Introduction
Sepsis is a type of systemic inflammatory response
syndrome (SIRS) and is mediated by an immune response triggered by
infection, which can progress from sepsis to severe sepsis and
septic shock (1). Sepsis can lead
to symptoms, including fever, increased heart rate, breathing rate
and confusion (2). In 2015, the
incidence rates of sepsis and severe sepsis in high-income
countries were 0.44 and 0.27%, respectively (3). Meanwhile, the mortality rates for
sepsis were reported to be 3 and 75 cases per 1,000 individuals in
two Chinese military hospitals (4). Sepsis remains difficult to predict,
diagnose and treat (5). Thus,
there is an urgent need to identify target genes and microRNAs
(miRNAs) that can seve as biomarkers for sepsis.
Previous studies have demonstrated that
proinflammatory cytokines, including interleukin-6 (IL-6) and tumor
necrosis factor-α (TNF-α), are key mediators of inflammation during
sepsis (6). A previous study
showed that IL-6, IL-1β, IL-8 and TNF-α levels were significantly
upregulated in culture-proven sepsis groups relative to those in
the control groups (7). Multiple
studies have reported that target genes and miRNAs are involved in
sepsis. For example, Nrf2 is a basic leucine zipper
transcription factor (TF) that mediates the response to
lipopolysaccharides (LPS) and TNF-α by activating NF-κB production
during experimental sepsis (8). In
addition, procalcitonin (PCT) is elevated in patients with SIRS,
and has been approved by the Food and Drug Administration (U.S.
FDA) for the assessment of risk for developing severe sepsis in
patients (9). Although PCT is
closely associated with inflammation, there are some limitations
specific for infection resulting in questionable efficacy as PCT
can also be increased in noninfectious disease conditions (10). Generally, the concentration value
of PCT <0.5 ng/ml indicates a low risk while values of 0.5–2.0
ng/ml represent an intermediate likelihood of sepsis and/or septic
shock. Wacker et al reported (11) that PCT had a modest diagnostic
performance with 77% sensitivity and 79% specificity. Therefore,
PCT is not specific for diagnosis in patients with values in the
intermediate range. Importantly, multiple miRNAs have various
biological functions in inflammation, metabolism and tumor
progression. These candidate miRNAs show high accuracy and
sensitivity, and are expected to be ideal biomarkers for sepsis.
Evidence suggests that the sensitivity and specificity of miR-223
for predicting the occurrence of sepsis after urinary operation
were higher than those of PCT (12,13).
In addition, miR-155 has been suggested to directly target key
genes that are involved in LPS signaling, such as Fas-associated
death domain protein, IκB kinase ε, and the receptor (TNFR
superfamily)-interacting serine-threonine kinase 1 to enhance TNF-α
production (14). Nevertheless,
miR-125b targets the 3′-untranslated region of the TNF-α transcript
(14). However, the fundamental
mechanisms underlying the pathogenesis of sepsis remain unclear.
Multiple mechanisms involving complex systemic inflammation
networks, genetic polymorphisms, immune dysfunction, abnormal
coagulant function, and host response to pathogenic microorganisms
and their toxins are likely to be involved in sepsis. Therefore,
the pathogenesis of sepsis warrants further investigation.
In the present study, we performed bioinformatics
analysis to identify the differentially expressed genes (DEGs) in
sepsis, as well as the TFs and miRNAs of these DEGs. Subsequently,
an integrated regulatory network was constructed based on the DEGs,
miRNAs and TFs. Finally, we investigated the interactions among the
DEGs and TFs/miRNAs and their corresponding functions. Our current
findings provided insights into the pathogenesis of sepsis and
identified novel targets for the treatment of sepsis.
Materials and methods
Microarray data
The GSE12624 dataset was downloaded from the GEO
database (http://www.ncbi.nlm.nih.gov/geo/) and contains gene
expression data of 34 sepsis patients and 36 healthy individuals
without sepsis. The inclusion criteria for the study are described
in (15). The microarray platform
was GPL4204 GE Healthcare/Amersham Biosciences CodeLink UniSet
Human I Bioarray. Raw data were available in TXT format.
Data preprocessing and identification
of DEGs
The probes corresponded to gene symbols according to
the latest annotation file from the NCBI gene database. When more
than one probe corresponded to the same gene symbol, the expression
level of the gene was calculated as the median of the two
expression values. Subsequently, the data were fitted to a
log-normal distribution using the log2 function, normalized using
the median function, and compared with septic samples and
non-septic samples using Bayesian methods from the limma package in
R (Linear Models for Microarray Data, http://www.bioconductor.org/packages/release/bioc/html/limma.html).
Finally, |log fold change (FC)| >0.585 and adjusted P-value
<0.05 were used as the threshold values for considering the
DEGs.
Identification of sepsis-related genes
and modules based on WGCNA
WGCNA is a systematic method for identifying
putative target genes involved in a disease. It is used to describe
the correlation among genes by finding significant modules from
high-throughput sequencing data (16). In the present study, WGCNA was
performed based on the following analysis workflow. i) The
correlations among the expression values of DEGs in the dataset
were determined. A higher correlation value indicates higher
consistency of gene expression in each dataset, which is a
prerequisite for the construction of a WGCNA network. ii) The
correlation matrix of gene co-expression values was constructed
based on Smn = |cor(m,n)|, where
Smn indicates the correlation coefficient of
co-expression patterns between genes m and n. iii) The adjacency is
defined as amn = power(Smn,β), which measures
the pairwise correlation between the expression levels of two
genes. iv) Adjacency functions for both weighted and unweighted
networks require the user to choose threshold parameters. The
threshold of ≥0.9 was considered for the correlation coefficient
between log2 k (node count) and log2 p(k)
(frequency of node). v) The correlation matrix Smn was
transformed to the adjacency matrix amn. Afterwards, the
adjacency matrix amn was transformed to a topological
matrix using the following equation:
lmn+amnmin{km,kn}+1-amn
where lmn indicates the sum of adjacency
coefficient of the common edge between genes m and n and
km indicates sum of connection strengths of m with the
other network genes. vi) Gene significance (GS) measures were used
to incorporate external information into the co-expression network.
Module significance was determined by calculating the average |GS|
for all genes in a module.
