics of HIV-1 attachment
inhibitor BMS-626529, the active component of the prodrug
BMS-663068. Antimicrob Agents Chemother. 56:3498–3507. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Li Z, Zhou N, Sun Y, Ray N, Lataillade M,
Hanna GJ and Krystal M: Activity of the HIV-1 attachment inhibitor
BMS-626529, the active component of the prodrug BMS-663068, against
CD4-independent viruses and HIV-1 envelopes resistant to other
entry inhibitors. Antimicrob Agents Chemother. 57:4172–4180. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Ray N, Hwang C, Healy MD, Whitcomb J,
Lataillade M, Wind-Rotolo M, Krystal M and Hanna GJ: Prediction of
virological response and assessment of resistance emergence to the
HIV-1 attachment inhibitor BMS-626529 during 8-day monotherapy with
its prodrug BMS-663068. J Acquir Immune Defic Syndr. 64:7–15. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Zhou N, Nowicka-Sans B, McAuliffe B, Ray
N, Eggers B, Fang H, Fan L, Healy M, Langley DR, Hwang C, et al:
Genotypic correlates of susceptibility to HIV-1 attachment
inhibitor BMS-626529, the active agent of the prodrug BMS-663068. J
Antimicrob Chemother. 69:573–581. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Nettles RE, Schürmann D, Zhu L, Stonier M,
Huang SP, Chang I, Chien C, Krystal M, Wind-Rotolo M, Ray N, et al:
Pharmacodynamics, safety, and pharmacokinetics of BMS-663068, an
oral HIV-1 attachment inhibitor in HIV-1-infected subjects. J
Infect Dis. 206:1002–1011. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Lalezari JP, Latiff GH, Brinson C,
Echevarría J, Treviño-Pérez S, Bogner JR, Thompson M, Fourie J,
Sussmann Pena OA, Mendo Urbina FC, et al: Safety and efficacy of
the HIV-1 attachment inhibitor prodrug BMS-663068 in
treatment-experienced individuals: 24 week results of AI438011, a
phase 2b, randomised controlled trial. Lancet HIV. 427–37. 2015.
View Article : Google Scholar
|
|
58
|
Vii V Healthcare: Attachment inhibitor
comparison in heavily treatment experienced patients.
ClinicalTrials.gov. Identifier: NCT02362503. First posted. February
13–2015.https://clinicaltrials.gov/ct2/show/NCT02362503
|
|
59
|
Xie D, Yao C, Wang L, Min W, Xu J, Xiao J,
Huang M, Chen B, Liu B, Li X, et al: An albumin-conjugated peptide
exhibits potent anti-HIV activity and long in vivo half-life.
Antimicrob Agents Chemother. 54:191–196. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Zhang H, Jin R, Yao C, Zhang T, Wang M,
Xia W, Peng H, Wang X, Lu R, Wang C, et al: Combination of
long-acting HIV fusion inhibitor albuvirtide and LPV/r showed
potent efficacy in HIV-1 patients. AIDS Res Ther. 13:82016.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Xie D: International congress of drug
therapy in HIV infection 23–26 October 2016, Glasgow, UK. J Int
AIDS Soc. 19:214872016.PubMed/NCBI
|
|
62
|
Yang W, Xiao Q, Wang D, Yao C and Yang J:
Evaluation of pharmacokinetic interactions between long-acting
HIV-1 fusion inhibitor albuvirtide and lopinavir/ritonavir, in
HIV-infected subjects, combined with clinical study and simulation
results. Xenobiotica. 47:133–143. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Song R, Franco D, Kao C-Y, Yu F, Huang Y
and Ho DD: Epitope mapping of ibalizumab, a humanized anti-CD4
monoclonal antibody with anti-HIV-1 activity in infected patients.
J Virol. 84:6935–6942. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Bruno CJ and Jacobson JM: Ibalizumab: An
anti-CD4 monoclonal antibody for the treatment of HIV-1 infection.
J Antimicrob Chemother. 65:1839–1841. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Merkenschlager M, Buck D, Beverley PC and
Sattentau QJ: Functional epitope analysis of the human CD4
molecule. The MHC class II-dependent activation of resting T cells
is inhibited by monoclonal antibodies to CD4 regardless whether or
not they recognize epitopes involved in the binding of MHC class II
or HIV gp120. J Immunol. 145:2839–2845. 1990.PubMed/NCBI
|
|
66
|
Delmonico FL, Knowles RW, Colvin RB,
Cavender DE, Kawai T, Bedle M, Stroka D, Preffer FI, Haug C and
Cosimi AB: Immunosuppression of cynomolgus renal allograft
recipients with humanized OKT4A monoclonal antibodies. Transplant
Proc. 25:784–785. 1993.PubMed/NCBI
|
|
67
|
Reimann KA, Burkly LC, Burrus B, Waite BC,
Lord CI and Letvin NL: In vivo administration to rhesus monkeys of
a CD4-specific monoclonal antibody capable of blocking AIDS virus
replication. AIDS Res Hum Retroviruses. 9:199–207. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Moore JP, Sattentau QJ, Klasse PJ and
Burkly LC: A monoclonal antibody to CD4 domain 2 blocks soluble
CD4-induced conformational changes in the envelope glycoproteins of
human immunodeficiency virus type 1 (HIV-1) and HIV-1 infection of
CD4+ cells. J Virol. 66:4784–4793. 1992.PubMed/NCBI
|
|
69
|
Jacobson JM, Kuritzkes DR, Godofsky E,
DeJesus E, Larson JA, Weinheimer SP and Lewis ST: Safety,
pharmacokinetics, and antiretroviral activity of multiple doses of
ibalizumab (formerly TNX-355), an anti-CD4 monoclonal antibody, in
human immunodeficiency virus type 1-infected adults. Antimicrob
Agents Chemother. 53:450–457. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Kuritzkes DR, Jacobson J, Powderly WG,
Godofsky E, DeJesus E, Haas F, Reimann KA, Larson JL, Yarbough PO,
Curt V, et al: Antiretroviral activity of the anti-CD4 monoclonal
antibody TNX-355 in patients infected with HIV type 1. J Infect
Dis. 189:286–291. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
71
|
Fessel WJ, Anderson B, Follansbee SE,
Winters MA, Lewis ST, Weinheimer SP, Petropoulos CJ and Shafer RW:
The efficacy of an anti-CD4 monoclonal antibody for HIV-1
treatment. Antiviral Res. 92:484–487. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Zhang X-Q, Sorensen M, Fung M and Schooley
RT: Synergistic in vitro antiretroviral activity of a humanized
monoclonal anti-CD4 antibody (TNX-355) and enfuvirtide (T-20).
Antimicrob Agents Chemother. 50:2231–2233. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
United Biomedicals: Study to evaluate
safety and efficacy of UB421 antibody in HIV1 infected adults.
ClinicalTrials.gov. Identifier: NCT01668043. First posted. August
17–2012.https://clinicaltrials.gov/ct2/show/NCT01668043
|
|
74
|
Sáez-Cirión A, Bacchus C, Hocqueloux L,
Avettand-Fenoel V, Girault I, Lecuroux C, Potard V, Versmisse P,
Melard A, Prazuck T, et al ANRS VISCONTI study group, :
Post-treatment HIV-1 controllers with a long-term virological
remission after the interruption of early initiated antiretroviral
therapy ANRS VISCONTI Study. PLoS Pathog. 9:e10032112013.
View Article : Google Scholar : PubMed/NCBI
|