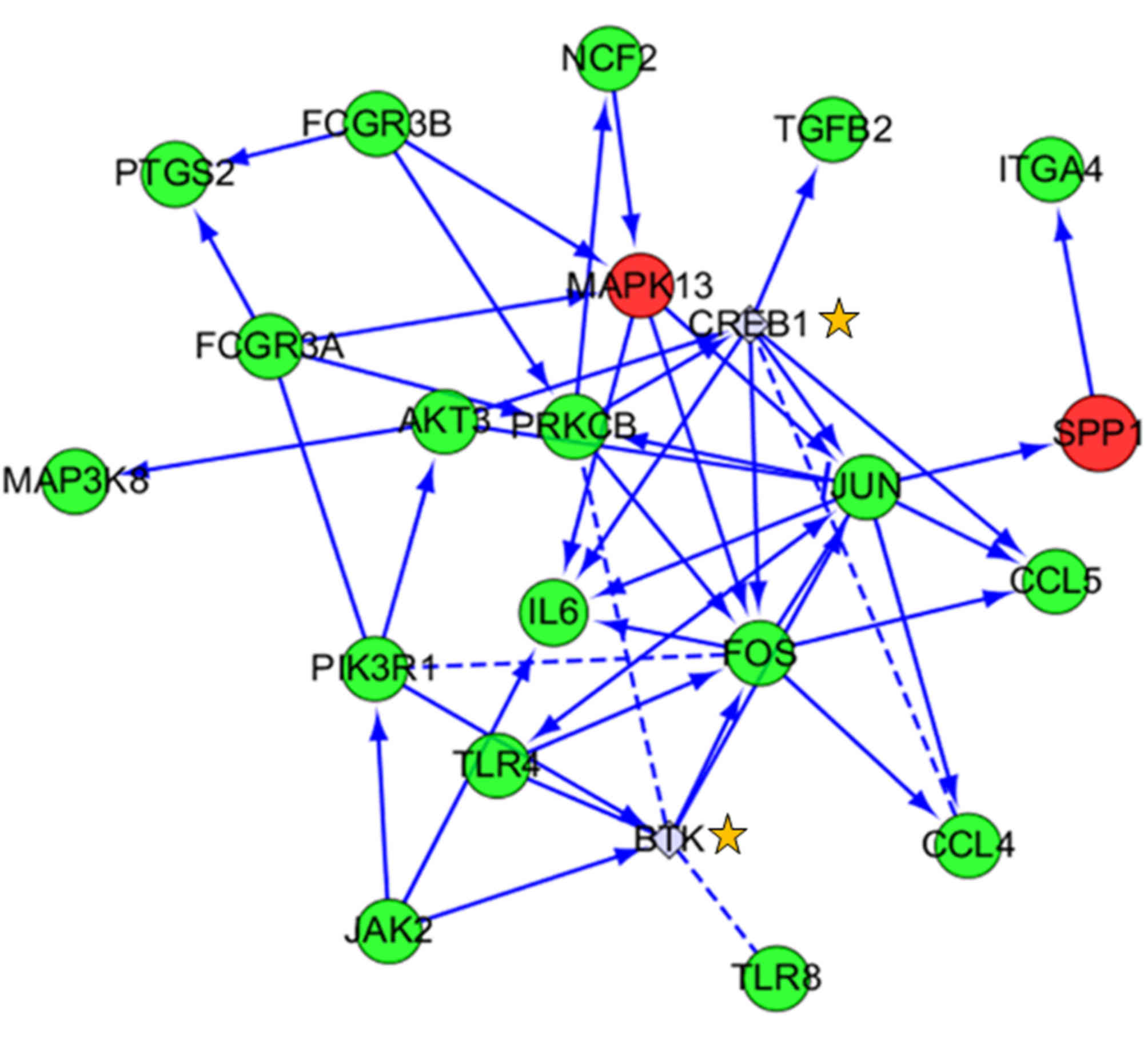
|
1
|
Cancer Genome Atlas Research Network:
Comprehensive molecular profiling of lung adenocarcinoma. Nature.
