Introduction
Lung cancer, which originates from the bronchial
epithelium, is one of the most malignant diseases, with high rates
of morbidity and mortality (1).
Among them, the non-small cell lung cancer (NSCLC) prevalence is
80%, but the 5-year survival is only ~15%, while the small cell
lung cancer has survival rates of 15–20%. Only 16% of patients with
lung cancer can be diagnosed at an early stage, and lung cancer
metastasis is the major reason for ineffective treatment and
patient mortality (1–3). Metastasis to different organs could
occur at later stages of lung cancer progression, which often
results in the patient experiencing profound pain and having an
increased risk of mortality (4).
MicroRNA (miRNA/miR)s have been shown to be
efficient regulators in a variety of disease models and thus, are
proposed to have great potential in therapeutic applications
(5). miR-338-3p exerts negative
effects on the proliferation of neuroblastoma cells, and it could
also inhibit the migration and invasion of human neuroblastoma
cells, which involves direct targeting of the 3′ untranslated
region of phosphhatidylinoitol-3,4,5-trisphosphate dependent Rac
exchange factor 2a mRNA while it affects the Phosphatase and Tensin
homolog (PTEN)/Akt signaling pathway (6). The phosphoinositide 3 kinase
(PI3K)/AKT signaling pathway is involved in the invasion of
squamous cell carcinoma (7–10),
while the association between miR-338-3p and AKT signaling pathway,
the role of PI3K/AKT and its downstream signaling pathway in lung
cancer cell invasion lacks further investigation. miR-338-3p is
capable of suppressing cell proliferation and of inducing apoptosis
of non-small-cell lung cancer, suppressing epithelial-mesenchymal
transition (EMT) and metastasis in lung cancer cells (11,12).
An RNA-Seq assay may help to get a better profile of the impact of
miR-338-3p on lung cancer cells. Furthermore, the suppressing
effect of miR-338-3p on lung cancer cells proliferation needs to be
evaluated in an in vivo model.
About 30% of human tumors carry a mutation of ras
gene (13–15). Of the three genes in this family
(composed of K-ras, N-ras and H-ras), K-ras is the most frequently
mutated member in human tumors, including adenocarcinomas of the
pancreas and the lung (16,17).
KrasG12D mice were used in the present study as an in
vivo tumor model, to investigate the efficiency of miR-338-3p
on tumor inhibition, in vivo.
To further elucidate the role of miR-338-3p in
inhibiting lung cancer cell invasion, in vitro and in
vivo assays were performed in the present study. The results of
the present study demonstrated that miR-338-3p inhibited lung
cancer cell invasion and proliferation by downregulating the
AKT/β-catenin signaling pathway. This finding partially determined
the underlying mechanism of the inhibitory role of miR-338-3p on
lung cancer cell invasion and proliferation. Additionally, these
data supported the potential use of miR-338-3p in the treatment of
lung cancer.
Materials and methods
Cell culture, transfection and
apoptosis assay
The A549 cell line was purchased from the American
Type Culture Collection (Manassas, VA, USA). The cells were
cultured in Dulbecco's modified Eagle's medium (Invitrogen; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) containing 10% fetal
bovine serum (Invitrogen; Thermo Fisher Scientific, Inc.) and were
maintained at 37°C in an atmosphere containing 5% CO2
and 95% humidity. Cell apoptosis was determined using an Annexin
V-FITC Apoptosis Detection Kit I (BD Biosciences, Franklin Lakes,
NJ, USA) with a FACS Influx flow cytometer (BD Biosciences)
according to the manufacturer's protocol. Briefly, cells were
re-suspended in a pre-cooled staining buffer and the cell density
was adjusted to 2×107 cells/ml. Then 50 µl
(~106 cells) of cell suspension was transferred to a 1.5
ml Eppendorf tube and 300 µl of 1X Binding Buffer was added.
Subsequently, 5 µl of Annexin V-FITC was added to the tube and
incubated at room temperature for 15 min. Finally, 5 ml of PI
reagent was added for 5 min at room temperature before analysis.