Gene Ontology (GO) enrichment and
Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses of
the key modules
The Database for Annotation, Visualization and
Integration Discovery (DAVID, a public high-throughput functional
annotation tool (version 6.8, http://david-d.ncifcrf.gov/) is an online
bioinformatics tool that can be used for functional annotation and
microarray analysis by integrating data mining environments and
analyzing gene lists (17). DEGs
in the modules were used as input for DAVID, and GO and KEGG
enrichment analyses were conducted using MEblue and MEyellow DEGs.
P-value <0.05 and the enriched gene count ≥2 were considered
significant.
Construction of the PPI network and
module analysis
The PPI network was constructed based on all the
DEGs using STRING from a well-known online server (version 10.0,
http://www.string-db.org/) (18). A combined score of >0.4 was
defined as the threshold value for constructing the PPI network.
The PPI network was visualized using Cytoscape software (version
3.2.0, http://cytoscape.org/) (19). In addition, MCODE (version 1.4.2,
http://apps.cytoscape.org/apps/MCODE)
in Cytoscape software was used to analyze the most significant
module, with the threshold value of 5 (20).
Construction of the TF-miRNA-target
DEGs regulatory network
The miRNA-target DEGs and TF-target DEGs were
predicted using Overrepresentation Enrichment Analysis enrichment
method in WebGestal (http://www.webgestalt.org/). Gene pairs with P-value
<0.05 were integrated into the TF-miRNA-target DEGs regulatory
network, which was visualized using Cytoscape.
Results
Sepsis-related genes and modules
A total of 407 DEGs, including 227 upregulated DEGs
and 180 downregulated DEGs, were identified. According to the
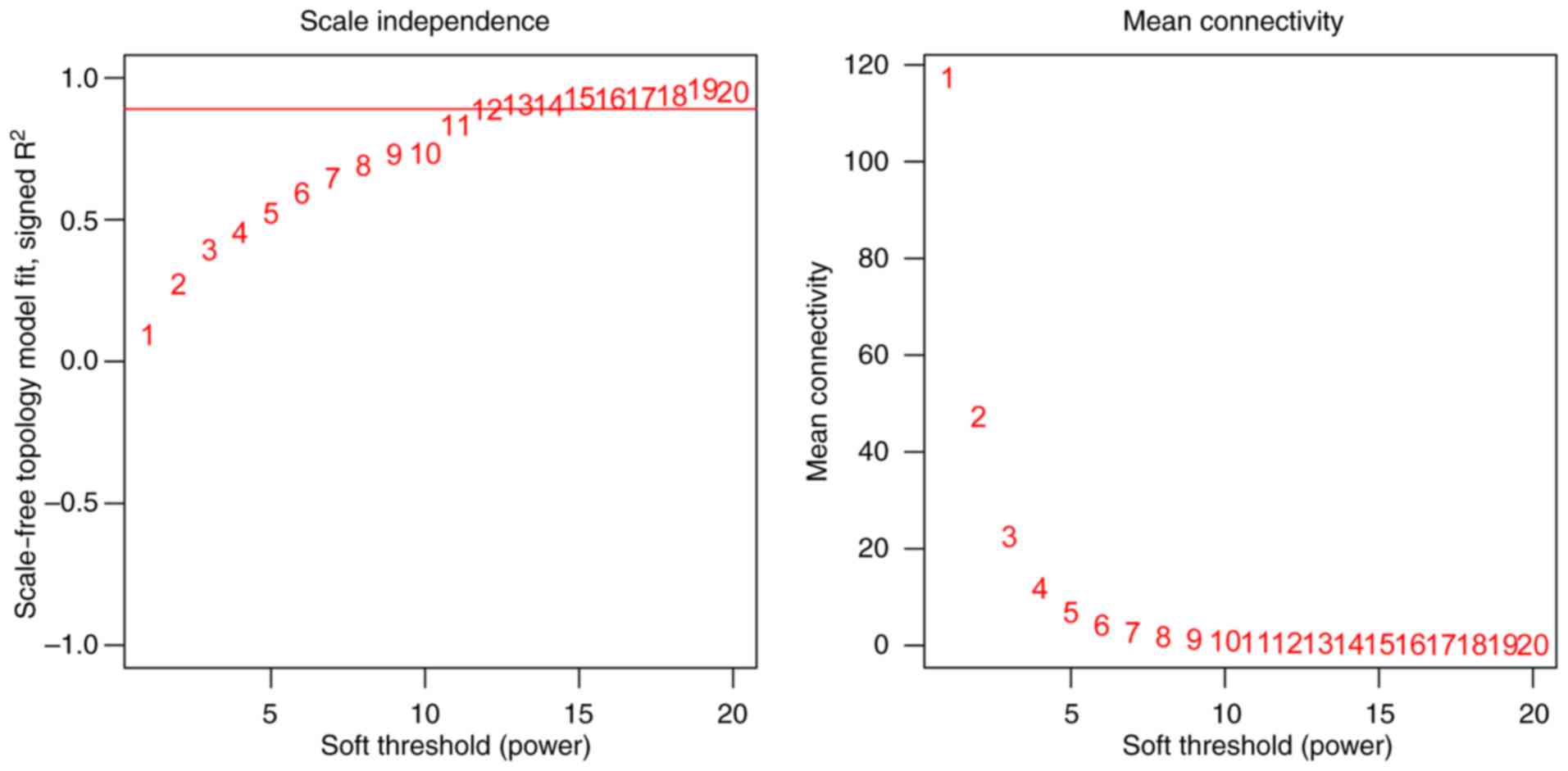
standard scale-free network model, the power value was set to 12
when the square of correlation coefficient was 0.9 (Fig. 1). The network conformed to a
scale-free model when the square of the correlation coefficient
square was set to the highest value. Subsequently, the WGCNA
network was constructed under power = 12. Gene cluster dendrogram
was obtained according to dissTOM using the hierarchical clustering
method. The dynamic tree cut method was employed to estimate the
number of clusters in the dataset. Finally, the DEGs were divided
into 13 co-expressed modules (Fig.