511:543–550. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cohen AJ, Brauer M, Burnett R, Anderson
HR, Frostad J, Estep K, Balakrishnan K, Brunekreef B, Dandona L,
Dandona R, et al: Estimates and 25-year trends of the global burden
of disease attributable to ambient air pollution: An analysis of
data from the Global Burden of Diseases Study 2015. Lancet.
389:1907–1918. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wang LQ, Zhao LH and Qiao YZ:
Identification of potential therapeutic targets for lung cancer by
bioinformatics analysis. Mol Med Rep. 13:1975–1982. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Aran V, Victorino AP, Thuler LC and
Ferreira CG: Colorectal cancer: Epidemiology, disease mechanisms
and interventions to reduce onset and mortality. Clin Colorectal
Cancer. 15:195–203. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhang M, Gao C, Yang Y, Li G, Dong J, Ai
Y, Ma Q and Li W: MiR-424 promotes non-small cell lung cancer
progression and metastasis through regulating the tumor suppressor
gene TNFAIP1. Cell Physiol Biochem. 42:211–221. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Singleton KR, Hinz TK, Kleczko EK, Marek
LA, Kwak J, Harp T, Kim J, Tan AC and Heasley LE: Kinome RNAi
screens reveal synergistic targeting of MTOR and FGFR1 pathways for
treatment of lung cancer and HNSCC. Cancer Res. 75:4398–4406. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liang B, Li C and Zhao J: Identification
of key pathways and genes in colorectal cancer using bioinformatics
analysis. Med Oncol. 33:1112016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Descotes F, Dessen P, Bringuier PP,
Decaussin M, Martin PM, Adams M, Villers A, Lechevallier E,
Rebillard X, Rodriguez-Lafrasse C, et al: Microarray gene
expression profiling and analysis of bladder cancer supports the
sub-classification of T1 tumours into T1a and T1b stages. BJU Int.
113:333–342. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sakashita H, Inoue H, Akamine S, Ishida T,
Inase N, Shirao K, Mori M and Mimori K: Identification of the
NEDD4L gene as a prognostic marker by integrated microarray
analysis of copy number and gene expression profiling in non-small
cell lung cancer. Ann Surg Oncol. 20 Suppl 3:S590–S598. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yu L, Wang L and Chen S: Dual character of
Toll-like receptor signaling: Pro-tumorigenic effects and
anti-tumor functions. Biochim Biophys Acta 1835. 144–154. 2013.
|
|
12
|
Pinto A, Morello S and Sorrentino R: Lung
cancer and Toll-like receptors. Cancer Immunol Immunother.
60:1211–1220. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lu TP, Tsai MH, Lee JM, Hsu CP, Chen PC,
Lin CW, Shih JY, Yang PC, Hsiao CK, Lai LC and Chuang EY:
Identification of a novel biomarker, SEMA5A, for non-small cell
lung carcinoma in nonsmoking women. Cancer Epidemiol Biomarkers
Prev. 19:2590–2597. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hu WP, Zeng YY, Zuo YH and Zhang J:
Identification of novel candidate genes involved in the progression
of emphysema by bioinformatic methods. Int J Chron Obstruct Pulmon
Dis. 13:3733–3747. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gautier L, Cope L, Bolstad BM and Irizarry
RA: Affy-analysis of Affymetrix GeneChip data at the probe level.
Bioinformatics. 20:307–315. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hardcastle TJ: Generalised empirical
Bayesian methods for discovery of differential data in
high-throughput biology. Bioinformatics. 32:195–202.
2016.PubMed/NCBI
|
|
17
|
Huang DW, Sherman BT, Tan Q, Kir J, Liu D,
Bryant D, Guo Y, Stephens R, Baseler MW, Lane HC and Lempicki RA:
DAVID Bioinformatics Resources: Expanded annotation database and
novel algorithms to better extract biology from large gene lists.