The cells were suspended with the appropriate amount of 1X Binding
buffer and the staining results were analyzed by flow cytometry
within 30 min. In all flow cytometry assay, 10,000 events were
analyzed by flow cytometry (BD Biosciences) using CellQuest™ Pro
software (version 5.1; BD Biosciences).
miR-338-3p mimic (miR-338-3p) and corresponding
miRNA negative control (miR-NC) were transfected into A549 cells
using Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol. In
total, 100 nmol/l miR-338-3p mimics was used for transfection at a
cell confluence of 50–60%. The cells were analyzed 24–48 h after
the transfection. The microRNA was synthesized by Sangon Biotech
Co., Ltd. (Shanghai, China). The microRNA sequences were as
follows: miR-338-3p mimics: 5′-AACAAUAUCCUGGUGCUGAGUG-3′; negative
control mimics:
5′-UUCUCCGAACGUGUCACGUTTACGUGACACGUUCGGAGAATT-3′.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
For the RT-qPCR analyses of miR-338-3p expressions,
total RNA of tissues and cells was extracted using
TRIzol® RNA reagent (Invitrogen; Thermo Fisher
Scientific, Inc.), and first-strand cDNA was prepared via RT with
Superscript II reverse transcriptase (Invitrogen; Thermo Fisher
Scientific, Inc.). The RT-PCR sample was incubated at 42°C for 50
min and heated to inactivate the enzyme at 70°C for 15 min. The
expression of miR-338-3p was quantified using Fast SYBR Green
Master Mix (Applied Biosystems; Thermo Fisher Scientific, Inc.)
under ABI 7300 Sequence Detection System (Life Technologies; Thermo
Fisher Scientific, Inc.), according to the manufacturer's protocol
and normalized to the expression of U6. The primers were as
follows: Forward (F)-miR-338-3p (5′-TGCGGTCCAGCATCAGTGAT-3′) and
reverse (R)-miR-338-3p (5′-CCAGTGCAGGGTCCGAGGT-3′), F-U6
(5′-TGCGGGTGCTCGCTTCGGCAGC-3′) and R-U6
(5′-CCAGTGCAGGGTCCGAGGT-3′). The PCR temperature protocol was set
as follows: The sample was incubated for 30 min at 16°C, followed
by 60 cycles at 30°C for 30 sec, 42°C for 30 sec and 50°C for 1
sec, and incubated at 85°C for 5 min to inactivate the reverse
transcriptase. The relative expression of each gene was calculated
and normalized by the 2−ΔΔCq method (18). Each sample was tested in
triplicate.
Reagents
Perifosine (AKT inhibitor; 2.5 µM for 24 h at 37°C),
LY294002 (PI3K inhibitor; 0.5 µM for 24 h at 37°C), BIO (GSK-3
inhibitor; 2.5 µM for 24 h at 37°C) XAV939 (Wnt/β-catenin signaling
inhibitor; 30 nM for 24 h at 37°C) were purchased from Selleck.cn
(Selleck Chemicals, Houston, TX, USA). The compound bpV was
purchased from BioVision (PTEN inhibitor; BioVision, Inc.,
Milpitas, CA, USA). The cell invasion assay was conducted using
24-well Transwell chambers with 8 µM pores (Corning, Inc., Corning,
NY, USA). In total, 1×105 cells in Dulbecco's modified
Eagel's medium (DMEM; Thermo Fisher Scientific, Inc.) without serum
were added to the Matrigel-coated chambers, and 1 ml DMEM was added
to the bottom chamber. A small amount of serum (0.5% fetal bovine
serum; Biological Industries, Kibbutz Beit Haemek, Israel) was
added to bottom of the chamber to stimulate cell invasion.
Following incubation for 24 h, the filters were fixed (4%
paraformaldehyde; 15 min at room temperature) and stained with 0.2%
crystal violet (10 min at room temperature). Invaded cells were
manually counted in five random fields under a light microscope
(Olympus Corporation, Tokyo, Japan), and images (magnification,
×200) were captured.
