Introduction
Ataxia-telangiectasia (A-T; OMIM no. 208900) is a
rare neurodegenerative disease inherited in an autosomal recessive
manner with great phenotype heterogeneity (1,2). It
is characterized by progressive cerebellar dysfunction,
oculocutaneous telangiectasias, immunodeficiency and cancer
predisposition (1). The estimated
incidence in live births is 1 in 40,000 to 100,000 worldwide
(2,3). The affected infant typically appears
normal in the first 2–3 years, then staggering (ataxia) occurs. The
majority of patients are wheelchair bound by 10 years old (4). The duration of disorder is associated
with the severity of cerebellar atrophy, but not all the patients
with severe cerebellar atrophy are unable to walk (5). The mildest atrophy has been observed
in young patients with an average age of 5 years (5). The clinical manifestations of ataxia
and oculocutaneous telangiectasia, combined with a series of
laboratory tests, are helpful for the diagnosis of A-T (6,7). In
most cases, A-T patients have elevated α-fetoprotein and
carcinoembryonic antigen expression, as well as abnormal levels of
serum-immunoglobulin. Although this disease cannot be cured at
present, early diagnosis is important for symptomatic treatment,
supportive care, genetic counseling and the avoidance of
unnecessary and costly diagnostic tests.
It is well known that mutations in the ATM
gene that result in complete inactivation or elimination of the ATM
protein will lead to A-T (8,9). ATM
protein has 3,050 amino acids and is a member of the
phosphoinositide 3-kinase-related protein kinase super family. It
serves important roles in regulating cell cycle, DNA alteration and
restoration and cell death via phosphorylation of its substrates
(10–12). As a redox thiol-sensitive protein
kinase, ATM functions by activating multiple redox or
phosphorylation sensitive mechanisms. During postnatal development,
ATM is responsible for maintaining genomic, telomeric and
chromosomal integrity under the conditions of genomic or redox
stress (13–15). At present, >900 phosphorylation
sites encompassing >700 proteins have been uncovered to be the
targets of ATM, and the majority of these targets are associated
with the DNA damage regulation (16).
The present study described a Chinese family which
had two affected siblings with A-T and urgently required prenatal
diagnosis of A-T on the third sibling. The clinical features of the
two live patients were described and compared. Targeted sequencing
was applied on the proband (the younger live sibling) for aiding
A-T diagnosis, which revealed one novel, likely pathogenic,
mutation c.5170G>T, as well as one known pathogenic mutation
c.748C>T. Further validations were conducted on the remaining
family members. The present study suggested that genetic testing is
of great importance for aiding clinical and prenatal diagnoses.
Materials and methods
Patients
The present study was approved by the Ethics
Committee of Wuhan Children's Hospital (Wuhan, China). Informed
written consent was obtained from the parents of the studied
family. The proband (II-2; age, 8) and his elder brother (II-1;
age, 13) from a family with Han ethnicity in southern China were
introduced to our clinic center due to signs of development
retrogression. Based on clinical diagnostic criteria, they were
initially diagnosed as A-T in our hospital. When the mother was
pregnant again, she visited the clinic center for prenatal
diagnosis.
Physical examination
Routine examination of general health as well as
neurological evaluations were performed on two patients. Blood
lymphocyte subsets (TBNK) was analyzed by flow cytometry (BD
FACSCanto™ II system). α-fetoprotein was evaluated by
electrochemiluminescence with the commercial kit (Roche Diagnostics
GmbH, Mannheim, Germany). Serum IgG, IgA, IgM, C3, and C4 were
determined by rate nephelometry. Sensory function was assessed with
the measure of vibrotactile perception. Motor coordination was
evaluated by finger-to-nose test and rapid alternating movement
test. Reflex tests were conducted on knee, ankle and other joints.
Muscular weakness was evaluated with common grading criteria
(17).
Electrophysiological assessments
The motor and sensory nerve conduction assessments
were performed by standard methods on the Natus Dantec™
Keypoint® G4 platform. The patients laid in a quiet,
shielded room with room temperature of 20–22°C and limb temperature
of 32–34°C. Surface electrodes were used for stimulation and
recording. Motor conduction velocity (MCV), distal motor latency
(DML) and compound muscle action potential (CMAP) were measured by
stimulating the nerve segments of the ankle to the fibulae
capitulum for the peroneal nerve, ankle to popliteal fossa for the
tibial nerve, wrist to elbow for the median nerve, and wrist to
elbow for the ulnar nerve, and recording from the extensor
digitorum brevis, abductor hallucis, abductor pollicis brevis, and
abductor digiti minim respectively. Sensory nerve conduction
velocity (SCV), amplitude (Amp) and sensory nerve action potential
(SNAP) were investigated through stimulating posterior leg (the
place with 10 cm apart from the recording electrode) for the sural
nerve, the median nerve and the ulnar nerve of the wrist, and then
antidromic recording at lower part of ankle for the sural nerve,
second digit for the median nerve and fifth digit for the ulnar
nerve. Normal values of electromyography were defined as the normal
values used in the Johns Hopkins Hospital in the United States
adjusted for the age under the guidance from Cornblath (18), i.e. parameters of nerve conduction
velocity are similar between adult and children older than 3 years
old.