Nucleic Acids Res. 35:W169–W175. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Franceschini A, Szklarczyk D, Frankild S,
Kuhn M, Simonovic M, Roth A, Lin J, Minguez P, Bork P, von Mering C
and Jensen LJ: STRING v9.1: Protein-protein interaction networks,
with increased coverage and integration. Nucleic Acids Res.
41:D808–D815. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bader GD and Hogue CW: An automated method
for finding molecular complexes in large protein interaction
networks. BMC Bioinformatics. 4:22003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Carter SL, Eklund AC, Kohane IS, Harris LN
and Szallasi Z: A signature of chromosomal instability inferred
from gene expression profiles predicts clinical outcome in multiple
human cancers. Nat Genet. 38:1043–1048. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dela Cruz CS, Tanoue LT and Matthay RA:
Lung cancer: Epidemiology, etiology, and prevention. Clin Chest
Med. 32:605–644. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang GZ, Cheng X, Li XC, Liu YQ, Wang XQ,
Shi X, Wang ZY, Guo YQ, Wen ZS, Huang YC and Zhou GB: Tobacco smoke
induces production of chemokine CCL20 to promote lung cancer.
Cancer Lett. 363:60–70. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Masuda K, Kuwano Y, Nishida K and Rokutan
K: Application of DNA microarray technology to gerontological
studies. Methods Mol Biol 1048. 285–308. 2013. View Article : Google Scholar
|
|
24
|
Chew SK, Lu D, Campos LS, Scott KL, Saci
A, Wang J, Collinson A, Raine K, Hinton J, Teague JW, et al:
Polygenic in vivo validation of cancer mutations using transposons.
Genome Biol. 15:4552014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhu X, Zhou L, Li R, Shen Q, Cheng H, Shen
Z and Zhu H: AGER promotes proliferation and migration in cervical
cancer. Biosci Rep. 38:BSR201713292018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kang R, Tang D, Lotze MT and Zeh HJ III:
AGER/RAGE-mediated autophagy promotes pancreatic tumorigenesis and
bioenergetics through the IL6-pSTAT3 pathway. Autophagy. 8:989–991.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Pan Z, Liu L, Nie W, Miggin S, Qiu F, Cao
Y, Chen J, Yang B, Zhou Y, Lu J and Yang L: Long non-coding RNA
AGER-1 functionally upregulates the innate immunity gene AGER and
approximates its anti-tumor effect in lung cancer. Mol Carcinog.
57:305–318. 2018. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yamaguchi K, Iwamoto H, Sakamoto S,
Horimasu Y, Masuda T, Miyamoto S, Nakashima T, Ohshimo S, Fujitaka
K, Hamada H and Hattori N: AGER rs2070600 polymorphism elevates
neutrophil-lymphocyte ratio and mortality in metastatic lung
adenocarcinoma. Oncotarget. 8:94382–94392. 2017.PubMed/NCBI
|
|
29
|
Wang X, Cui E, Zeng H, Hua F, Wang B, Mao
W and Feng X: RAGE genetic polymorphisms are associated with risk,
chemotherapy response and prognosis in patients with advanced
NSCLC. PLoS One. 7:e437342012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Evan GI and Vousden KH: Proliferation,
cell cycle and apoptosis in cancer. Nature. 411:342–348. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kolch W, Halasz M, Granovskaya M and
Kholodenko BN: The dynamic control of signal transduction networks
in cancer cells. Nat Rev Cancer. 15:515–527. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lv M and Wang L: Comprehensive analysis of
genes, pathways, and TFs in nonsmoking Taiwan females with lung
cancer. Exp Lung Res. 41:74–83. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ajona D, Pajares MJ, Corrales L,
Perez-Gracia JL, Agorreta J, Lozano MD, Torre W, Massion PP,
de-Torres JP, Jantus-Lewintre E, et al: Investigation of complement
activation product c4d as a diagnostic and prognostic biomarker for
lung cancer. J Natl Cancer Inst. 105:1385–1393. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Fonseca FL, Azzalis LA, Feder D, Nogoceke
E, Junqueira VB, Valenti VE and de Abreu LC: Adhesion molecules
affected by treatment of lung cancer cells with epidermal growth