2), and genes in the grey module contained genes that could not
clustered under the other modules. Subsequently, the most highly
connected intramodular hub gene in each module was considered as
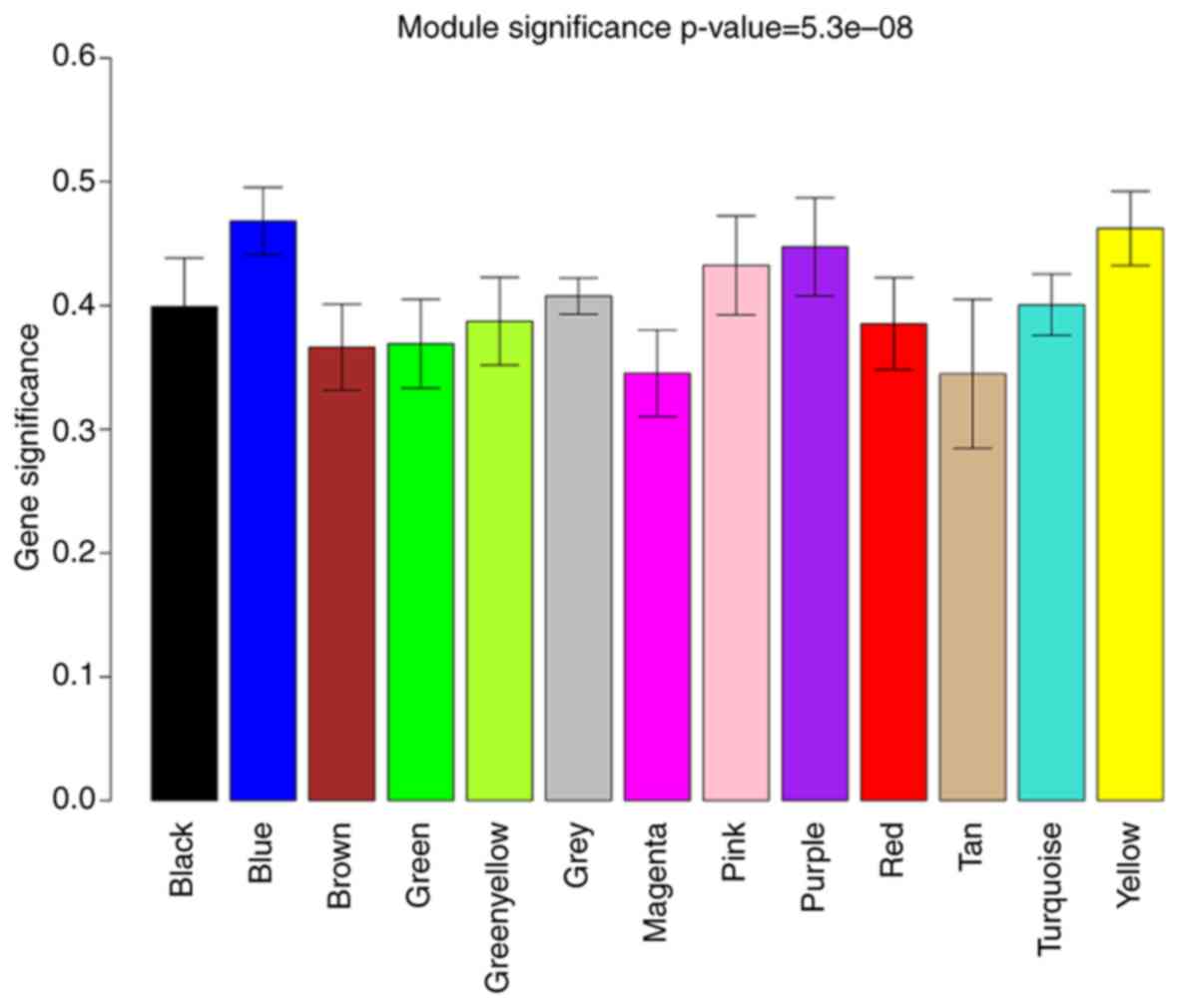
the module representative. The analysis identified a total of 7
modules with correlation coefficients >0.5. The correlation
coefficients of the MEblue and MEyellow modules were higher than
0.6. To ensure the reliability of the key network module, the |GS|
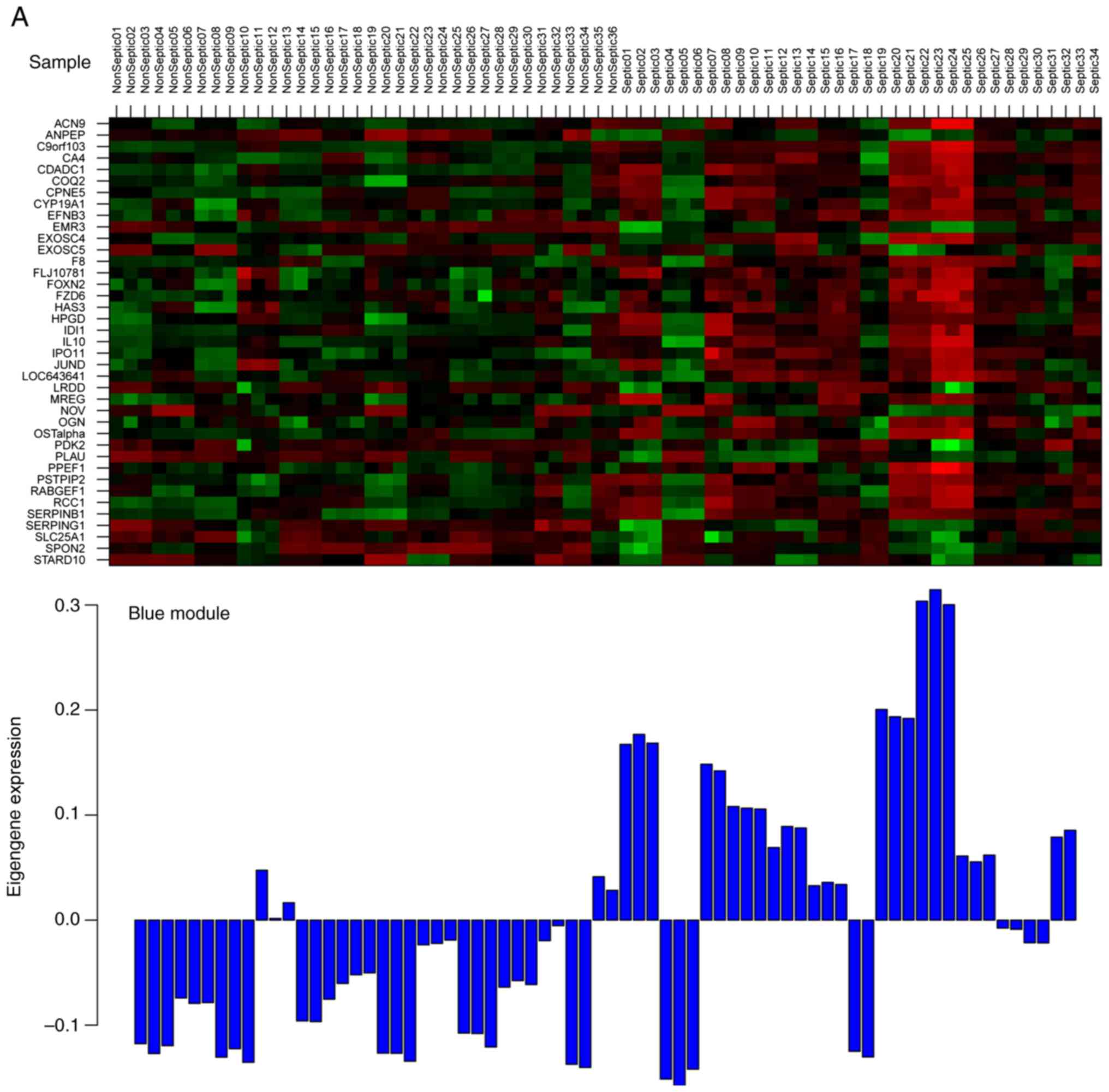
was used to further identify two key modules (Fig. 3). Finally, the MEblue (Fig. 3A) and MEyellow (Fig. 3B) modules were defined as the key
modules. All of DEGs in the two modules are shown in the Table I.