Standard intensity and duration of stimulation were
applied firstly. For the motor nerve conduction stimulation, the
intensity was 20–40 mA and the duration was 0.1 ms. If three
consecutive stimulations leaded to stable waves with no more than
10% amplitude fluctuation, then the middle value of CMAP was
recorded and used for calculating MCV. For the sensory nerve
conduction stimulation, the intensity was 20–30 mA and the duration
was 0.1 ms. The SNAP was generated by the equipment with the method
of successive averages and recorded when there was no more than 10%
amplitude fluctuation in the wave with stable shape. Then SNAP as
well as the distance between stimulation and recording electrodes
were used for calculating SCV.
Providing examinations failed with the
aforementioned parameters, higher intensities and longer durations
of stimulation were adopted, that is, intensities of 60–80 mA and a
duration of 0.5 ms for the motor nerve conduction stimulation, and
intensities of 20–40 mA and a duration of 0.5 ms for the sensory
nerve conduction stimulation. If the SNAP wave fluctuated with
>10% amplitude and unstable shape when the intensity increased
to 40 mA and duration extended to 1.0 ms, then this examination was
recorded as ‘-’.
Magnetic resonance imaging (MRI)
material
MRI was performed with a GE Signa Excite 1.5T HD
Echospeed platform according to the manufacture's manual. Analyzed
sequences included T1WI FSE [fast spin echo; repetition time (TR) =
500–600 ms, echo time (TE) = 8–12 ms), T2WI FSE (TR = 3,000–4,000
ms, TE = 90–110 ms), T2 FLAIR (fluid-attenuated inversion recovery;
TR = 8,000–9,000 ms, TE = 100–120 ms)] acquired in the axial,
sagittal and coronal planes respectively. The parameters used in
the DWI (diffusion weighted imaging) were as follows: TR = 5000 ms,
TE = 82 ms, slice thickness = 6 mm, slice gap = 1 mm, field of view
(FOV) = 24×24–36×36 cm, matrix = 256×256, and number of excitations
= 2–4. The scanning results were confirmed by a board-certified
neuroradiologist.
Genetic analysis
Targeted sequencing of genes associated with
hereditary ataxias, including KCNA1, CACNA1A, CACNB4, SLC1A3,
SACS, ABCB7, ATM, APTX and TTPA, was conducted on the
proband (II-2), as described previously (19). Sanger sequencing of the identified
pathogenic mutations was conducted on the parents, brother and
fetus (II-3). Sanger sequencing for the fetus was performed at the
Wuhan Children's Hospital. Remaining genetic testing and validation
procedures were carried out in BGI Genomics (Shenzhen, China).
Variant interpretation adhered to the Standards and
Guidelines for the interpretation of sequence variants: A Joint
Consensus Recommendation of the American College of Medical
Genetics And Genomics (ACMG) and the Association for Molecular
Pathology (AMP), 2015 (20).
According to dbSNP database (www.ncbi.nlm.nih.gov/SNP/), HapMap database
(ftp.ncbi.nlm.nih.gov/hapmap/), HGMD
(www.hgmd.cf.ac.uk/), 1000 genomes
project database (www.1000genomes.org/), Exome Sequencing Project 6500
(evs.gs.washington.edu/EVS/), the
Exome Aggregation Consortium (exac.broadinstitute.org/), local SNP databases of 100
normal Chinese (in-house) and available literature, the frequency
and novelty of the variants were consequently determined (21). PolyPhen-2 (22) and SIFT programs (23) were used to evaluate the potential
deleterious effect.
Results
The two live patients were from a Han family with
unaffected parents. The 8-year-old proband (patient II-2) was born
at full term without suffocation. At 1 year of age, he was observed
to have normal development and intelligence, but weak limbs and
poor memory. Typical symptoms of ataxia were noticed at 5 years old
when he presented with slurred speech and evident regression of
movement coordination, including unstable walking, trembling hands
and clumsy action, as well as positive results in the
finger-to-nose and rapid alternating movement tests. Conjunctival
hyperemia was found in both eyes and hair was dry and dull.
