Introduction
As the average age of the population increase, the
prevalence of neurodegenerative disorders including Alzheimer's
disease (AD), Parkinson's disease (PD), amyotrophic lateral
sclerosis (ALS) and multiple sclerosis (MS) increases, while an
increased prevalence of osteoporosis (OP) has paralleled the
increase in these neurodegenerative disorders over previous decades
(1). A prospective study in China
has demonstrated that in males and females, decreased bone mineral
density (BMD) and an increased rate of bone loss were associated
with a higher risk of AD (2).
Compared with the general population, BMD appeared to be decreased
and the OP rate was increased in patients suffering from PD
(3). Although studies
investigating OP in patients with ALS are scarce, bone mineral loss
has been noted in ALS (4). In a
cohort of young men with MS, 80% presented with bone mass loss and
of these, 37% exhibited overt OP (5). Although evidence has indicated that
OP is closely associated with the progression of neurodegenerative
diseases, evidence to confirm the causes is lacking. In the present
study, the association between AD and OP was investigated and the
aim was to examine how amyloid-β1–42
(Aβ1–42), the typical pathological product of AD,
induced a negative effect on the proliferation of BMSCs.
AD and OP are 2 slowly-progressing but common
age-associated diseases primarily affecting the elderly worldwide,
and severely decreasing their quality of life. Decreases in
cognitive competence, behavioral disorders and gradual loss of
autonomy are frequently observed in patients suffering from AD.
While OP is a systemic disease caused by a number of etiological
factors, a decrease in BMD, impaired bone microstructure, increased
bone fragility and fracture risk are frequently observed. AD and OP
appear to be 2 independent diseases, but they share certain common
risk factors including alcohol and tobacco consumption (6–8).
Increasing evidence has indicated that the decrease in BMD is
associated with the development of AD (2)and that OP and hip fractures are common
complications observed in patients with AD, but whether these
phenomena are part of a pathological process during the development
of AD or are a ‘by-product’ of disuse OP caused by neurological
function disorders of patients with AD remains poorly
understood.
A previous study based on amyloid precursor protein
(APP)/PS1 transgenic mice has demonstrated that bone microstructure
was poorer in these AD model mice compared with a negative control
(9), and mRNA and protein levels
of Aβ were increased in the bone tissue of patients with OP
(10), indicating that dementia
may result in adverse effects to the skeletal system. Amyloid-β
(Aβ) peptides are typical pathological products of AD and serve an
important role in the development of AD; the toxic effect of
Aβ1–42 is the more notable (11). Bone marrow mesenchymal stem cells
(BMSCs), possessing key properties including self-renewal and
pluripotency, have been extensively studied and are acknowledged to
serve a key role in bone metabolism. Proliferation of BMSCs,
independent of their differentiation potential, is also associated
with the bone formation processes essential for repair and renewal
of old and dead cells. At present, the effect of Aβ1–42
on the proliferation of BMSCs remains unclear and requires
additional study.
Autophagy, which depends upon the formation of
autophagosomes, is regarded as an essential process for the
elimination of damaged organelles and biomacromolecules to maintain
cellular homeostasis. As a cell regulatory process, autophagy
serves an important role in regulating BMSC function (12,13).
Autophagy has been demonstrated not only to participate in the
formation, but also the elimination, of Aβ (14). However, the effect of autophagy on
the proliferation of Aβ1–42-treated BMSCs remains
unclear. Protein kinase B (AKT) and mechanistic target of rapamycin
(mTOR), key regulatory factors within the AKT/mTOR signaling
pathway may be phosphorylated and serve a critical role in
regulating multiple cell functions. The AKT/mTOR signaling pathway
is associated with cell growth (15) and autophagy (16), but whether this pathway
participates in the regulation of autophagy in BMSCs following
treatment with Aβ1–42 remains unknown.
The aim of the present study was to determine the
effect on proliferation of BMSCs treated with Aβ1–42
in vitro, and the potential role of the AKT/mTOR signaling
pathway and autophagy in this process.
Materials and methods
Cell line and primary reagents
Sprague-Dawley rat BMSCs were purchased from Cyagen
Biosciences (RASMX-01001, Guangzhou, China) Inc. Based on the cell
descriptions provided by the supplier, the BMSCs were positive for
the cell surface markers cluster of differentiation (CD)29, CD44
and CD90, and negative for CD11, CD34 and protein tyrosine
phosphatase receptor type, C. BMSCs were cultured with L-Dulbecco's
modified Eagle's medium (10567-014; Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) containing 10% fetal bovine
serum (SH30070.03; Hyclone; GE Healthcare Life Sciences, Logan, UT,
USA) and 1% penicillin-streptomycin (SV30010.10, Hyclone; GE
Healthcare Life Sciences), and were placed in a cell incubator with
a humidified 5% CO2 atmosphere at 37°C. The media was
changed every other day. Only cells in the 6th generation or
younger were used in the present study. Aβ1–42 peptide
freeze-dried powder (A9810), the autophagy inducer rapamycin (RAPA;
V900930) and the inhibitor 3-methyladenine (3-MA; M9281) were all
obtained from Sigma-Aldrich; Merck KGaA (Darmstadt, Germany). The
cell cycle analysis kit (C1052) was purchased from Beyotime
Institute of Biotechnology (Shanghai, China) and the
5-ethynyl-2′-deoxyuidine (EdU) cell proliferation kit (C-10310-3)
was obtained from Guangzhou RiboBio Co., Ltd. (Guangzhou, China).
All autophagy-associated antibodies used in the present study,
including microtubule-associated proteins 1A/1B light chain 3B
(LC3B-II; ab48394; 1: 3,000) and sequestosome 1 (p62; ab56416;
1:1,000), were purchased from Abcam (Cambridge, MA, USA), and the
AKT/mTOR signaling pathway-associated antibodies AKT (2920;
1:1,000), mTOR (2983; 1:1,000), phosphorylated (p)-AKT (4060;
1:1,000) and p-mTOR (5536; 1:1,000), were obtained from Cell
Signaling Technology, Inc., (Danvers, MA, USA). The AKT/mTOR
signaling pathway activator SC79 (HY-18749) was obtained from
MedChemExpress, Monmouth Junction, NJ, USA. Each experiment in the
present study was repeated independently 3 times.
Preparation of Aβ1–42
The Aβ1–42 used in the present study was
prepared as previously described (17).
Cell viability assay
Cell viability of BMSCs was determined using the
Cell Counting Kit-8 (CCK-8) assay (C0037; Beyotime Institute of
Biotechnology). Cells were seeded into 96-well plates at a density
of 5×103 cells/well and cultured for 48 h, then divided
into different groups, and each group consisted of 3 wells in
parallel. Following addition of the 10 µl CCK-8 reagent and
incubation at 37°C for 1 h, optical density was evaluated using a
microplate reader at a wavelength of 450 nm.