Proliferation assay
An MTT assay was used to analyze cell proliferation.
A549 cells were transfected with either miR-338-3p mimic
(miR-338-3p) or mimic control (NC). After 24 h transfection, cells
were seeded into 96-well plate at 5.0×103 cells/ml and
cultured for 24 h. At each time point, 10 µl MTT reagent (5 mg/ml;
Sigma-Aldrich; Merck KGaA) was added to each well, successive
incubated for 4 h at 37°C. The supernatant was removed and 200 µl
DMSO (Invitrogen; Thermo Fisher Scientific, Inc.) was added to
dissolve the formazan crystals for 30 min. Spectrometric absorbance
at a wavelength of 570 nm was measured on microplate reader
(Spectra Max M5; Molecular Devices, LLC, Sunnyvale, CA, USA). Each
sample was tested in triplicate and all experiments were performed
three times.
Animal model
Primary lung cancer was induced in
KrasG12D mice as previously described (19). Briefly, 7–8-week-old mice (C57/B6
background; n=6; female; weight, 20±2 g, kept at a temperature of
21±2°C, with free access to water/food and kept in a 8/16
dark/light cycle) were anaesthetized with diethyl ether and
received 64 µl of cre-advenovirus (AdCre) solution (Biowit
Technologies, Ltd., Shenzen, China) through the nasal cavity. The
virus solution was prepared by mixing 30 µl of AdCre
(1010 pfu), 70 µl of Eagle's minimum essential medium
(Sigma-Aldrich; Merck KGaA), and 0.5 µl of 2 M CaCl2 and
placed in room temperature for 20 min. The weight of mice was
determined every two days. Subsequently, 35 days after the virus
treatment, the mice were euthanatized via CO2 (in a 3 L
chamber for 3 min), the lung tissues were harvested, and their size
and weight were determined. All procedures were performed with the
approval of the Animal Ethics Committee of Shenzhen Jiake
Biotechnology Co., Ltd. (Shenzhen, China). Lung cancer tissues were
washed with PBS and fixed with 4% paraformaldehyde (12 h at room
temperature), and the sections were paraffin-embedded, cut into 6
µm sections, and stained with routine H&E. The sections were
deparaffinized with xylene for 10 min at room temperature and
rehydrated in absolute alcohol for 5 min, 95% alcohol for 2 min and
70% alcohol for 2 min. The sections were briefly washed in
distilled water and stained in Harris hematoxylin solution for 8
min at room temperature. Subsequently, the sections were washed in
running tap water for 5 min and differentiated in 1% acid alcohol
for 30 sec. The sections were washed with running tap water for 1
min, placed in 0.2% ammonia water for 30 sec, washed in running tap
water for 5 min and rinsed in 95% alcohol. The sections were
counterstained in eosin-phloxine B solution for 30 sec at room
temperature, followed by dehydration with 95% alcohol, two changes
of absolute alcohol, 5 min each. They were cleared in xylene for 5
min and mounted with xylene based mounting medium. The sections
were visualized using a light microscope (magnification, ×200;
CKX53; Olympus Corporation).
In vivo delivery of miR-338-3p
A mPEG-PLGA-PLL-LA (PEAL-LA; Sigma-Aldrich; Merck
KGaA) nanoparticle- based microRNA in vivo delivery was
applied as previously described (20). One nanomole of miR-338-3p was
dissolved in 50 µl RNase-free water and emulsified in 500 µl
dichloromethane solution containing 5 mg of PEAL-LA copolymer by
sonication (Scientz sonicator probe, Ningbo Scientz Biotechnology,
Co., Ltd., Zhejiang, China) at 400 W for 1 min on ice to obtain a
water/oil emulsion. Subsequently, 2 ml PluronicTM F68 water
solution (1 mg/ml) was added to the water/oil emulsion and
sonicated again to obtain a water/oil/water emulsion. The resulting
water/oil/water emulsion was transferred to a rotary evaporator
(150 g; 20°C) to evaporate the organic solvent and obtain the
miR-338-3p-PEAL-LA nanoparticles. One week following AdCre
administration in KrasG12D mice, a single dose of
miR-338-3p-PEAL-LA nanoparticles (at a dose of 0.53 mg/kg
miR-338-3p) formulated above were administered via tail vein
injection every 2 days for 4 weeks.