 | Table I.Differentially expressed genes in the
MEblue and MEyellow modules. |
Table I.
Differentially expressed genes in the
MEblue and MEyellow modules.
| Genes | Module | Description | Genes | Module | Description |
|---|
| ACN9 | blue | up | ADAMTS5 | yellow | down |
| ANPEP | blue | down | CALCRL | yellow | up |
| CA4 | blue | up | DGKG | yellow | down |
| CDADC1 | blue | up | DHRS9 | yellow | up |
| COQ2 | blue | up | ERLIN1 | yellow | up |
| CPNE5 | blue | up | FAM105A | yellow | up |
| CYP19A1 | blue | up | FAR2 | yellow | up |
| EFNB3 | blue | up | GADD45A | yellow | up |
| EMR3 | blue | down | HMGB2 | yellow | up |
| EXOSC4 | blue | up | IL18RAP | yellow | up |
| EXOSC5 | blue | down | KLHL2 | yellow | up |
| F8 | blue | up | LEPROT | yellow | up |
| FOXN2 | blue | up | LRPPRC | yellow | down |
| FZD6 | blue | up | MAP3K8 | yellow | up |
| HAS3 | blue | up |
NAALADL1 | yellow | down |
| HPGD | blue | up | PECR | yellow | up |
| IDI1 | blue | up | PROS1 | yellow | up |
| IDNK | blue | up | RPS6KA5 | yellow | down |
| IL10 | blue | up | SAMSN1 | yellow | up |
| IPO11 | blue | up | SDPR | yellow | up |
| JUND | blue | up | SIPA1L2 | yellow | up |
|
LOC643641 | blue | up | SP140 | yellow | down |
| MREG | blue | up | SPIB | yellow | down |
| NOV | blue | down | UBE2H | yellow | up |
| OGN | blue | up | URGCP | yellow | down |
| PDK2 | blue | down | USP46 | yellow | down |
| PIDD | blue | down | VNN1 | yellow | up |
| PLAU | blue | down |
|
|
|
| PNMAL1 | blue | up |
|
|
|
| PPEF1 | blue | up |
|
|
|
| PSTPIP2 | blue | up |
|
|
|
| RABGEF1 | blue | up |
|
|
|
| RCC1 | blue | up |
|
|
|
|
SERPINB1 | blue | up |
|
|
|
|
SERPING1 | blue | down |
|
|
|
| SLC25A1 | blue | down |
|
|
|
| SLC51A | blue | up |
|
|
|
| SPON2 | blue | down |
|
|
|
| STARD10 | blue | down |
|
|
|
GO function and KEGG pathway
analysis
A total of 66 DEGs were identified in the MEblue and
MEyellow modules, including 46 upregulated DEGs and 20
downregulated DEGs. F8, PLAU and SERPING1 in the
MEblue module were enriched with complement and coagulation
cascades. EXOSC4 and EXOSC5 in the MEblue module were
enriched in the RNA degradation pathway. MAP3K8 and
RPS6KA5 in the MEyellow module were enriched in the MAPK and
TNF signaling pathways. The GO functions of the genes in the two
modules are shown in Table
II.
| Table II.Gene Ontology functions for genes in
the two modules. |
Table II.
Gene Ontology functions for genes in
the two modules.