Cell cycle analysis
BMSCs were seeded into 6-well plates at a density of
1×104 cells/well. When cell confluence reached 40%,
media with or without increasing concentrations of
Aβ1–42 (1, 2.5 and 5 µM/l) and media with or without
Aβ1–42 (5 µM/l), Aβ1–42 (5 µM/l) + 3-MA (2
mM/l), Aβ1–42 (5 µM/l) + SC79 (4 µg/ml) and
Aβ1–42 (5 µM/l) + SC79 (4 µg/ml) + RAPA (3 µM/l) were
added. After 48 h; culture, cells were collected. Following washing
with cold PBS twice, 70% alcohol was added for fixation for 2 h at
4°C. Subsequent to the addition of 500 µl pre-prepared operating
fluid consisting of RNase A and propidium iodide (5 µg/ml, Beyotime
Institute of Biotechnology) and incubation for 60 min in the dark
at room temperature, samples were then immediately analyzed using a
flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA) at a
wavelength of 488 nm and ModFit LT version 5 (Verity Software
House, Topsham, ME, USA) was used to analyze the data.
EdU cell proliferation analysis
As an efficient and well-known method for detecting
cell proliferation, EdU analysis was used in the present study.
BMSCs were seeded into 96-well plates at a density of
5×103 cells/well and divided into different groups.
According to the protocol of the manufacturer, cells were incubated
with the pre-prepared EdU solution for 2 h, then following washing
with PBS twice, 4% paraformaldehyde was used for fixation for 15
min at room temperature, followed by washing with 2 mg/ml glycine
solution. Preparation of Apollo staining was performed and cells
were incubated for 30 min at room temperature in the dark. Then 100
µl 0.5% Triton X-100 solution was used for permeation. Subsequent
to washing with PBS, 100 µl 1X Hoechst33342 solution was added for
staining of DNA for 30 min at room temperature in the dark. The
results were then immediately detected using a wide-field
fluorescence microscope (IX71; Olympus Corporation, Tokyo, Japan;
magnification, ×400) and the number of EdU positive cells was
counted.
Western blot analysis
BMSCs were collected following treatment with or
without increasing concentrations of Aβ1–42 (1, 2.5 and
5 µM/l) and with or without Aβ1–42 (5 µM/l),
Aβ1–42 (5 µM/l) + 3-MA (2 mM/l), Aβ1–42 (5
µM/l) + SC79 (4 µg/ml) and Aβ1–42 (5 µM/l) + SC79 (4
µg/ml) + RAPA (3 µM/l) for 48 h, lysed with RIPA lysis buffer for
30 min on ice, and centrifuged at 12,000 × g for 30 min at 4°C.
Following total protein quantification using a bicinchoninic acid
protein assay (P0010s; Beyotime Institute of Biotechnology),
samples containing 30 µg total protein were resolved by SDS-PAGE
(5% stacking gel and 10% separating gel) under a voltage of 80 V,
and transferred onto polyvinylidene difluoride membranes by
electroblotting at 110 mA for 60 min. Membranes were blocked by
incubating with 5% bovine serum albumin (BSA; Beyotime Institute of
Biotechnology) for 2 h at room temperature, and then membranes were
incubated with anti LC3B-II (ab48394; 1:3,000; Abcam, Cambridge,
MA, USA), p62 (ab56416; 1:1,000 Abcam), AKT (2920; 1:1,000 Cell
Signaling Technology, Inc., Danvers, MA, USA), mTOR (2983; 1:1,000;
Cell Signaling Technology, Inc.), p-AKT (4060; 1:1,000; Cell
Signaling Technology, Inc.) and p-mTOR (5536; 1:1,000; Cell
Signaling Technology, Inc.) antibodies at 4°C overnight followed by
incubation with secondary antibodies conjugated to horseradish
peroxidase (ZB-2306; 1:1,000; OriGene Technologies, Inc., Beijing,
China) for 2 h at room temperature. The EC3 imaging system (UVP;
Analytik Jena AG, Jena, Germany) was used to detect the proteins
and ImageJ version 1.44P (National Institutes of Health, Bethesda,
MD, USA) was used to perform the densitometric analysis.
Autophagosome analysis with
transmission electron microscopy (TEM) and immunofluorescence
For TEM (H-7650; Hitachi, Ltd., Tokyo, Japan), BMSCs
from the control and 5 µM/l Aβ1–42-treated groups were
collected using a cell scraper following culture for 48 h, and
cells were washed twice with cold PBS and then fixed in 5%
glutaraldehyde at 4°C overnight. Dehydration, saturation,
sectioning at 70 nm and staining of the samples were conducted
according to standard procedures by the electron microscopy room of
Department of Cell Biology, China Medical University, and
autophagosomes were observed using a transmission electron
microscope and counted in every 10 fields.
For immunofluorescent staining, 4% paraformaldehyde
(P0099; Beyotime Institute of Biotechnology) was used for the
fixation of cells from different groups at room temperature for 15
min. Following washing with PBS, 0.2% Triton X-100 was added to
permeabilize the cells for 10 min, then they were blocked in 5% BSA
in blocking buffer for 1 h at 37°C followed by incubation with
anti-LC3 antibody (ab48394; 1:200; Abcam) overnight at 4°C. The
goat anti-rabbit secondary antibody labeled with fluorescein
(ZF-0511; 1:500; OriGene Technologies, Inc.) was then applied for 2
h. Following staining with 10 µg/ml 4′,6-diamidino-2-phenylindole
(C1006; Beyotime Institute of Biotechnology) for 10 min in the dark
at room temperature and rinsing with PBS again, a wide-field
fluorescence microscope (IX71; Olympus Corporation; magnification,
×600) was used to detect the autophagosomes. The number of LC3
puncta was counted visually among 3 randomly-selected fields.
Statistical analysis
Data are presented as the mean ± standard error of
the mean of 3 independent experiments performed in triplicate and
the GraphPad Prism 5.0 software (GraphPad Software, Inc., La Jolla,
CA, USA) was used for statistical analysis. Statistically
significant differences between two groups were analyzed using
Student's t-test. Differences between multiple groups were analyzed
with one-way analysis of variance, followed by a
Student-Newman-Keuls post-hoc test. P<0.05 was considered to
indicate a statistically significant difference.