Proprioceptive sensibility was normal, but vibration sense was
absent. The knee reflex was normal; however, the ankle reflex was
not elicited. Muscle tensions of four limbs were normal, and muscle
strength was graded as level V. Some abnormal results were found in
the electromyography (EMG) examination (Tables I and II): i) The amplitude (AMP) of peroneal
nerves and ulnar nerves was decreased on both sides; ii) the ulnar
nerves were not elicited; and iii) the values of sensory conduction
velocity (SCV) and AMP of median nerves on both sides were smaller
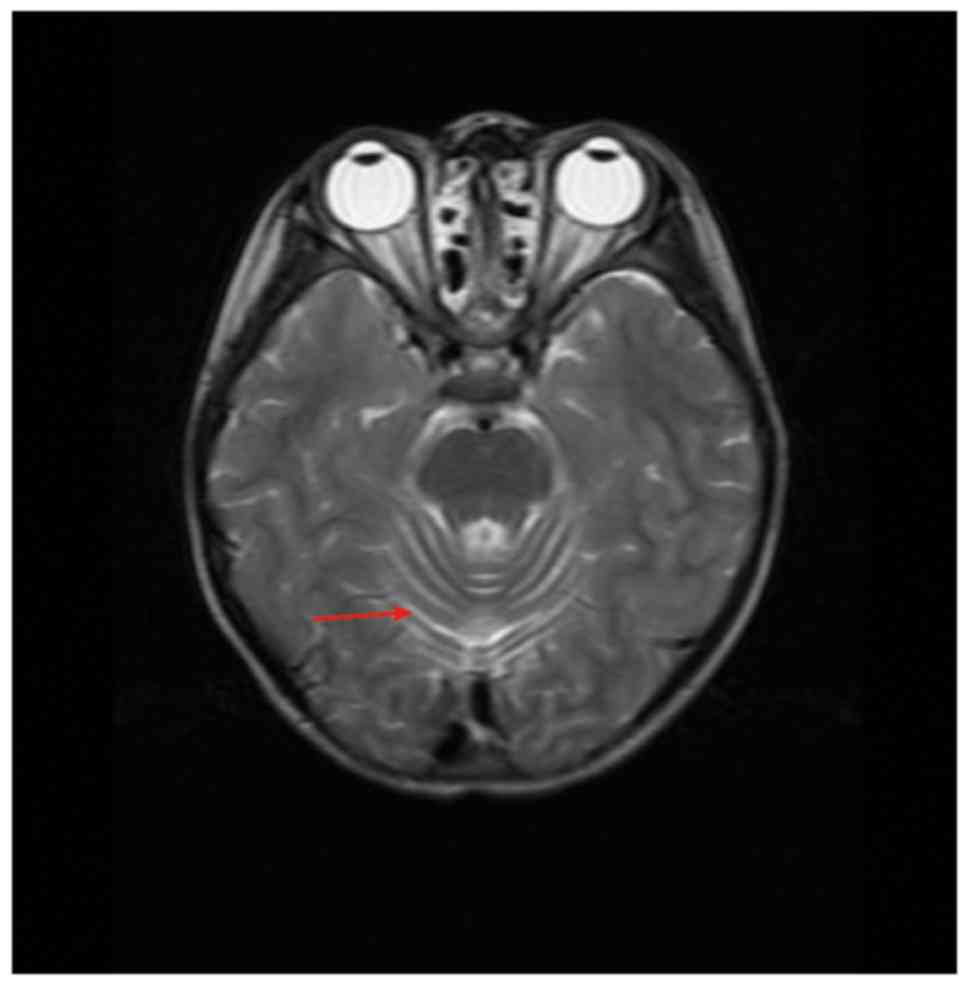
than the normal limits. In addition, brain MRI examinations showed
that the proband had enlarged cerebellar sulci (Fig. 1). According to descriptions from
the parents, the proband was not susceptible to infectious
diseases. However, significantly elevated serum α-fetoprotein (AFP;
170 IU/ml; normal range 0–3.07 IU/ml) (Table III) and slightly decreased
CD4+/CD8+ T lymphocyte ratio (Table IV) was detected in the blood test,
implying hepatic dysplasia and immunodeficiency in the patient.
Other indicators in the blood test were normal or slightly
decreased (Tables III and
IV).
 | Table I.Electromyography results of the
common peroneal, tibial and sural nerves for the two patients. |
Table I.