| Module | GO-ID-Name | Count | P-value | Genes |
|---|
| MEblue |
|
GO_BP | GO:0007596~blood
coagulation | 3 | 1.75E-02 | F8, SERPING1,
PLAU |
|
|
GO:0050817~coagulation | 3 | 1.75E-02 | F8, SERPING1,
PLAU |
|
|
GO:0007599~hemostasis | 3 | 1.95E-02 | F8, SERPING1,
PLAU |
|
|
GO:0050878~regulation of body fluid
levels | 3 | 3.19E-02 | F8, SERPING1,
PLAU |
|
|
GO:0008299~isoprenoid biosynthetic
process | 2 | 3.92E-02 | COQ2,
IDI1 |
|
|
GO:0032101~regulation of response to
external stimulus | 3 | 3.98E-02 | SERPING1, IL10,
PLAU |
|
| GO:0045861~negative
regulation of proteolysis | 2 | 4.30E-02 | SERPING1,
IL10 |
|
GO_CC | GO:0031983~vesicle
lumen | 3 | 4.84E-03 | F8, ANPEP,
SERPING1 |
|
|
GO:0044421~extracellular region part | 7 | 1.88E-02 | NOV, OGN, F8,
SERPING1, EMR3, SPON2, IL10 |
|
| GO:0000178~exosome
(RNase complex) | 2 | 2.69E-02 | EXOSC4,
EXOSC5 |
|
GO_MF |
GO:0000175~3′-5′-exoribonuclease
activity | 2 | 2.74E-02 | EXOSC4,
EXOSC5 |
|
|
GO:0004532~exoribonuclease activity | 2 | 2.96E-02 | EXOSC4,
EXOSC5 |
|
|
GO:0016896~exoribonuclease activity,
producing 5′-phosphomonoesters | 2 | 2.96E-02 | EXOSC4,
EXOSC5 |
|
|
GO:0016796~exonuclease activity, active
with either ribo- or deoxyribonucleic acids and producing
5′-phosphomonoesters | 2 | 4.52E-02 | EXOSC4,
EXOSC5 |
| MEyellow |
|
GO_BP |
GO:0006508~proteolysis | 6 | 1.64E-02 | RPS6KA5, USP46,
ERLIN1, UBE2H, NAALADL1, ADAMTS5 |
|
|
GO:0006511~ubiquitin-dependent protein
catabolic process | 3 | 4.90E-02 | USP46, ERLIN1,
UBE2H |
|
GO_MF |
GO:0008237~metallopeptidase activity | 3 | 2.89E-02 | RPS6KA5,
NAALADL1, ADAMTS5 |
|
|
GO:0070011~peptidase activity, acting on
L-amino acid peptides | 4 | 4.40E-02 | RPS6KA5, USP46,
NAALADL1, ADAMTS5 |
|
|
GO:0008233~peptidase activity | 4 | 4.92E-02 | RPS6KA5, USP46,
NAALADL1, ADAMTS5 |
PPI network based on the MEblue and
MEyellow modules
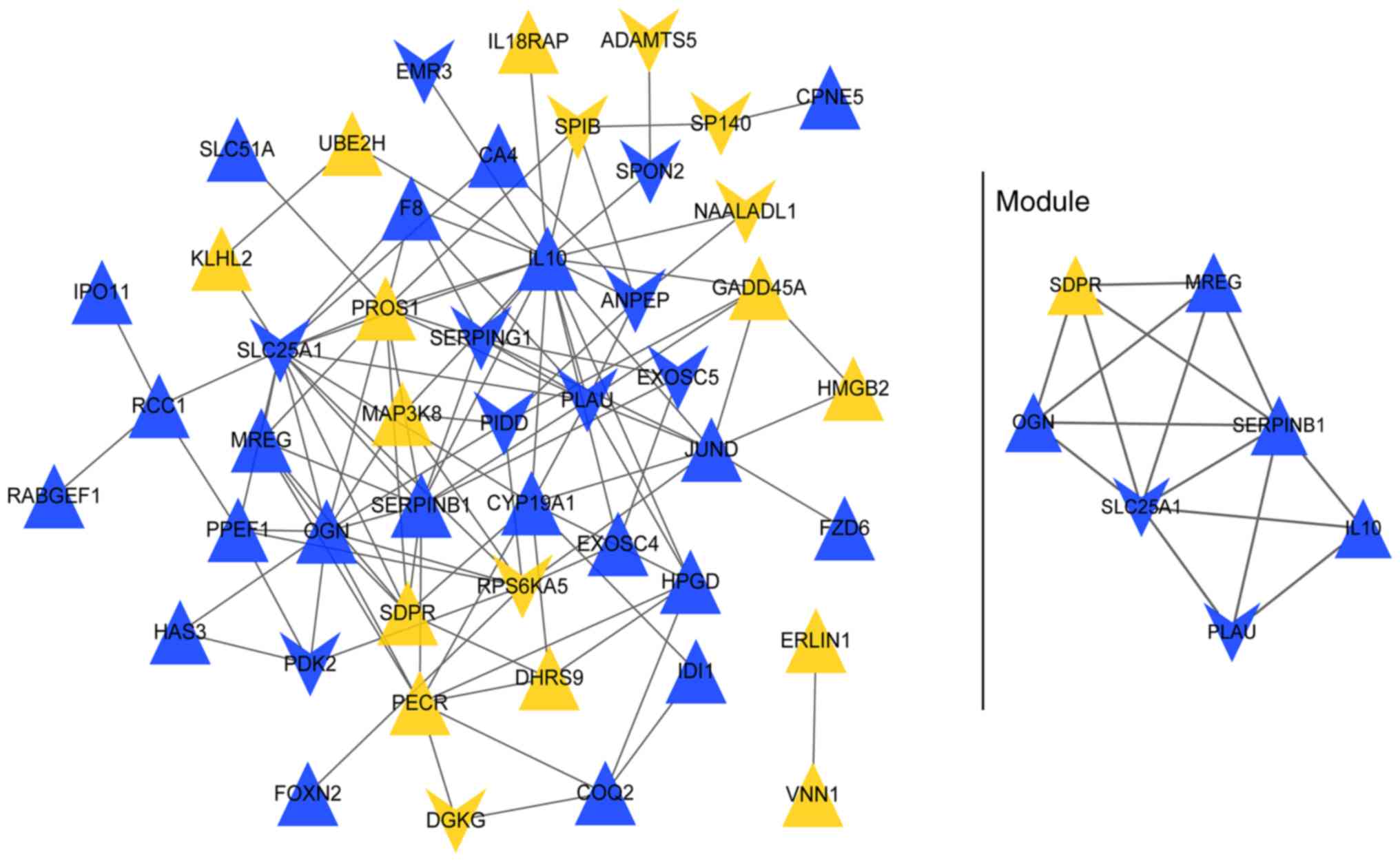
The PPI network (Fig.
4) contained 48 nodes (genes) and 112 edges (protein-protein
interrelations), such as MAP3K8-RPS6KA5, MAP3K8-IL10,
RPS6KA5-EXOSC4 and EXOSC4-EXOSC5). One sub-network (hub module) had
an MCODE score ≥5 and comprised 7 nodes (e.g. IL10) and 15 edges
(Fig. 4).