Results
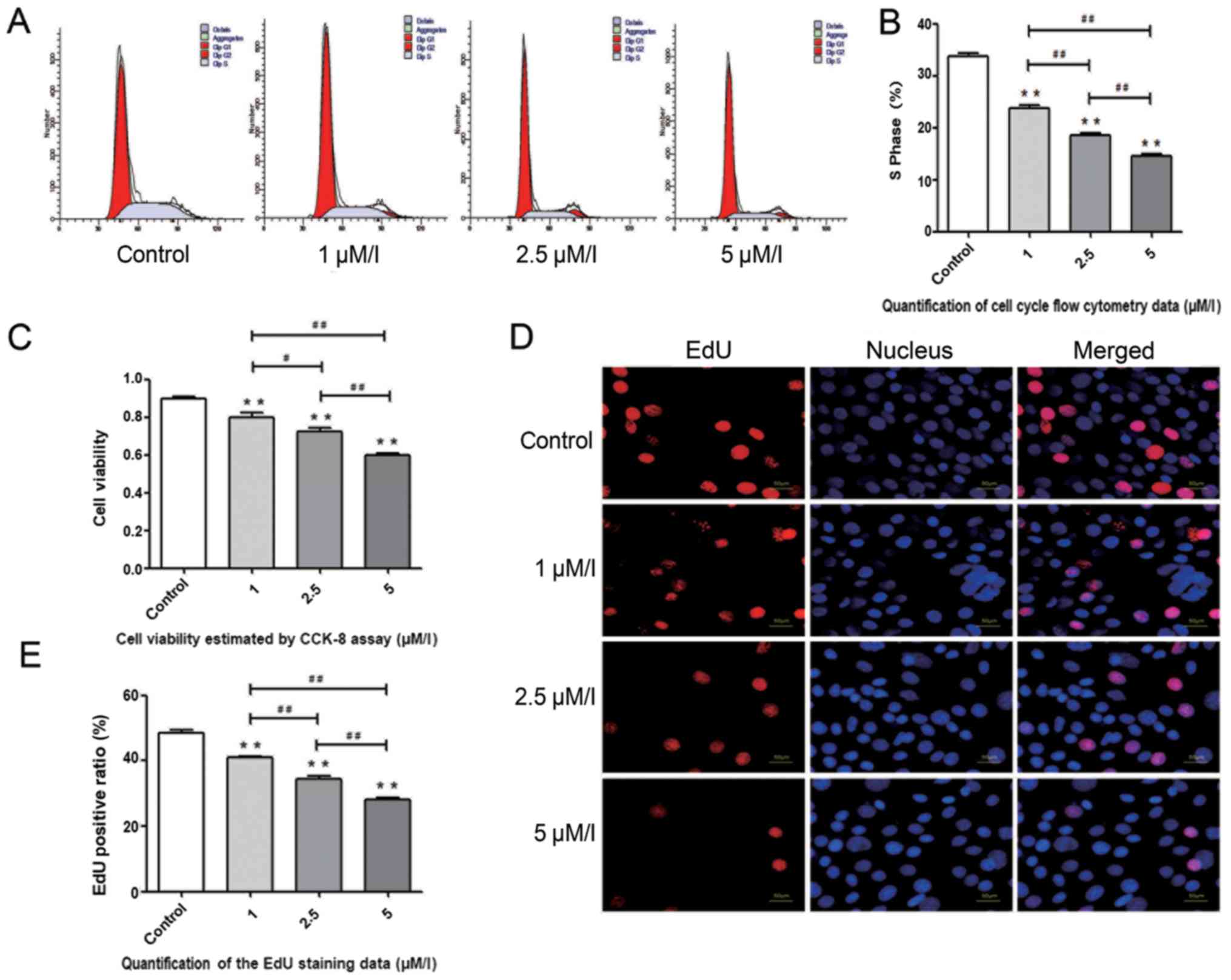
Aβ1–42 exhibits an adverse
effect on the proliferation of BMSCs
To determine the effect of Aβ1–42 on the
proliferation of BMSCs, cells were divided into different groups as
follows: Control, 1, 2.5 and 5 µM/l Aβ1–42, and treated
for 48 h. The proliferation of BMSCs was measured by cell cycle
analysis, CCK-8 assay and EdU staining. It was demonstrated that
the S phase BMSCs detected by flow cytometry decreased gradually
with the increasing concentrations of Aβ1–42 (Fig. 1A and B). The CCK-8 assay indicated
that the viability of BMSCs was impaired directly by
Aβ1–42 (Fig. 1C). EdU
staining, a rapid and sensitive method for detecting proliferation,
demonstrated a similar result to the cell cycle analysis and CCK-8
assay (Fig. 1D and E).
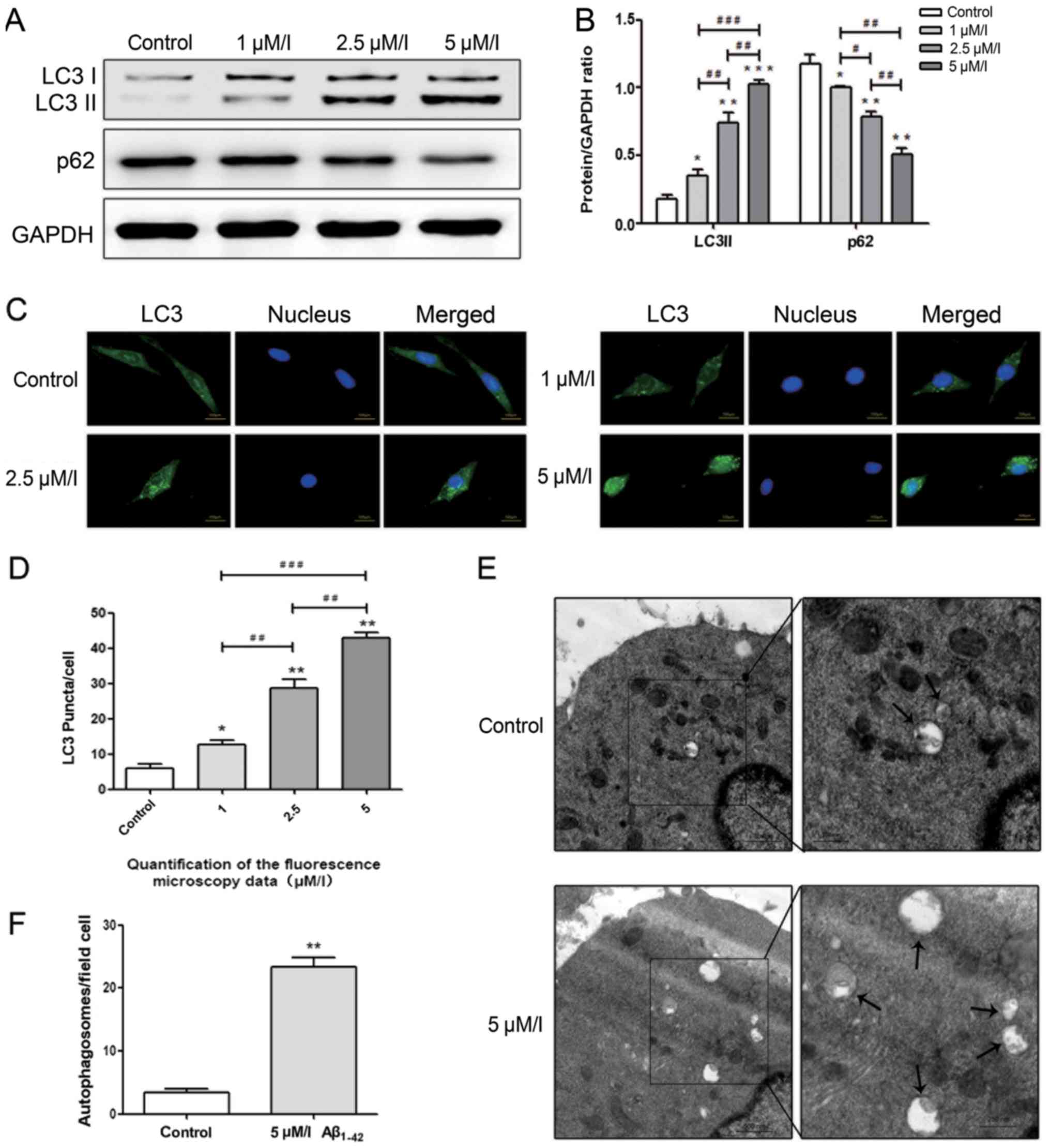
Autophagy level increases in BMSCs
treated with Aβ1–42
As demonstrated, LC3 and p62 are positively and
negatively associated with the level of autophagy, respectively.