Electromyography results of the
common peroneal, tibial and sural nerves for the two patients.
|
| Left common
peroneal nerve | Right common
peroneal nerve | Left tibial
nerve | Right tibial
nerve | Left sural
nerve | Right sural
nerve |
|---|
|
|
|
|
|
|
|
|
|---|
| Patient | MCV (m/s) | AMP (mv) | MCV (m/s) | AMP (mv) | MCV (m/s) | AMP (mv) | MCV (m/s) | AMP (mv) | SCV (m/s) | AMP (mv) | SCV (m/s) | AMP (mv) |
|---|
| II:1 | 44.2 (n) | 1.2 (l) | 44.4 (n) | 1.9 (l) | 43.1 (n) | 8.4 (l) | 41.9 (nl) | 6.6 (l) | – | – | – | – |
| II:2 | 48.4 (n) | 1.2 (l) | 48.7 (n) | 0.8 (l) | 47.6 (n) | 21.5 (n) | 49.2 (nl) | 19.2 (n) | 67.6 (n) | 13.3 (n) | 54.6 (n) | 8.4 (n) |
| Table II.Electromyography results of the
median and ulnar nerves for the two patients. |
Table II.
Electromyography results of the
median and ulnar nerves for the two patients.
|
| Left median
nerve | Right median
nerve | Left ulnar
nerve | Right ulnar
nerve |
|---|
|
|
|
|
|
|
|---|
| Patient | MCV (m/s) | AMP (ms) | SCV (m/s) | AMP (µv) | MCV (m/s) | AMP (ms) | SCV (m/s) | AMP (µv) | MCV (m/s) | AMP (mv) | SCV (m/s) | MCV (m/s) | AMP (mv) | SCV (m/s) |
|---|
| II:1 | 58.5 (n) | 2.8 (n) | 47.9 (n) | 11 (l) | 59.5 (n) | 3.8 (n) | 43 (n) | 9.7 (l) | 56.8 (n) | 7.1 (n) | 35.5 (l) | 59.3 (nl) | 4.6 (l) | 40.7 (l) |
| II:2 | 50.4 (n) | 5.7 (n) | 42.7 (l) | 15 (l) | 52.6 (n) | 8.8 (n) | 30.3 (l) | 5.7 (l) | 48.5 (n) | 5.3 (l) | – | 45.6 (nl) | 4.8 (l) | – |
| Table III.Major clinical and laboratory
features of two patients. |
Table III.
Major clinical and laboratory
features of two patients.
|
|
|
|
|
|
|
|
Immunoglobulins | Complement |
|---|
|
|
|
|
|
|
|
|
|
|
|---|
| Patient | Sex | Age (years,
months) | Age of ataxia onset
(months) | Age of
telangiectasia onset (years) | Cerebellar
atrophy | α-fetoprotein
(IU/ml)a | IgG (g/l) | IgA (g/l) | IgM (g/l) | C3c (g/l) | C4 (g/l) |
|---|
| II-1 | M | 13. 5 | 2 | Unknown | Yes | 234.8a | 9.68 | 1.74 | 2.39 | 0.76a | 0.15 |
| II-2 | M | 8.
1 | 1 | Unknown | Yes | 170a | 6.78 | – | – | – | – |
| Table IV.Blood lymphocyte subsets (TBNK)
detected result of two patients. |
Table IV.
Blood lymphocyte subsets (TBNK)
detected result of two patients.
| Patient | CD3+ TLs
(%) | CD3+ TL
count (per µl) | CD8+ TLs
(%) | CD8+ TL
count (per µl) | CD4+ TLs
(%) | CD4+ TL
count (per µl) | NK cells (%) | NK cell count (per
µl) | CD19+
BLs (%) | CD19+ BL
count (per µl) |
CD4+/CD8+ TL
ratio |
|---|
| II-1 | 76.32a | 1,183 | 56.15a | 883 | 16.19 | 255a | 11.55 | 176a | 10.83a | 165a | 0.29a |
| II-2 | 63.72 | 577a | 32.66 | 297 | 24.2 | 220a | 24.44 | 220 |
8.94a |
81a | 0.74a |
The proband's 13-year-old brother (patient II-1) had
all typical symptoms of ataxia, as the proband did, as well as some
additional clinical features. Patient II-1 presented earlier
regression of movement coordination at 2 years old. The symptoms
gradually progressed and as a result, he could not walk at 8 years
old. Brain MRI showed cerebellar atrophy (data not shown), and
intellectual retrogression was confirmed. Both eyes had
conjunctival hyperemia and difficulties in seeing objects on the
left, suggesting oculomotor apraxia. The head and neck had
abnormally slow movement. Muscle strength of the upper and lower
limbs were grade IV and III respectively. EMG results (Tables I and II) were similar in patients II-1 and
II-2, but patient II-1's sural nerve, rather than ulnar nerve, was
not elicited on both sides. Furthermore, AMPs of the tibial nerve
were decreased on both sides. As shown in Tables II and III, serum AFP (234.8 IU/ml; normal
range 0–3.07 IU/ml) was significantly increased and
CD4+/CD8+ T lymphocyte ratio (0.29; normal
range 0.96–2.05) was significantly decreased, implying severe
immunodeficiency.