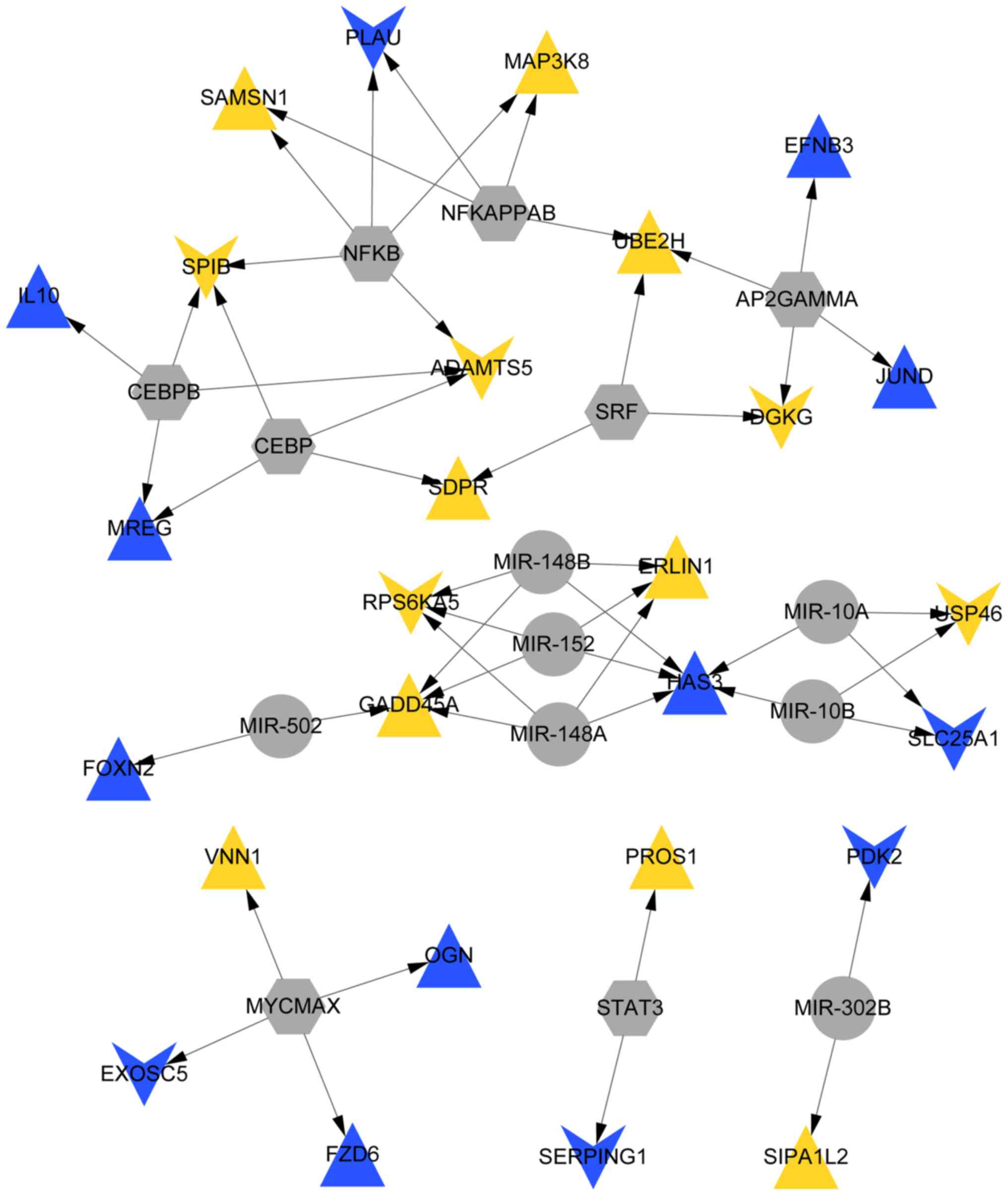
miRNA-TF-target gene regulatory
network
Overall, the analysis identified 8 TFs (NF-κB) and 7
miRNAs (miR152 and miR-148A/B), which comprised 52 TF-miRNA-target
gene pairs (17 upregulated genes, such as MAP3K8 and 10
downregulated genes, such as RPS6KA5) and were used to
construct an miRNA-TF-target gene regulatory network (Fig. 5).
Discussion
In the present study, we identified a total of 407
DEGs in the sepsis samples, including 227 upregulated DEGs and 180
downregulated DEGs. Subsequently, these DEGs were grouped into 13
co-expressed modules after WGCNA. Additionally, MEblue and MEyellow
modules with a correlation coefficient >0.6 were defined as the
key modules; these modules included 6 upregulated and 20
downregulated DEGs. EXOSC4 and EXOSC5 in the MEblue
module were enriched in the RNA degradation pathway. MAP3K8
and RPS6KA5 in the MEyellow module were enriched in the MAPK
and TNF signaling pathways. In addition, the resulting PPI network
comprised 48 nodes and 112 edges such as MAP3K8-RPS6KA5,
MAP3K8-IL10, RPS6KA5-EXOSC4 and EXOSC4-EXOSC5. Finally, the
analysis identified 8 TFs (NF-κB) and 7 miRNAs (miR-152 and
miR-148A/B) that corresponded to 52 TF-miRNA-target gene pairs (17
upregulated genes, such as MAP3K8 and 10 downregulated
genes, such as RPS6KA5).
MAP3K8 is a serine-threonine kinase that
plays a critical role in innate immunity and is known to induce
tumor necrosis factor (TNF) production by activating ERK (21). In addition, TNF-α has been
implicated as a key mediator in inflammation, morbidity and
mortality associated with sepsis. TNF-α has been demonstrated to be
responsible for the initial hypothermia and lethality in septic
mice. (22). In addition, host
reactions during sepsis, septic shock, and multiple organ failure
are associated with increased TNF production in humans (23). TNF-α is a strong pro-inflammatory
cytokine associated with septic patients and has been considered as
a target for the treatment of sepsis (24). In the present study, MAP3K8
expression levels were found to be upregulated in sepsis samples
relative to those of the control samples. As indicated above,
MAP3K8 induces TNF production, consistent with increased TNF
levels in the sepsis samples in the present study. Importantly,
MAP3K8 and RPS6KA5 in the MEyellow module were
enriched in the MAPK and TNF signaling pathways. In addition,
MAP3K8 interacts with IL10 based on the constructed PPI network.