According to the results of the present study, the protein level of
LC3 increased gradually with increasing concentrations of
Aβ1–42 (1, 2.5 and 5 µM/l), and the expression of p62
decreased accordingly (Fig. 2A and
B). Fluorescence microscopy was also used to detect the number
of LC3 puncta at the cellular level and the results indicated that
the number of LC3 puncta increased with the increasing
concentrations of Aβ1–42 (Fig. 2C and D). TEM, a reliable method for
detecting autophagy, was used to demonstrate the occurrence of
autophagy induced by 5 µM/l Aβ1–42 compared with the
control group, and an increased number of autophagosomes were
detected in 5 µM/l Aβ1–42 group compared with the
control group (Fig. 2E and F).
Autophagy of BMSCs induced by
Aβ1–42 is mediated via the AKT/mTOR signaling
pathway
The present study demonstrated that the AKT/mTOR
signaling pathway, which is negatively associated with autophagy,
was suppressed following treatment with 5 µM/l Aβ1–42
following 48 h culture. To additionally assess whether the AKT/mTOR
signaling pathway participated in the regulation of autophagy in
Aβ1–42-treated BMSCs, 2 mM/l 3-MA, 4 µg/ml SC79, and 4
µg/ml SC79 +3 µM/l RAPA was added following treatment with 5 µM/l
Aβ1–42, and western blot analysis was performed 48 h
later. The results indicated that, compared with the
Aβ1–42 group, the activation of AKT and inhibition of
autophagy initiated the AKT/mTOR signaling pathway, while the
activation of autophagy using RAPA suppressed the expression of
mTOR compared with the Aβ1–42 + SC79 group (Fig. 3A and B). Accordingly, the
expression of LC3 and p62 demonstrated that the autophagy level
decreased in the Aβ1–42 +3-MA and Aβ1–42 +
SC79 groups compared with the Aβ1–42 group, and
increased in Aβ1–42 + SC79 + RAPA group compared with
the Aβ1–42 + SC79 group (Fig. 3A and C). These results indicated
that the AKT/mTOR signaling pathway was directly involved in the
regulation of autophagy induced by Aβ1–42.
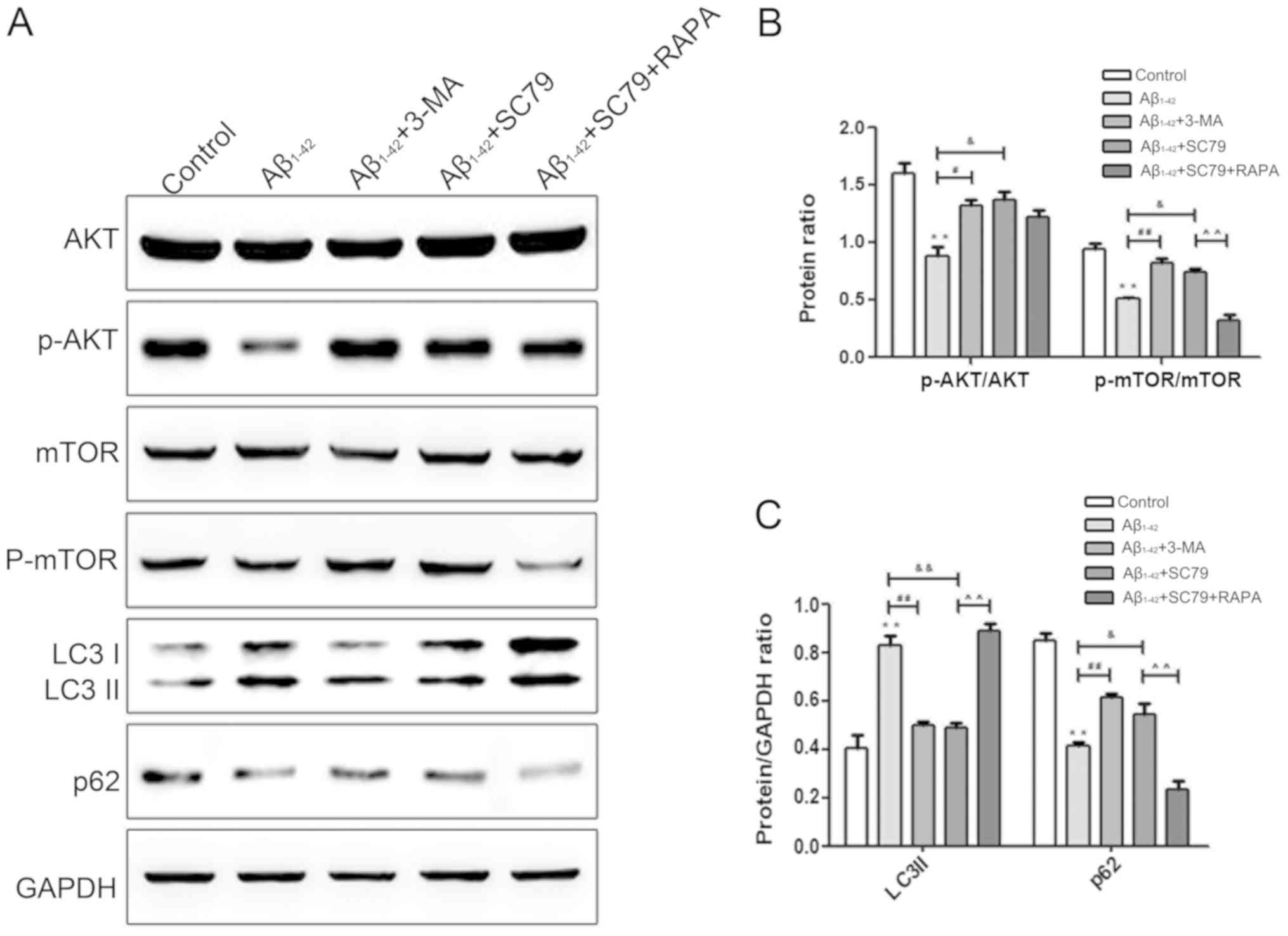 | Figure 3.Autophagy is mediated via the
AKT/mTOR signaling pathway in Aβ1–42-treated BMSCs. (A)
Western blot analysis demonstrating the protein levels of LC3, p62,
AKT, p-AKT, mTOR and p-mTOR following treatment with
Aβ1–42, 3-MA, SC79 and SC79 + RAPA for 48 h, and the
expression of the AKT/mTOR signaling pathway-associated proteins of
AKT, p-AKT, mTOR and p-mTOR following treatment with
Aβ1–42, 3-MA, SC79 and SC79 + RAPA for 48 h. (B)
Quantification of the AKT, p-AKT, mTOR and p-mTOR western blot
analysis data. **P<0.01 vs. the control group.