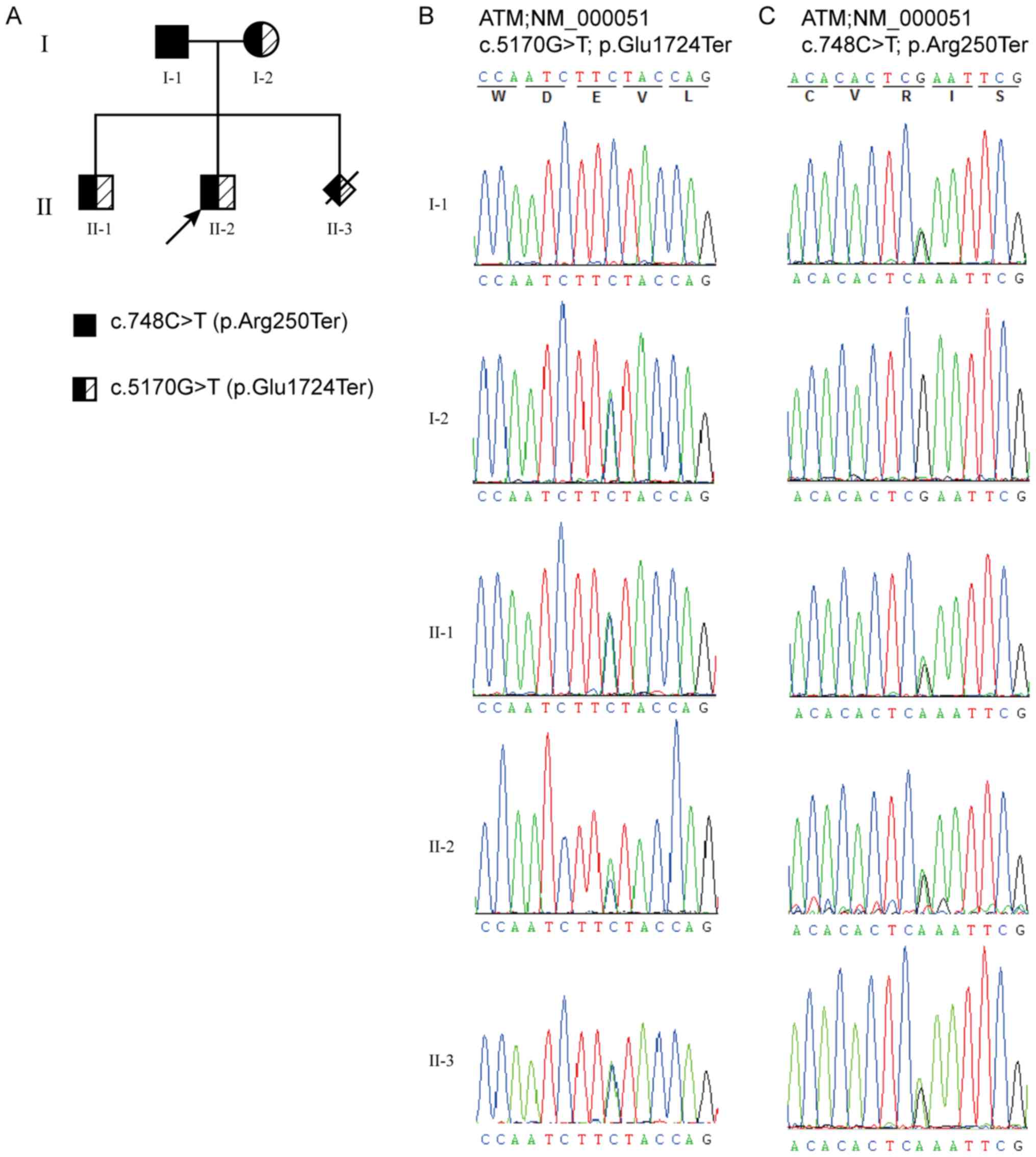
Targeted sequencing was performed on the proband.