factor. Lung. 189:383–389. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang H, Zhu Y, Zhao M, Wu C, Zhang P, Tang
L, Zhang H, Chen X, Yang Y and Liu G: miRNA-29c suppresses lung
cancer cell adhesion to extracellular matrix and metastasis by
targeting integrin β1 and matrix metalloproteinase 2 (MMP2). PLoS
One. 8:e701922013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ungewiss C, Rizvi ZH, Roybal JD, Peng DH,
Gold KA, Shin DH, Creighton CJ and Gibbons DL: The
microRNA-200/Zeb1 axis regulates ECM-dependent β1-integrin/FAK
signaling, cancer cell invasion and metastasis through CRKL. Sci
Rep. 6:186522016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Rakoff-Nahoum S and Medzhitov R: Toll-like
receptors and cancer. Nat Rev Cancer. 9:57–63. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kawai T and Akira S: The role of
pattern-recognition receptors in innate immunity: Update on
Toll-like receptors. Nat Immunol. 11:373–384. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Chen R, Alvero AB, Silasi DA, Steffensen
KD and Mor G: Cancers take their Toll-the function and regulation
of Toll-like receptors in cancer cells. Oncogene. 27:225–233. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Huang B, Zhao J, Unkeless JC, Feng ZH and
Xiong H: TLR signaling by tumor and immune cells: A double-edged
sword. Oncogene. 27:218–224. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Li D, Jin Y, Sun Y, Lei J and Liu C:
Knockdown of toll-like receptor 4 inhibits human NSCLC cancer cell
growth and inflammatory cytokine secretion in vitro, in
vivo. Int J Oncol. 45:813–821. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Jo U, Park KH, Whang YM, Sung JS, Won NH,
Park JK and Kim YH: EGFR endocytosis is a novel therapeutic target
in lung cancer with wild-type EGFR. Oncotarget. 5:1265–1278. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Sequist LV and Lynch TJ: EGFR tyrosine
kinase inhibitors in lung cancer: An evolving story. Annu Rev Med.
59:429–442. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Fang S and Wang Z: EGFR mutations as a
prognostic and predictive marker in non-small-cell lung cancer.
Drug Des Devel Ther. 8:1595–1611. 2014.PubMed/NCBI
|
|
45
|
Moschini I, Dell'Anna C, Losardo PL, Bordi
P, D'Abbiero N and Tiseo M: Radiotherapy of non-small-cell lung
cancer in the era of EGFR gene mutations and EGF receptor tyrosine
kinase inhibitors. Future Oncol. 11:2329–2342. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Li Y, Yang Z, Li W, Xu S, Wang T, Wang T,
Niu M, Zhang S, Jia L and Li S: TOPK promotes lung cancer
resistance to EGFR tyrosine kinase inhibitors by phosphorylating
and activating c-Jun. Oncotarget. 7:6748–6764. 2016.PubMed/NCBI
|
|
47
|
Durchdewald M, Angel P and Hess J: The
transcription factor Fos: A Janus-type regulator in health and
disease. Histol Histopathol. 24:1451–1461. 2009.PubMed/NCBI
|
|
48
|
Ataie-Kachoie P, Pourgholami MH and Morris
DL: Inhibition of the IL-6 signaling pathway: A strategy to combat
chronic inflammatory diseases and cancer. Cytokine Growth Factor
Rev. 24:163–173. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Kumari N, Dwarakanath BS, Das A and Bhatt
AN: Role of interleukin-6 in cancer progression and therapeutic
resistance. Tumour Biol. 37:11553–11572. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Nilsson MB, Langley RR and Fidler IJ:
Interleukin-6, secreted by human ovarian carcinoma cells, is a
potent proangiogenic cytokine. Cancer Res. 65:10794–10800. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Heikkilä K, Ebrahim S and Lawlor DA:
Systematic review of the association between circulating
interleukin-6 (IL-6) and cancer. Eur J Cancer. 44:937–945. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Tai MC, Kajino T, Nakatochi M, Arima C,
Shimada Y, Suzuki M, Miyoshi H, Yatabe Y, Yanagisawa K and
Takahashi T: miR-342-3p regulates MYC transcriptional activity via
direct repression of E2F1 in human lung cancer. Carcinogenesis.