#P<0.05 and ##P<0.01 vs. the
Aβ1–42 group, &P<0.05 vs. the
Aβ1–42 group. ^^P<0.01 vs. the
Aβ1–42 + SC79 group. n=10 per group. (C) Quantification
of the western blot analysis data demonstrating the expression of
the AKT/mTOR signaling pathway-associated proteins AKT, p-AKT, mTOR
and p-mTOR. **P<0.01 vs. the control group.
##P<0.01 vs. the Aβ1–42 group.
&P<0.05 and &&P<0.01 vs.
the Aβ1–42 group. ^^P<0.01 vs. the
Aβ1–42 + SC79 group. n=10 per group. All values are
presented as the mean ± standard error of the mean from 3
independent experiments. Aβ, amyloid β; BMSC, bone mesenchymal stem
cells; mTOR, mammalian target of rapamycin; AKT, protein kinase B;
p, phosphorylated; RAPA, rapamycin; LC3, microtubule-associated
proteins 1A/1B light chain 3B; LC3 II, lipid-modified LC3; p62,
sequestosome 1; 3-MA, 3-methyladenine. |
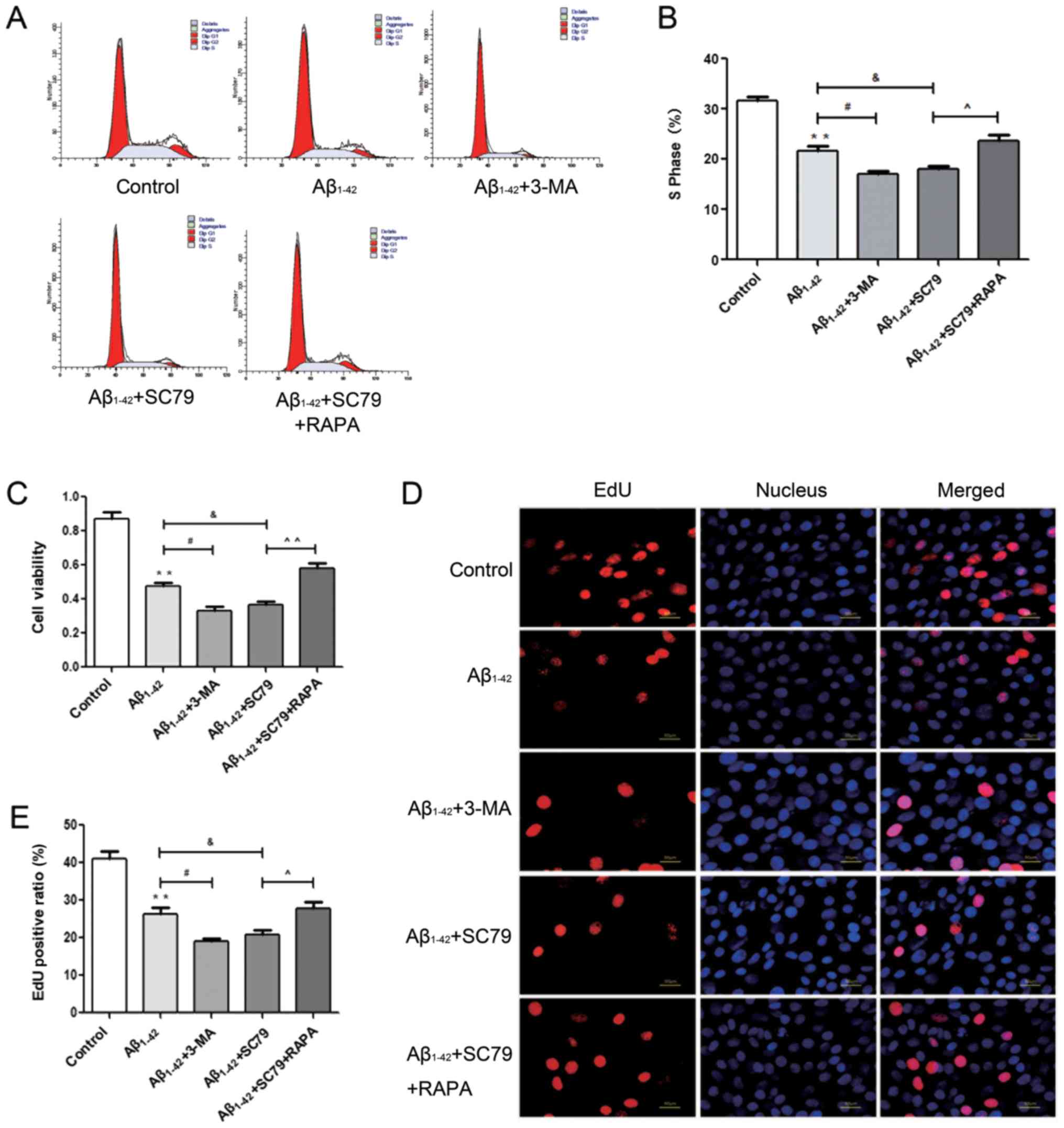
Autophagy alleviates the decrease in
proliferation of BMSCs treated with Aβ1–42
To determine the potential role of autophagy induced
by Aβ1–42, the proliferation of BMSCs was examined
accordingly. Based on the inhibitory effect of Aβ1–42 on
the proliferation of BMSCs, fewer S phase cells were detected in
the 2 Mm/l autophagy inhibitor 3-MA group and 4 µg/ml AKT agonist
SC79 group by flow cytometry, but this decrease was partly reversed
by 3µM/l RAPA, the autophagy inducer (Fig. 4A and B). The CCK-8 assay
demonstrated that the cell viability of BMSCs was decreased
following treatment with 3-MA and SC79 compared with the
Aβ1–42 group, while RAPA inhibited this decrease
(Fig. 4C). EdU staining revealed
that the suppression of DNA replication was more pronounced when
autophagy was inhibited or when AKT was activated compared with the
effect of Aβ1–42 alone, while the activation of
autophagy with RAPA alleviated the decrease in DNA replication
induced by SC79 (Fig. 4D and
E).