The generated data had a mean depth of 285.1-fold and a coverage of
99.71% across the targeted regions (Table V). In total, four non-synonymous
and 10 synonymous variants were identified in nine genes associated
with hereditary ataxias. Once filtered, two nonsense mutations
c.748C>T (p.Arg250Ter) and c.5170G>T (p.Glu1724Ter) in the
ATM gene were deemed to be pathogenic and likely pathogenic,
respectively, according to the guidelines of ACMG/AMP (20). Both mutations were absent in 1000
Genomes Project database, Exome Sequencing Project 6500, The Exome
Aggregation Consortium and local SNP database of normal Chinese.
The mutation c.748C>T occurring in exon 7 converted arginine to
stop codon at amino acid position 250, which has previously been
reported as pathogenic (24–26).
The c.5170G>T mutation located on exon 34 changed glutamic acid
to stop codon at amino acid position 1,724. It is worth noting that
no pathogenicity association between this mutation and A-T has been
identified in the previous literature. Further Sanger sequencing
was performed on the extensive family members to verify whether the
two mutations c.748C>T and c.5170G>T segregated with the
disease. It was found that all three siblings (patient II-1, II-2
and II-3) carried the same compound heterozygous mutations of
c.748C>T and c.5170G>T, which were inherited from their
father and mother, respectively (Fig.
2). These results confirmed A-T in all siblings and the parents
decided to terminate the pregnancy (patient II-3).
| Table V.Bioinformatics quality control
matrices of the proband's targeted next generation sequencing
data. |
Table V.
Bioinformatics quality control
matrices of the proband's targeted next generation sequencing
data.
| Measure | Result |
|---|
| Number of
genes | 9 |
| Length of target
region (bp) | 65,439 |
| Coverage of target
region (%) | 99.71 |
| Average depth of
target region (-fold) | 285.1 |
| Proportion of
target region with sequencing depth of >30-fold (%) | 97.50 |
Discussion
Ataxia-telangiectasia, characterized by progressive
difficulty with coordinating movement, is a rare inherited disorder
that affects the nervous and immune system, as well as other
processes (1). A series of
clinical criteria for A-T diagnosis have been identified (7), but there are still limitations to
prenatal diagnosis, and cases with variable phenotypes or late
onset. By conducting targeted sequencing and Sanger sequencing on
an A-T family with variable clinical signs, novel nonsense ‘likely
pathogenic’ and known nonsense ‘pathogenic’ mutations were found in
the ATM gene, which confirmed the A-T in two patients and
then aided the prenatal diagnosis of A-T in the third child of this
family. Therefore, genetic testing is of crucial importance for
confirming A-T, particularly in the initial phases of the
disease.
The clinical features of patients II-1 and II-2 were
similar with those of the A-T patients in previous reports, such as
a combination of progressive cerebellar ataxia, dysarthria,
conjunctival hyperemia and elevated serum AFP levels (27–29).
However, it was found that the two patients distinguished
themselves with onset, severity and development of the disease. For
example, symptoms of A-T had presented since 2 years old in patient
II-1, but at 5 years old in patient II-2; at 8 years old, patient
II-1 was not able to walk, whereas patient II-2 could walk slowly;
further, patient II-1 had obvious intellectual retrogression, while
patient II-2 had normal intelligence. According to the clinical
examinations, it was speculated that the more severe symptoms in
patient II-1 might be explained by the following findings: i)
Patient II-1's sural nerve, rather than ulnar nerve, was not
elicited in the EMG, therefore disrupting his walking ability; ii)
while patient II-2 had wide cerebellar sulci, the initial stage of
cerebellar atrophy, patient II-1 had cerebellar atrophy, thus
causing more critical consequences; and iii) the indexes of serum
AFP and CD4+/CD8+ T lymphocyte ratio in
patient II-1 were more severely shifted away from the normal
ranges, indicating a more serious immunodeficiency. Taken together,
these findings demonstrated that the phenotypes of A-T were quite
heterogeneous, especially in the initial phases of the disease,
which presents difficulties in making accurate clinical
diagnoses.
The majority of ATM mutations causing A-T are
nonsense and frameshift mutations, resulting in truncation of ATM
protein (30–33). Consistent with these previously
findings, the present study detected two nonsense mutations. The
mutation c.748C>T has been reported to be pathogenic in A-T
patients (24–26). To the best of knowledge, this is
the first paper to identify c.5170G>T to be associated with A-T
pathogenicity. This mutation produces a truncated premature protein
at amino acid position 1724, which is conserved in multiple
species, including rhesus, mouse, dog, elephant, wild yak, and
bonobo.