Microarray assay
For the microarray assay, total RNA of tissues and
cells (1×106 at a confluence of 95%) was extracted using
TRIzol® RNA reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) and RNA was reversed to cDNA with a cDNA RT kit
(Agilent Technologies, Inc., Santa Clara, CA, USA). The RT sample
was incubated at 42°C for 50 min and heated to inactivate the
enzyme at 70°C for 15 min. A total of 10 µg of labeled cRNA was
hybridized at 45°C for 16 h to the mouse genome array MOE430A2.0
(Affymetrix; Thermo Fisher Scientific, Inc.). Processed chips were
read with an Agilent G2565CA Microarray Scanner (Agilent
Technologies, Inc.). Scanned microarray images were imported into
GeneSpring GX (v12.0) software to generate signal values and
absent/present calls for each probe-set using the MAS 5.0
statistical expression algorithm; this process identified genes
that were differentially expressed.
Immunofluorescence
Cells (at the confluence of 80–90%) were fixed with
4% formaldehyde for 15 min at room temperature and permeabilized
with 0.2% (v/v) Triton X-100 in PBS for 10 min at room temperature.
Specimens were incubated with anti-Ki67 (cat. no. GTX16667; 1:100;
GeneTex, Inc., Irvine, CA, USA), anti-serine-473-AKT (cat. no.
GTX121936; 1:100; GeneTex, Inc.) and anti-β-catenin antibodies
(cat. no. GTX101254; 1:100; GeneTex, Inc.) at 4°C overnight and
then incubated with Alexa Fluor-488/Cy3-conjugated anti-rabbit IgG
at 37°C for 1.5 h (cat. nos. 406416 and 406402; 1:200; BioLegend,
Inc., San Diego, CA, USA). To stain the nuclei, the specimens were
incubated with DAPI solution (2.5 mg/ml−1;
Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) for 10 min at room
temperature. The fluorescence signal was visualized using a
confocal microscope (magnification, ×600; Olympus Corporation).
Western blotting (WB)
Subconfluent cell cultures were washed with D-PBS,
and cell lysates were prepared in a lysis buffer containing 1%
(v/v) Triton X-100, 1% (v/v) deoxycholic acid, 2 mM
CaCl2 and protease inhibitors (10 µg/ml leupeptin, 10
µg/ml aprotinin, 1.8 mg/ml iodoacetamide and 1 mmol/l phenylmethyl
sulfonyl fluoride) and quantified using a bicinchoninic protein
assay kit (Pierce; Thermo Fisher Scientific, Inc.). Equal amounts
of total protein (30 µg whole cell lysate) were subjected to
electrophoresis on 12% Bis/Tris gels, transblotted onto
nitrocellulose membranes, and probed with different primary
antibodies. The membranes were blocked for 1 h at room temperature
using blocking buffer containing 3% bovine serum albumin (BSA;
Sigma-Aldrich; Merck KGaA) in PBS. Primary antibodies for AKT (cat.
no. GTX121936), β-catenin (cat. no. GTX101254), phosphorylated
(p)-β-catenin (Ser33/47/Thr41; cat. no. GTX132605), p-β-catenin
(Ser45; cat. no. GTX50180), p-β-catenin (Ser675; cat. no.
GTX123611), AKT (Ser473; cat. no. GTX128414), p-AKT (Thr308; cat.
no. GTX28933), E-cadherin (cat. no. GTX100443), intercellular
adhesion molecule-1 (ICAM-1; cat. no. GTX64322), matrix
metalloproteinase-1 (MMP-1; cat. no. GTX100534), MMP-2 (cat. no.