Discussion
The causes of neurodegenerative disorders and OP
remain unclear, and the association between neurodegenerative
diseases and OP is also unknown. It has been hypothesized that an
environmental toxicant may contribute to the development of
neurodegenerative disorders, for example, free copper (Cu) ions may
mediate the aggregation of Aβ in AD brains (18), and overexposure to Cu from the
environment is a risk for AD (19). Iron (Fe) has been demonstrated to
participate in the pathological process of PD (20). Aluminum, Cu, zinc and a number of
other ions have been demonstrated to be significantly increased in
the cerebrospinal fluid of patients with ALS (21), and accumulation of Fe is also an
early event in MS (22). In the
pathology of OP, environmental cadmium exposure is associated with
an increased loss of BMD in males and females, leading to OP and
increased risk of fractures, particularly in the elderly and
females (23,24). All the aforementioned evidence has
indicated that the external environment, in particular metal ions,
participate in the pathological processes of these two
neurodegenerative diseases and OP, but whether the internal factors
of neurodegenerative diseases, including typical pathological
products, affect the process of OP remains unknown. In the present
study, it was demonstrated that Aβ1–42, an endogenous
pathological product of AD, inhibited the proliferation of BMSCs,
which provided additional evidence for the occurrence of
AD-associated OP.
Clinically, OP is frequently perceived to occur
concurrently with the development of AD. Previous studies have
demonstrated that the level of hip BMD is decreased and risk of hip
fracture is increased in patients with AD (25,26).
A study involving an AD mouse model expressing a Swedish mutation
of APP indicated that impaired bone mass was detected (27), and that such suppression of
osteoblastogenesis and bone formation in Tg2576 mice, a breed of AD
model mice, was triggered by reactive oxygen species induced by
mutant APP (28). Furthermore, our
previous study also demonstrated that excessive Aβ was identified
in the bone tissue of APP/PS1 transgenic mouse, and bone mineral
loss was more serious compared with the control group (9). In addition, Aβ has been suggested to
enhance the function of osteoclasts (OCs) (10), and gene knockout experiments and
the use of Tg2576 mice have identified a role for Aβ in the
activation of OCs (29,30), Aβ also enhanced receptor activator
of nuclear factor kappa-light-chain-enhancer of activated B cells
ligand-induced OC activation through calcium oscillation signaling
pathways (31). An OC is a
regulatory cell in bone resorption, and serves a key role in the
development of OP. These data have demonstrated that OP may occur
secondary to AD. AD is characterized pathologically by synapse loss
and the presence of Aβ plaques and tau tangles (32). Aβ, a peptide consisting of 36–43
amino acids, is generated via sequential proteolysis of APP by
β-secretase and γ-secretase. Aβ is known to be specifically toxic
to neurons (33), while the
noxious effect of Aβ1–42, the major component of senile
plaques, is the most remarkable. Despite this, conclusive evidence
to demonstrate the effect of Aβ on bone metabolism is lacking.
BMSCs, the progenitor cells of osteoblasts, participate indirectly
in the homeostasis of bone formation and absorption. In addition to
differentiation, proliferation is also an important function of
BMSCs and it is required for BMSCs to expand cell populations to
perform certain functions. As demonstrated previously, Aβ inhibits
the proliferation of neural stem cells (NSCs) (34) and serves a crucial role in the
development of AD due to its toxic effects. In the present study,
it was demonstrated that Aβ1–42 decreased cell
viability, the number of cells in S phase and the level of DNA
replication of BMSCs in a dose-dependent manner; these results
provided direct evidence that Aβ1–42 may exert a
negative effect on the proliferation of cells from the brain,
particularly on cells from the skeletal system, and that they had a
similar effect to that of Aβ on the proliferation of NSCs,
indicating that Aβ1–42 may also serve a critical role in
the development of AD-associated OP.
Autophagy, since its identification, has been
recognized as an essential process by which damaged organelles and
biomacromolecules are eliminated (35,36).
This degradation pathway depends upon the formation of
autophagosomes with double-layered membranes, which combine with
lysosomes and result in degradation of the contents, and is
associated with various human disorders, including
neurodegenerative diseases, cancer and infectious diseases
(37). Autophagy may be activated
in response to adverse environmental conditions including nutrient
deprivation, exposure to toxic agents and a number of other stress
signals (38–41) and serves as a survival mechanism to
maintain cell functions. As recommended techniques for detecting
autophagy (42), western blot
analysis, immunofluorescence and TEM were employed, and
demonstrated that the autophagy level increased with increasing
concentration of Aβ1–42. These results were similar to
the phenomenon that Aβ upregulated the autophagy level in the brain
and PC12 cells (43). Notably,
this upregulation in autophagy level was accompanied by a decrease
in proliferation in BMSCs following treatment with
Aβ1–42. However, additional studies are required to
investigate the underlying mechanism of autophagy induced by
Aβ1–42 and the role of autophagy. mTOR, in particular
the mTOR complex 1, is a key regulator of autophagy and cell
proliferation. mTOR receives inputs from different signaling
pathways. In the present study, it was demonstrated that
alternations to AKT, an upstream modulator, were consistent with
the variation tendency of mTOR. The phosphorylation of AKT and mTOR
decreased following treatment with Aβ1–42, suggesting
that the AKT/mTOR signaling pathway was involved in this regulatory
process. Furthermore, the use of autophagy inhibitor 3-MA and AKT
activator SC79 increased the suppression of the AKT/mTOR signaling
pathway induced by Aβ1–42. Accordingly, the autophagy
level also decreased, while treatment with RAPA, an autophagy
inducer, resulted in marked decreases in the level of p-mTOR, while
the level of autophagy increased. As a result, it was determined
that autophagy induced by Aβ1–42 was mediated via the
AKT/mTOR signaling pathway. The phosphoinositol 3-kinase/AKT/mTOR
signaling pathway serves a critical role in the central nervous
system, particularly in the pathology of AD (44,45);
the results of the present study provided evidence that this
pathway may also serve a role in AD-associated OP.
The effects of autophagy may be two-fold: Knockout
of autophagy-related gene resulted in a higher rate of cell death
(46), while it has been
demonstrated that autophagy plays a regulatory role in human tumor
cell death and cell death control in numerous studies (47,48)
As an essential and highly-conserved intracellular degradation
process, autophagy serves a significant role in eukaryotic cell
growth, cell death, infection and homeostasis (49,50).
Autophagy may increase cell proliferation in conditions of external
stress, including hypoxia (51).