In conclusion, one novel mutation and one known
disease-inducing mutation of A-T was identified. The present study
not only expanded the mutation spectrum of ATM-associated
A-T, but also contributed valuable guidance on the genetic
diagnosis and the prenatal screening of A-T.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed in the current
study are available in the CNGB Nucleotide Sequence Archive (CNSA,
http://db.cngb.org./cnsa) with accession number
CNP0000265.
Authors' contributions
AZ and XT conceived the study. JC and BM analyzed
the patients and collected clinical data. JC, RS, WZ, BM, QS and RZ
performed data analyses and prepared the manuscript. ZL, BZ, XC,
CZ, ML, PH and JW conducted the genetic experiments. All authors
reviewed and approved the manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of the Children's Hospital of Wuhan (Wuhan, China). The
parents of the patients provided written informed consent.
Patient consent for publication
Not applicable
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Gilad S, Chessa L, Khosravi R, Russell P,
Galanty Y, Piane M, Gatti RA, Jorgensen TJ, Shiloh Y and Bar-Shira
A: Genotype-phenotype relationships in ataxia-telangiectasia and
variants. Am J Hum Genet. 62:551–561. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Schoenaker MH, Suarez F, Szczepanski T,
Mahlaoui N and Loeffen JL: Treatment of acute leukemia in children
with ataxia telangiectasia (A-T). Eur J Med Genet. 59:641–646.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
National Cancer Institute. Ataxia
telangiectasia, . Fact sheet. https://www.cancer.gov/about-cancer/causes-prevention/genetics/ataxia-fact-sheetDecember
18–2018
|
|
4
|
Chun HH and Gatti RA:
Ataxia-telangiectasia, an evolving phenotype. DNA Repair (Amst).
3:1187–1196. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tavani F, Zimmerman RA, Berry GT, Sullivan
K, Gatti R and Bingham P: Ataxia-telangiectasia: The pattern of
cerebellar atrophy on MRI. Neuroradiology. 45:315–319. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Waldmann TA and McIntire KR:
Serum-alpha-fetoprotein levels in patients with
ataxia-telangiectasia. Lancet. 2:1112–1115. 1972. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jason JM and Gelfand EW: Diagnostic
considerations in ataxia-telangiectasia. Arch Dis Child.
54:682–686. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gatti RA, Berkel I, Boder E, Braedt G,
Charmley P, Concannon P, Ersoy F, Foroud T, Jaspers NG, Lange K, et
al: Localization of an ataxia-telangiectasia gene to chromosome
11q22-23. Nature. 336:577–580. 1988. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chun HH, Sun X, Nahas SA, Teraoka S, Lai
CH, Concannon P and Gatti RA: Improved diagnostic testing for
ataxia-telangiectasia by immunoblotting of nuclear lysates for ATM
protein expression. Mol Genet Metab. 80:437–443. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Xu Y, Gao P, Lv X, Zhang L and Zhang J:
The role of the ataxia telangiectasia mutated gene in lung cancer:
Recent advances in research. Ther Adv Respir Dis. 11:375–380. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Goodarzi AA, Jonnalagadda JC, Douglas P,
Young D, Ye R, Moorhead GB, Lees-Miller SP and Khanna KK:
Autophosphorylation of ataxia-telangiectasia mutated is regulated
by protein phosphatase 2A. EMBO J. 23:4451–4461. 2014. View Article : Google Scholar
|
|
12
|
Kozlov SV, Graham ME, Jakob B, Tobias F,
Kijas AW, Tanuji M, Chen P, Robinson PJ, Taucher-Scholz G, Suzuki
K, et al: Autophosphorylation and ATM activation: Additional sites
add to the complexity. J Biol Chem. 286:9107–9119. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Barlow C, Dennery PA, Shigenaga MK, Smith
MA, Morrow JD, Roberts LJ II, Wynshaw-Boris A and Levine RL: Loss
of the ataxia-telangiectasia gene product causes oxidative damage
in target organs. Proc Natl Acad Sci USA. 96:9915–9919. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yan M, Qiang W, Liu N, Shen J, Lynn WS and
Wong PK: The ataxia-telangiectasia gene product may modulate DNA
turnover and control cell fate by regulating cellular redox in
lymphocytes. FASEB J. 15:1132–1138. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yan M, Zhu C, Liu N, Jiang Y, Scofield VL,
Riggs PK, Qiang W, Lynn WS and Wong PK: ATM controls c-Myc and DNA
synthesis during postnatal thymocyte development through regulation
of redox state. Free Radic Biol Med. 41:640–648. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Matsuoka S, Ballif BA, Smogorzewska A,
McDonald ER III, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini
N, Lerenthal Y, et al: ATM and ATR substrate analysis reveals
extensive protein networks responsive to DNA damage. Science.