36:1464–1473. 2015.PubMed/NCBI
|
|
53
|
Gabay M, Li Y and Felsher DW: MYC
activation is a hallmark of cancer initiation and maintenance. Cold
Spring Harb Perspect Med. 4:a0142412014. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Sica A, Allavena P and Mantovani A: Cancer
related inflammation: The macrophage connection. Cancer Lett.
267:204–215. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Yu Q, Guo Q, Chen L and Liu S:
Clinicopathological significance and potential drug targeting of
CDH1 in lung cancer: A meta-analysis and literature review. Drug
Des Devel Ther. 9:2171–2178. 2015.PubMed/NCBI
|
|
56
|
Thomas SM and Brugge JS: Cellular
functions regulated by Src family kinases. Annu Rev Cell Dev Biol.
13:513–609. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Zhen Y, Liu J, Huang Y, Wang Y, Li W and
Wu J: miR-133b inhibits cell growth, migration, and invasion by
targeting MMP9 in non-small cell lung cancer. Oncol Res.
25:1109–1116. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Zhang C, Elkahloun AG, Robertson M, Gills
JJ, Tsurutani J, Shih JH, Fukuoka J, Hollander MC, Harris CC,
Travis WD, et al: Loss of cytoplasmic CDK1 predicts poor survival
in human lung cancer and confers chemotherapeutic resistance. PLoS
One. 6:e238492011. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Liu X, Su L and Liu X: Loss of CDH1
up-regulates epidermal growth factor receptor via phosphorylation
of YBX1 in non-small cell lung cancer cells. FEBS Lett.
587:3995–4000. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Behrens C, Lin HY, Lee JJ, Raso MG, Hong
WK, Wistuba II and Lotan R: Immunohistochemical expression of basic
fibroblast growth factor and fibroblast growth factor receptors 1
and 2 in the pathogenesis of lung cancer. Clin Cancer Res.
14:6014–6022. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Ribatti D, Vacca A, Rusnati M and Presta
M: The discovery of basic fibroblast growth factor/fibroblast
growth factor-2 and its role in haematological malignancies.
Cytokine Growth Factor Rev. 18:327–334. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Zhang L, Yu M, Deng J, Lv X, Liu J, Xiao
Y, Yang W, Zhang Y and Li C: Chemokine signaling pathway involved
in CCL2 expression in patients with rheumatoid arthritis. Yonsei
Med J. 56:1134–1142. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
González-Motos V, Jürgens C, Ritter B,
Kropp KA, Durán V, Larsen O, Binz A, Ouwendijk WJD, Lenac Rovis T,
Jonjic S, et al: Varicella zoster virus glycoprotein C increases
chemokine-mediated leukocyte migration. PLoS Pathog.
13:e10063462017. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Griffith JW, Sokol CL and Luster AD:
Chemokines and chemokine receptors: Positioning cells for host
defense and immunity. Annu Rev Immunol. 32:659–702. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Franciszkiewicz K, Boissonnas A, Boutet M,
Combadière C and Mami-Chouaib F: Role of chemokines and chemokine
receptors in shaping the effector phase of the antitumor immunonse.
Cancer Res. 72:6325–6332. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Hsiao CJ, Hsiao G, Chen WL, Wang SW,
Chiang CP, Liu LY, Guh JH, Lee TH and Chung CL: Cephalochromin
induces G0/G1 cell cycle arrest and apoptosis in A549 human
non-small-cell lung cancer cells by inflicting mitochondrial
disruption. J Nat Prod. 77:758–765. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Shcherba M, Liang Y, Fernandes D,
Perez-Soler R and Cheng H: Cell cycle inhibitors for the treatment
of NSCLC. Expert Opin Pharmacother. 15:991–1004. 2014. View Article : Google Scholar : PubMed/NCBI
|