Although conflicting results exist, the majority of studies
consider autophagy as a protective mechanism in AD pathology
(52,53). In the present study, when autophagy
induced by Aβ1–42 was suppressed using 3-MA, or the
AKT/mTOR signaling pathway was activated by SC79, the proliferation
of BMSCs increased. Following treatment with the autophagy inducer
RAPA, it was demonstrated that the proliferation of BMSCs increased
even following treatment with SC79. These results indicated that
autophagy was likely to be beneficial for Aβ1–42-treated
BMSCs. The results of the present study are consistent with
previous studies that focused on the role of autophagy induced by
Aβ in brain tissue; for example, autophagy enhanced by RAPA rescued
dysfunctions in AD model mice (52,54).
The present study hypothesized that autophagy may also exhibit a
protective role in AD-associated OP.
In conclusion, the present study demonstrated that
Aβ1–42 inhibited the proliferation of BMSCs and
upregulated the autophagy level simultaneously. The present study
also suggested that the AKT/mTOR signaling pathway was involved in
Aβ1–42-induced autophagy, and that this autophagy served
a protective role in confronting the negative effects of
Aβ1–42. These data provide an improved understanding of
the pathogenesis of AD-associated OP, and regulating the autophagy
level may be a novel therapeutic target.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Chinese
National Natural Science Foundation project (grant nos., 81471094
and 81170808), the Fund of Liaoning Province Department of
Education (grant no., L2013301), the Liaoning Province Natural
Science Foundation (grant no., 2015020725) and the Shenyang
Municipal Science and Technology Fund (grant no.,
F12-277-1-47).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
BY and MY conceived the present study. BY and ZC
performed the experiments and wrote the paper. WZhang, DY and WZhao
helped with data analysis. All the authors have read and approved
the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Roos PM: Osteoporosis in
neurodegeneration. J Trace Elem Med Biol. 28:418–421. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhou R, Deng J, Zhang M, Zhou HD and Wang
YJ: Association between bone mineral density and the risk of
Alzheimer's disease. J Alzheimers Dis. 24:101–108. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zha Y, Shen L and Ji HF: Osteoporosis risk
and bone mineral density levels in patients with Parkinson's
disease: A meta-analysis. Bone. 52:498–505. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sato Y, Honda Y, Asoh T, Kikuyama M and
Oizumi K: Hypovitaminosis D and decreased bone mineral density in
amyotrophic lateral sclerosis. Eur Neurol. 37:225–229. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Weinstockguttman B, Gallaghe E, Baier M,
Green L, Feichter J, Patrick K, Miller C, Wrest K and Ramanathan M:
Risk of bone loss in men with multiple sclerosis. Mult Scler.
10:170–175. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tysiewicz-Dudek M, Pietraszkiewicz F and
Drozdzowska B: Alzheimer's disease and osteoporosis: Common risk
factors or one condition predisposing to the other? Ortop Traumatol
Rehabili. 10:315–323. 2008.(In English, Polish).
|
|
7
|
Peters R, Peters J, Warner J, Beckett N
and Bulpitt C: Alcohol, dementia and cognitive decline in the
elderly: A systematic review. Age Ageing. 37:505–512. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cataldo JK, Prochaska JJ and Glantz SA:
Cigarette smoking is a risk factor for Alzheimer's Disease: An
analysis controlling for tobacco industry affiliation. J Alzheimers
Dis. 19:465–480. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yang MW, Wang TH, Yan PP, Chu LW, Yu J,
Gao ZD, Li YZ and Guo BL: Curcumin improves bone microarchitecture
and enhances mineral density in APP/PS1 transgenic mice.
Phytomedicine. 18:205–213. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li S, Liu B, Zhang L and Rong L: Amyloid
beta peptide is elevated in osteoporotic bone tissues and enhances
osteoclast function. Bone. 61:164–175. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kuperstein I, Broersen K, Benilova I,
Rozenski J, Jonckheere W, Debulpaep M, Vandersteen A, Segers-Nolten
I, Van Der Werf K, Subramaniam V, et al: Neurotoxicity of
Alzheimer's disease Aβ peptides is induced by small changes in the
Aβ42 to Aβ40 ratio. EMBO J. 29:3408–3420. 2014. View Article : Google Scholar
|
|
12
|
Song C, Song C and Tong F: Autophagy
induction is a survival response against oxidative stress in bone
marrow-derived mesenchymal stromal cells. Cytotherapy.
16:1361–1370. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wan Y, Zhuo N, Li Y, Zhao W and Jiang D:
Autophagy promotes osteogenic differentiation of human bone marrow
mesenchymal stem cell derived from osteoporotic vertebrae. Biochem
Biophys Res Commun. 488:46–52. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Barbero-Camps E, Roca-Agujetas V,
Bartolessis I, de Dios C, Fernandez-Checa JC, Mari M, Morales A,
Hartmann T and Colell A: Cholesterol impairs autophagy-mediated
clearance of amyloid beta while promoting its secretion. Autophagy.
14:1129–1154. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Schmelzle T and Hall MN: TOR, a central
controller of cell growth. Cell. 103:253–262. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hu B, Zhang Y, Jia L, Wu H, Fan C, Sun Y,
Ye C, Liao M and Zhou J: Binding of the pathogen receptor HSP90AA1
to avibirnavirus VP2 induces autophagy by inactivating the AKT-MTOR
pathway. Autophagy. 11:503–515. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lee EO, Kang JL and Chong YH: The
amyloid-beta peptide suppresses transforming growth
factor-beta1-induced matrix metalloproteinase-2 production via
Smad7 expression in human monocytic THP-1 cells. J Biol Chem.
280:7845–7853. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Squitti R: Metals in Alzheimer's disease:
A systemic perspective. Front Biosci. 17:451–472. 2012. View Article : Google Scholar
|
|
19
|
Brewer GJ: Alzheimer's disease causation
by copper toxicity and treatment with zinc. Front Aging Neurosci.
6:922014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Dusek P, Roos PM, Litwin T, Schneider SA,
Flaten TP and Aaseth J: The neurotoxicity of iron, copper and
manganese in Parkinson's and Wilson's diseases. J Trace Elem Med
Biol. 31:193–203. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Roos PM, Vesterberg O, Syversen T, Flaten
TP and Nordberg M: Metal concentrations in cerebrospinal fluid and
blood plasma from patients with amyotrophic lateral sclerosis. Biol
Trace Elem Res. 151:159–170. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
LeVine SM, Bilgen M and Lynch SG: Iron
accumulation in multiple sclerosis: An early pathogenic event.