GTX104577), GAPDH (cat. no. GTX100118) were purchased from GeneTex,
Inc. and p-β-catenin (Ser552; cat. no. 956; Cell Signaling
Technology, Inc., Danvers, MA, USA) were all diluted (1:1,000) with
blocking agent and applied to membranes for 24 h at 4°C. Membranes
were washed with PBST and incubated with horseradish peroxidase
(HRP)-conjugated secondary antibodies (1:200; cat. no. sc-2357;
Santa Cruz Biotechnology, Inc., Dallas, TX, USA) for 2 h at room
temperature and washed with PBST. A chemiluminescent detection
(Thermo Fisher Scientific, Inc.) of HRP activity was used to detect
the signal in the membranes, and images were taken with
chemiluminescence apparatus (Quantity One® software;
Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Statistical analysis
Each experiment was repeated at least three times
independently. The results are expressed as the mean ± standard
error, unless stated otherwise. Statistical comparisons among
multiple groups were conducted using one-way analysis of variance
with the Student-Newman-Keuls test, or a Student's t-test
(unpaired, two-tailed) was used when making comparisons between two
groups. P<0.05 was considered to indicate a statistically
significant difference.
Results
miR-338-3p inhibits the proliferation
and invasion of A549 cells
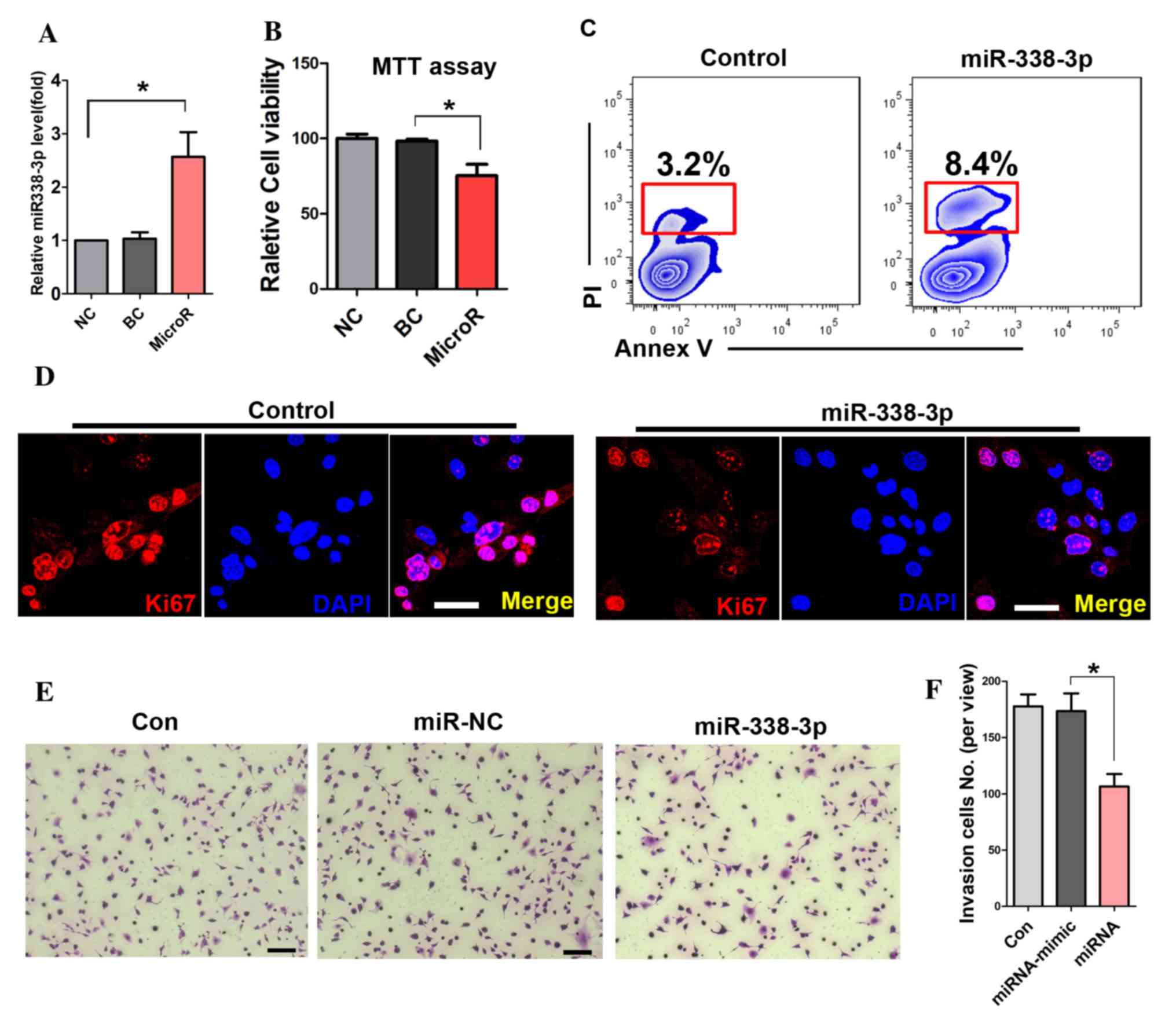
To detect the transfection efficiency of miR-338-3p,
RT-PCR was performed. The result showed that miR-338-3p expression
levels were significantly increased in the miR-338-3p transfected
cells (Fig. 1A). The MTT assay
showed that miR-338-3p attenuated the viability of A549 cells
(Fig. 1B). Further cell apoptotic
analysis with flow cytometry showed that miR-338-3p enhanced the
apoptotic rate of A549 cells compared with that in the miR-NC group
(8.4 and 3.2%; Fig. 1C). In
addition, the expression of Ki67 was significantly reduced in
miR-338-3p-treated cells (Fig.
1D). Furthermore, the number of migrated cells in the transwell
assay was significantly reduced in the miR-338-3p-treated group
during the cell invasion assay (Fig.
1E and F).
The effect of miR-338-3p on the
transcriptome of A549 cells
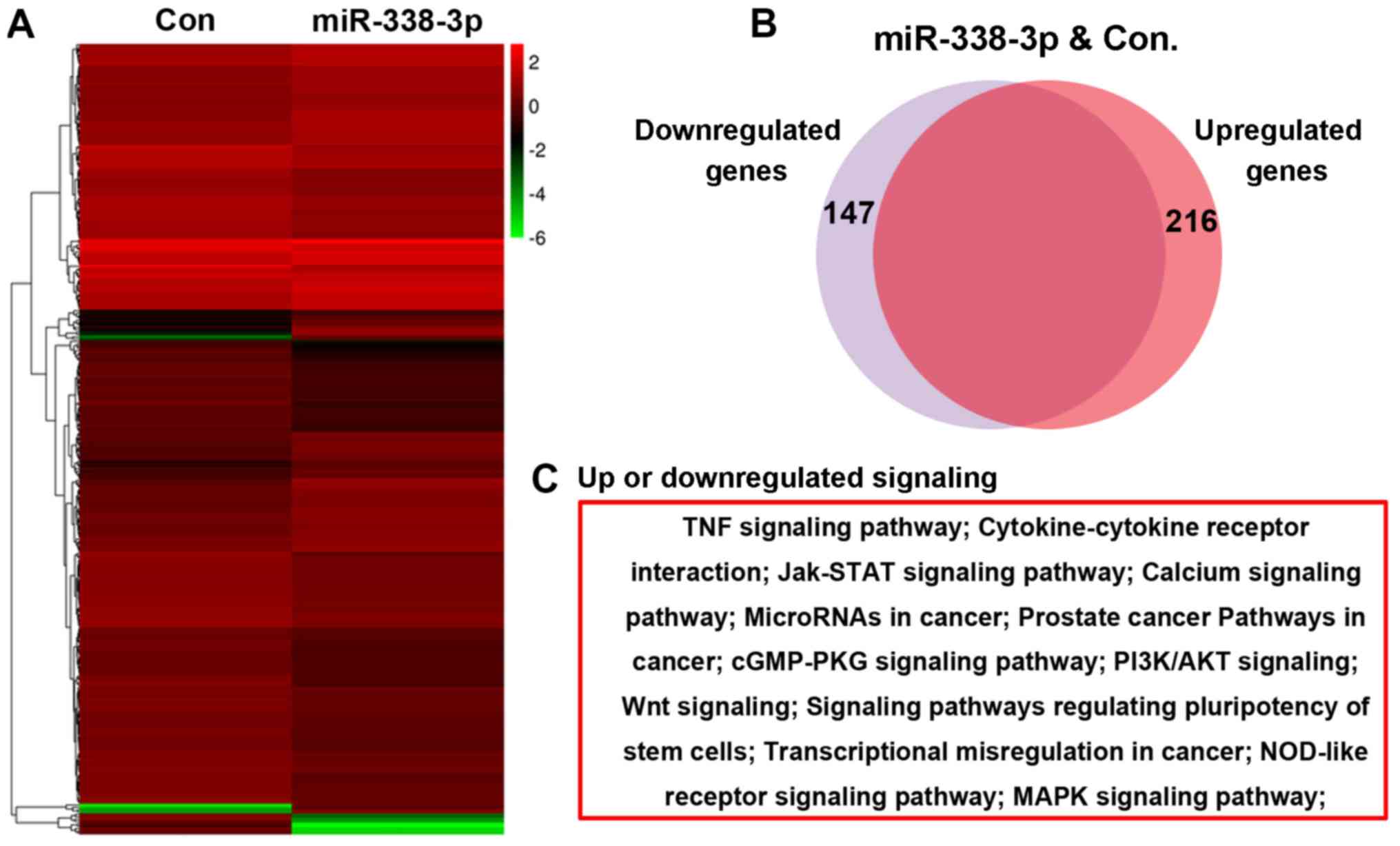
A microarray assay was performed to evaluate the
effect of miR-338-3p on the A549 cell transcriptome, and the
bioinformatic analysis result showed that 216 genes were
upregulated and 147 genes were downregulated in miR-338-3p-treated
cells (Fig. 2A and B). Further
bioinformatics analysis indicated that these altered genes were
enriched in several signaling pathways, including the Janus