IL10 is an anti-inflammatory agent that can improve disease outcome
in the model of sepsis syndrome (25). Therefore, these findings indicate
that MAP3K8 is involved in sepsis through the MAPK and TNF
signaling pathways. The nuclear transcription factor NF-κB is known
to be activated following hemorrhagic shock and sepsis (26). In the present study, NF-κB acts as
the upstream TF of the MAP3K8 gene. Proinflammatory
cytokines, such as TNF-α and IL-1, activate important signaling
pathways. In particular, cytokines activate members of the NF-κB
group of TFs, which play central roles in inflammation and innate
immunity (27). Activation of
NF-κB and other TFs involved in the innate immune/inflammatory
response can upregulate the expression of various genes, such as
MMP-9, VEGF and TNF (28). Therefore, MAP3K8 is
potentially involved in sepsis through the activation of NF-κB and
is likely to be involved in the MAPK and TNF signaling
pathways.
In the PPI network, RPS6KA5 interacted with MAP3K8,
and these two genes were enriched in the MAPK and TNF signaling
pathways. RPS6KA5, also known as mitogen- and
stress-activated protein kinase 1 (MSK1), is a downstream
target of both p38 and ERK1/2 (29). RPS6KA5 stimulates the
transcription of various pro-inflammatory genes, such as IL-6, IL-8
and TNF-α, by activating TFs (30). Therefore, RPS6KA5 was
associated with sepsis through the MAPK and TNF signaling pathways.
In the miRNA-TF-target gene regulatory network, RPS6KA5 was
the target gene of miR-152, miR-148A and miR-148B. A previous study
indicated that members of the miR-148 family (miR-148A, miR-148B
and miR-152) negatively regulated antigen presentation and
Toll-like receptor (TLR)-triggered cytokine secretion in dendritic
cells (31). TLRs are a class of
proteins that play key roles in the innate immune system and
secrete proinflammatory cytokines, such as TNF-α, IL-6 and IL-12
(32,33). In addition, soluble TLR2 is a
biomarker for sepsis in critically ill patients with multi-organ
failure within 12 h of ICU admission (34). Although there was no direct
evidence to identify that miR-148 is better than PCT or TLR2,
miRNAs with high accuracy and sensitivity, are expected to be ideal
biomarkers for sepsis (13). Thus,
the receiver operating characteristic (ROC) curve of the miR-148
family (including sensitivity and specificity) should be compared
with those of PCT or TLR2 in diagnostic performance of sepsis
patients. It is one of the limitation of the present study.
Therefore, members of the miR-148 family (miR-148A, miR-148B and
miR-152) may be candidate biomarkers for sepsis.
The present study has certain limitations. First,
limited samples were collected from the sepsis patients, and
experimental validation of the results was not performed. PCR or
western blotting will be performed in subsequent studies to verify
the findings. In addition, experiments should be conducted to
verify whether RPS6KA5 is a target of miR-148A/B and miR-152
in sepsis. In addition, the microarray dataset GSE12624 from the
Gene Expression Omnibus only included 34 patients with sepsis and
36 healthy individuals without sepsis. Therefore, correlation among
the miR-148 family (miR-148A/B and miR-152), and the type of
infection was not performed. In addition, an ROC curve of the
miR-148 family should be assayed in the diagnostic performance of
sepsis patients. However, the present results will not be affected
by these limitations.
Therefore, MAP3K8 is potentially induced
during sepsis through NF-κB activation and is potentially involved
in the MAPK and TNF signaling pathways. Meanwhile, RPS6KA5
interacted with MAP3K8 in the PPI network and was also found to be
enriched in the MAPK and TNF signaling pathways. Members of the
miR-148 family (miR-148A/B and miR-152) are candidate biomarkers
for sepsis.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
All data generated or analyzed during this study are
included in the published article.
Authors' contributions
Conception and design of the research were carried
out by LD, HL and GY. Data collection, analysis and interpretation
were conducted by SZ. Drafting of the manuscript was performed by
LD and HL. Revision of the manuscript for important intellectual
content was conducted by GY. All authors read and approved the
manuscript and agree to be accountable for all aspects of the
research in ensuring that the accuracy or integrity of any part of
the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Rhodes A, Evans LE, Alhazzani W, Levy MM,
Antonelli M, Ferrer R, Kumar A, Sevransky JE, Sprung CL, Nunnally
ME, et al: Surviving sepsis campaign: International guidelines for
management of sepsis and septic shock: 2016. Crit Care Med.
45:486–552. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tsai D, Stewart P, Goud R, Gourley S,
Hewagama S, Krishnaswamy S, Wallis SC, Lipman J and Roberts JA:
Total and unbound ceftriaxone pharmacokinetics in critically ill
Australian Indigenous patients with severe sepsis. Int J Antimicrob
Agents. 48:748–752. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fleischmann C, Scherag A, Adhikari NK,
Hartog CS, Tsaganos T, Schlattmann P, Angus DC and Reinhart K;
International Forum of Acute Care Trialists, : Assessment of global
incidence and mortality of hospital-treated sepsis.current
estimates and limitations. Am J Respir Crit Care Med. 193:259–272.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cheng W, Wang S, Shen C, Zhao D, Li D and
Shang Y: Epidemiology of hospitalized burns patients in china: A
Systematic Review. Burn Open. 2:8–16. 2017. View Article : Google Scholar
|
|
5
|
Mitra P, Guha D, Nag SS, Mondal BC and
Dasgupta S: Role of plasma fibrinogen in diagnosis and prediction
of short term outcome in neonatal sepsis. Indian J Hematol Blood
Transfus. 33:195–199. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kang S, Tanaka T, Masuda K and Kishimoto
T: Implications of IL-6 Targeting Therapy for Sepsis. Immunotherapy
(Los Angel). 3:1382017. View Article : Google Scholar
|
|
7
|
Kurt AN, Aygun AD, Godekmerdan A, Kurt A,
Dogan Y and Yilmaz E: Serum IL-1beta, IL-6, IL-8, and TNF-alpha
levels in early diagnosis and management of neonatal sepsis.