316:1160–1166. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Medical Research Council, . Aids to
examination of the peripheral nervous system. Memorandum No. 45.
Medical Research Council; London: 1976
|
|
18
|
Cornblath D: Electrophysiology in
Guillain-Barre syndrome. Ann Neurol. 27 (Suppl):S17–S20. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhou X, Liao Y, Xu M, Ji Z, Xu Y, Zhou L,
Wei X, Hu P, Han P, Yang F, et al: A novel mutation R190H in the
AT-hook 1 domain of MeCP2 identified in an atypical Rett syndrome.
Oncotarget. 8:82156–82164. 2017.PubMed/NCBI
|
|
20
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the american college
of medical genetics and genomics and the association for molecular
pathology. Genet Med. 17:405–424. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wei X, Ju X, Yi X, Zhu Q, Qu N, Liu T,
Chen Y, Jiang H, Yang G, Zhen R, et al: Identification of sequence
variants in genetic disease-causing genes using targeted
next-generation sequencing. PLoS One. 6:e295002013. View Article : Google Scholar
|
|
22
|
Adzhubei IA, Schmidt S, Peshkin L,
Ramensky VE, Gerasimova A, Bork P, Kondrashov AS and Sunyaev SR: A
method and server for predicting damaging missense mutations. Nat
Methods. 7:248–249. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kumar P, Henikoff S and Ng PC: Predicting
the effects of coding non-synonymous variants on protein function
using the SIFT algorithm. Nat Protoc. 4:1073–1081. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Moulton J: Functional characterization and
targeted correction of ATM mutations identified in Japanese
patients with ataxia-telangiectasia. Hum Mutat. 33:198–208.
2015.
|
|
25
|
Teraoka SN, Telatar M, Becker-Catania S,
Liang T, Onengüt S, Tolun A, Chessa L, Sanal O, Bernatowska E,
Gatti RA and Concannon P: Splicing defects in the
ataxia-telangiectasia gene, ATM: Underlying mutations and
consequences. Am J Hum Genet. 64:1617–1631. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Buzin CH, Gatti RA, Nguyen VQ, Wen CY,
Mitui M, Sanal O, Chen JS, Nozari G, Mengos A, Li X, Fujimura F and
Sommer SS: Comprehensive scanning of the ATM gene with DOVAM-S. Hum
Mutat. 21:123–131. 2010. View Article : Google Scholar
|
|
27
|
Lavin MF and Shiloh Y: The genetic defect
in ataxia-telangiectasia. Ann Rev Immunol. 15:177–202. 2003.
View Article : Google Scholar
|
|
28
|
Taylor AM, Byrd PJ, McConville CM and
Thacker S: Genetic and cellular features of ataxia telangiectasia.
Int J Radiat Biol. 65:65–70. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kraus M, Lev A, Simon AJ, Levran I,
Nissenkorn A, Levi YB, Berkun Y, Efrati O, Amariglio N, Rechavi G
and Somech R: Disturbed B and T cell homeostasis and neogenesis in
patients with ataxia telangiectasia. J Clin Immunol. 34:561–572.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wright J, Teraoka S, Onengut S, Tolun A,
Gatti RA, Ochs HD and Concannon P: A high frequency of distinct ATM
gene mutations in ataxia-telangiectasia. Am J Hum Genet.
59:839–846. 1996.PubMed/NCBI
|
|
31
|
Concannon P and Gatti RA: Diversity of ATM
gene mutations detected in patients with ataxia-telangiectasia. Hum
Mutat. 10:100–107. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li A and Swift M: Mutations at the
ataxia-telangiectasia locus and clinical phenotypes of A-T
patients. Am J Med Genet. 92:170–177. 2010. View Article : Google Scholar
|
|
33
|
Jacquemin V, Rieunier G, Jacob S,
Bellanger D, d'Enghien CD, Laugé A, Stoppa-Lyonnet D and Stern MH:
Underexpression and abnormal localization of ATM products in ataxia
telangiectasia patients bearing ATM missense mutations. Eur J Hum
Genet. 20:305–312. 2012. View Article : Google Scholar : PubMed/NCBI
|