Expert Rev Neurother. 13:247–250. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhu G, Wang H, Shi Y, Weng S, Jin T, Kong
Q and Nordberg GF: Environmental cadmium exposure and forearm bone
density. Biometals. 17:499–503. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Akesson A, Bjellerup P, Lundh T, Lidfeldt
J, Nerbrand C, Samsioe G, Skerfving S and Vahter M: Cadmium-induced
effects on bone in a population-based study of women. Environ
Health Perspect. 114:830–834. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang HK, Hung CM, Lin SH, Tai YC, Lu K,
Liliang PC, Lin CW, Lee YC, Fang PH and Chang LC: Increased risk of
hip fractures in patients with dementia: A nationwide
population-based study. Bmc Neurol. 14:1752014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhao Y, Shen L and Ji HF: Alzheimer's
disease and risk of hip fracture: A meta-analysis study.
ScientificWorldJournal. 2012:8721732012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhao L, Liu S, Wang Y, Zhang Q, Zhao W,
Wang Z and Yin M: Effects of Curculigoside on Memory Impairment and
Bone Loss via Anti-Oxidative Character in APP/PS1 Mutated
Transgenic Mice. PLoS One. 10:e01332892015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Xia WF, Jung JU, Cui S, Xiong S, Xiong L,
Shi XM, Mei L and Xiong WC: Swedish mutant APP suppresses
osteoblast differentiation and causes osteoporotic deficit, which
are ameliorated by N-acetyl-L-cysteine. J Bone Miner Res.
28:2122–2135. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhou Z, Immel D, Xi CX, Bierhaus A, Feng
X, Mei L, Nawroth P, Stern DM and Xiong WC: Regulation of
osteoclast function and bone mass by RAGE. J Exp Med.
203:1067–1080. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cui S, Xiong F, Hong Y, Jung JU, Li XS,
Liu JZ, Yan R, Mei L, Feng X and Xiong WC: APPswe/Aβ regulation of
osteoclast activation and RAGE expression in an age-dependent
manner. J Bone Miner Res. 26:1084–1098. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
L S, Yang B, Teguh D, Zhou L, Xu J and
Rong L: Amyloid β peptide enhances RANKL-induced osteoclast
activation through NF-κB, ERK, and calcium oscillation signaling.
International J Mol Sci. 17:16832016. View Article : Google Scholar
|
|
32
|
Hardy J: Alzheimer's disease: The amyloid
cascade hypothesis: An update and reappraisal. J Alzheimers Dis. 9
(3 Suppl):S151–S153. 2006. View Article : Google Scholar
|
|
33
|
Haass C and Selkoe DJ: Soluble protein
oligomers in neurodegeneration: lessons from the Alzheimer's
amyloid beta-peptide. Nat Rev Mol Cell Biol. 8:101–112. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lee IS, Jung K, Kim IS and Park KI:
Amyloid-β oligomers regulate the properties of human neural stem
cells through GSK-3β signaling. Exp Mol Med. 45:e602013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Levine B: Autophagy in the pathogenesis of
disease. Cell. 132:27–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Autophagy fights disease through cellular
self-digestion. Nature. 451:10692008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Rubinsztein DC, Codogno P and Levin B:
Autophagy modulation as a potential therapeutic target for diverse
diseases. Nat Rev Drug Dis. 11:709–730. 2012. View Article : Google Scholar
|
|
38
|
Kim I and Lemasters JJ: Mitochondrial
degradation by autophagy (mitophagy) in GFP-LC3 transgenic
hepatocytes during nutrient deprivation. Am J Physiol Cell Physiol.
300:C3082011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lee J: Neuronal Autophagy: A housekeeper
or a fighter in neuronal cell survival? Exp Neurobiol. 21:1–8.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Shenab HM: Autophagy is a survival force
via suppression of necrotic cell death. Exp Cell Res.
318:1304–1308. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Loos B, Engelbrecht AM, Lockshin RA,
Klionsky DJ and Zakeri Z: The variability of autophagy and cell
death susceptibility. Autophagy. 9:1270–1285. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Klionsky DJ, Abdelmohsen K, Abe A, Abedin
MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD,
Adeli K, et al: Guidelines for the use and interpretation of assays
for monitoring autophagy. (3rd). Autophagy. 12:1–222. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Pajak B, Songin M, Strosznajder JB,
Orzechowski A and Gajkowska B: Ultrastructural evidence of amyloid
β-induced autophagy in PC12 cells. Folia Neuropathol. 47:252–258.
2009.PubMed/NCBI
|
|
44
|
Neill CO: PI3-kinase/Akt/mTOR signaling:
Impaired on/off switches in aging, cognitive decline and
Alzheimer's disease. Exp Gerontol. 48:647–653. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Griffin RJ, Moloney A, Kelliher M,
Johnston JA, Ravid R, Dockery P, O'Connor R and O'Neill C:
Activation of Akt/PKB, increased phosphorylation of Akt substrates
and loss and altered distribution of Akt and PTEN are features of
Alzheimer's disease pathology. J Neurochem. 93:105–117. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Levine B and Yuan J: Autophagy in cell
death. An innocent convict J Clin Invest. 115:2679–2688. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Alva AS, Gultekin SH and Baehrecke EH:
Autophagy in human tumors: Cell survival or death? CellDeath
Differ. 11:1046–1048. 2004.
|
|
48
|
Platini F, Pérez-tomás R, Ambrosio S and
Tessitore L: Understanding autophagy in cell death control. Curr
Pharm Des. 16:101–113. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Denton D, Xu T and Kumar S: Autophagy as a
pro-death pathway. Immunol Cell Biol. 93:35–42. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Mariño G, Madeo F and Kroemer G: Autophagy
for tissue homeostasis and neuroprotection. Curr Opin Cell Biol.
23:198–206. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Annabi B, Lee YT, Turcotte S, Naud E,
Desrosiers RR, Champagne M, Eliopoulos N, Galipeau J and Béliveau
R: Hypoxia Promotes Murine Bone-Marrow-Derived Stromal Cell
Migration and Tube Formation. Stem Cells. 21:337–347. 2010.
View Article : Google Scholar
|
|
52
|
Wang S, Zhou SL, Min FY, Ma JJ, Shi XJ,
Bereczki E and Wu J: mTOR-mediated hyperphosphorylation of tau in
the hippocampus is involved in cognitive deficits in
streptozotocin-induced diabetic mice. Metab Brain Dis. 29:729–736.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Ronsisvalle N, Di Benedetto G, Parenti C,
Amoroso S, Bernardini R and Cantarella G: CHF5074 protects SH-SY5Y
human neuronal-like cells from amyloidbeta 25–35 and tumor necrosis
factor related apoptosis inducing ligand toxicity in vitro. Current
Alzheimer Res. 11:714–724. 2014. View Article : Google Scholar
|
|
54
|
Caccamo A, Majumder S, Richardson A,
Strong R and Oddo S: Molecular Interplay between Mammalian Target
of Rapamycin (mTOR), Amyloid-beta, and Tau: Effects On Cognitive
Impairments. J Biol Chem. 285:13107–13120. 2010. View Article : Google Scholar : PubMed/NCBI
|