Mediators Inflamm. 2007:313972007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Thimmulappa RK, Lee H, Rangasamy T, Reddy
SP, Yamamoto M, Kensler TW and Biswal S: Nrf2 is a critical
regulator of the innate immune response and survival during
experimental sepsis. J Clin Invest. 116:984–995. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Schuetz P, Birkhahn R, Sherwin R, Jones
AE, Singer A, Kline JA, Runyon MS, Self WH, Courtney DM, Nowak RM,
et al: Serial procalcitonin predicts mortality in severe sepsis
patients: Results from the multicenter procalcitonin monitoring
sepsis (MOSES) Study. Crit Care Med. 45:781–789. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Riedel S: Procalcitonin and the role of
biomarkers in the diagnosis and management of sepsis. Diagn
Microbiol Infect Dis. 73:221–227. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wacker C, Prkno A, Brunkhorst FM and
Schlattmann P: Procalcitonin as a diagnostic marker for sepsis: A
systematic review and meta-analysis. Lancet Infect Dis. 13:426–435.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wu X, Yang J, Yu L and Long D: Plasma
miRNA-223 correlates with risk, inflammatory markers as well as
prognosis in sepsis patients. Medicine (Baltimore). 97:e113522018.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bao YYX and Chen Z: The early diagnostic
value of microRNA-223 for patients with complication of sepsis
after ureteroscopic lithotrity. Chin J Integr Tradit West Med
Intensive Crit Care. 24:465–468. 2017.(In Chinese).
|
|
14
|
Tili E, Michaille JJ, Cimino A, Costinean
S, Dumitru CD, Adair B, Fabbri M, Alder H, Liu CG, Calin GA and
Croce CM: Modulation of miR-155 and miR-125b levels following
lipopolysaccharide/TNF-alpha stimulation and their possible roles
in regulating the response to endotoxin shock. J Immunol.
179:5082–5089. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Menges T, König IR, Hossain H, Little S,
Tchatalbachev S, Thierer F, Hackstein H, Franjkovic I, Colaris T,
Martens F, et al: Sepsis syndrome and death in trauma patients are
associated with variation in the gene encoding tumor necrosis
factor. Crit Care Med. 36:1456–1462, e1-e6. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Langfelder P and Horvath S: WGCNA: An R
package for weighted correlation network analysis. BMC
Bioinformatics. 9:5592008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Huang W, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Szklarczyk D, Franceschini A, Wyder S,
Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos
A, Tsafou KP, et al: STRING v10: Protein-protein interaction
networks, integrated over the tree of life. Nucleic Acids Res.
43(D1): D447–D452. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T: Cytoscape: A
software environment for integrated models of biomolecular
interaction networks. Genome Res. 13:2498–2504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bandettini WP, Kellman P, Mancini C,
Booker OJ, Vasu S, Leung SW, Wilson JR, Shanbhag SM, Chen MY and
Arai AE: MultiContrast Delayed Enhancement (MCODE) improves
detection of subendocardial myocardial infarction by late
gadolinium enhancement cardiovascular magnetic resonance: A
clinical validation study. J Cardiovasc Magn Reson. 14:832012.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mielke LA, Elkins KL, Wei L, Starr R,
Tsichlis PN, O'Shea JJ and Watford WT: Tumor progression locus 2
(Map3k8) is critical for host defense against Listeria
monocytogenes and IL-1 beta production. J Immunol. 183:7984–7993.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Leon LR, White AA and Kluger MJ: Role of
IL-6 and TNF in thermoregulation and survival during sepsis in
mice. Am J Physiol. 275:R269–R277. 1998.PubMed/NCBI
|
|
23
|
Stuber F, Udalova IA, Book M, Drutskaya
LN, Kuprash DV, Turetskaya RL, Schade FU and Nedospasov SA: −308
tumor necrosis factor (TNF) polymorphism is not associated with
survival in severe sepsis and is unrelated to lipopolysaccharide
inducibility of the human TNF promoter. J Inflamm. 46:42–50.
1995-1996.
|
|
24
|
Riedemann NC, Guo RF and Ward PA: Novel
strategies for the treatment of sepsis. Nat Med. 9:517–524. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Oberholzer C, Oberholzer A, Bahjat FR,
Minter RM, Tannahill CL, Abouhamze A, LaFace D, Hutchins B,
Clare-Salzler MJ and Moldawer LL: Targeted adenovirus-induced
expression of IL-10 decreases thymic apoptosis and improves
survival in murine sepsis. Proc Natl Acad Sci USA. 98:11503–11508.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Filgueiras LR Jr, Martins JO, Serezani CH,
Capelozzi VL, Montes MBA and Jancar S: Sepsis-induced acute lung
injury (ALI) is milder in diabetic rats and correlates with
impaired NFkB activation. PLoS One. 7:e449872012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li Q and Verma IM: NF-kappaB regulation in
the immune system. Nat Rev Immunol. 2:725–734. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Karin M: Nuclear factor-kappaB in cancer
development and progression. Nature. 441:431–436. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jiang C, Yu L, Tu Q, Zhao Y, Zhang H and
Zhao S: Assignment of a member of the ribosomal protein S6 kinase
family, RPS6KA5, to human chromosome 14q31-->q32.1 by radiation
hybrid mapping. Cytogenet Cell Genet. 87:261–262. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Funding AT, Johansen C, Kragballe K,
Otkjaer K, Jensen UB, Madsen MW, Fjording MS, Finnemann J,
Skak-Nielsen T, Paludan SR and Iversen L: Mitogen- and
stress-activated protein kinase 1 is activated in lesional
psoriatic epidermis and regulates the expression of
pro-inflammatory cytokines. J Invest Dermatol. 126:1784–1791. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Liu X, Zhan Z, Xu L, Ma F, Li D, Guo Z, Li
N and Cao X: MicroRNA-148/152 impair innate response and antigen
presentation of TLR-triggered dendritic cells by targeting CaMKIIα.
J Immunol. 185:7244–7251. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Aslam R, Speck ER, Kim M, Crow AR, Bang
KW, Nestel FP, Ni H, Lazarus AH, Freedman J and Semple JW: Platelet
Toll-like receptor expression modulates lipopolysaccharide-induced
thrombocytopenia and tumor necrosis factor-alpha production in
vivo. Blood. 107:637–641. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Eidson LN, Inoue K, Young LJ, Tansey MG
and Murphy AZ: Toll-like receptor 4 mediates morphine-induced
neuroinflammation and tolerance via soluble tumor necrosis factor
signaling. Neuropsychopharmacology. 42:661–670. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Holst B, Szakmany T, Raby AC, Hamlyn V,
Durno K, Hall JE and Labéta MO: Soluble Toll-like receptor 2 is a
biomarker for sepsis in critically ill patients with multi-organ
failure within 12 h of ICU admission. Intensive Care Med Exp.
5:22017. View Article : Google Scholar : PubMed/NCBI
|