kinase-signal transducer and activator of transcription, PI3K/AKT
and Wnt signaling pathways (Fig.
2C).
miR-338-3p downregulates AKT and
β-catenin signaling pathways
To further evaluate the effect of miR-338-3p on AKT
and β-catenin signaling pathway, immunostaining and WB were
performed. First, immunostaining showed that the expression levels
of (p-AKT at Ser473) were reduced in miR-338-3p cells (Fig. 3A), and similar results were
observed in the β-catenin staining (Fig. 3B). The WB results showed that both
p-AKT(Ser473) and p-AKT(Thr308) were downregulated with miR-338-3p
treatment (Fig. 3C), while
p-β-catenin (Ser552) but not p-β-catenin (Ser33/47/Thr41),
p-β-catenin (Ser45) or p-β-catenin (Ser675) was downregulated
(Fig. 3D). In addition, inhibiting
AKT signaling with perifosine attenuated the activity of both
p-β-catenin (Ser552) and β-catenin (Fig. 3E), suggesting that AKT signaling
positively regulated β-catenin signaling. Further assay of the
expression of cell invasion markers showed that activation of both
β-catenin (with BIO) and AKT (with bpV) signaling in
miR-338-3p-treated A549 cells resulted in a decrease in E-cadherin
expression and an increase in intercellular adhesion molecule-1,
matrix metalloproteinase (MMP)-1 and MMP-2 expression (Fig. 3F).
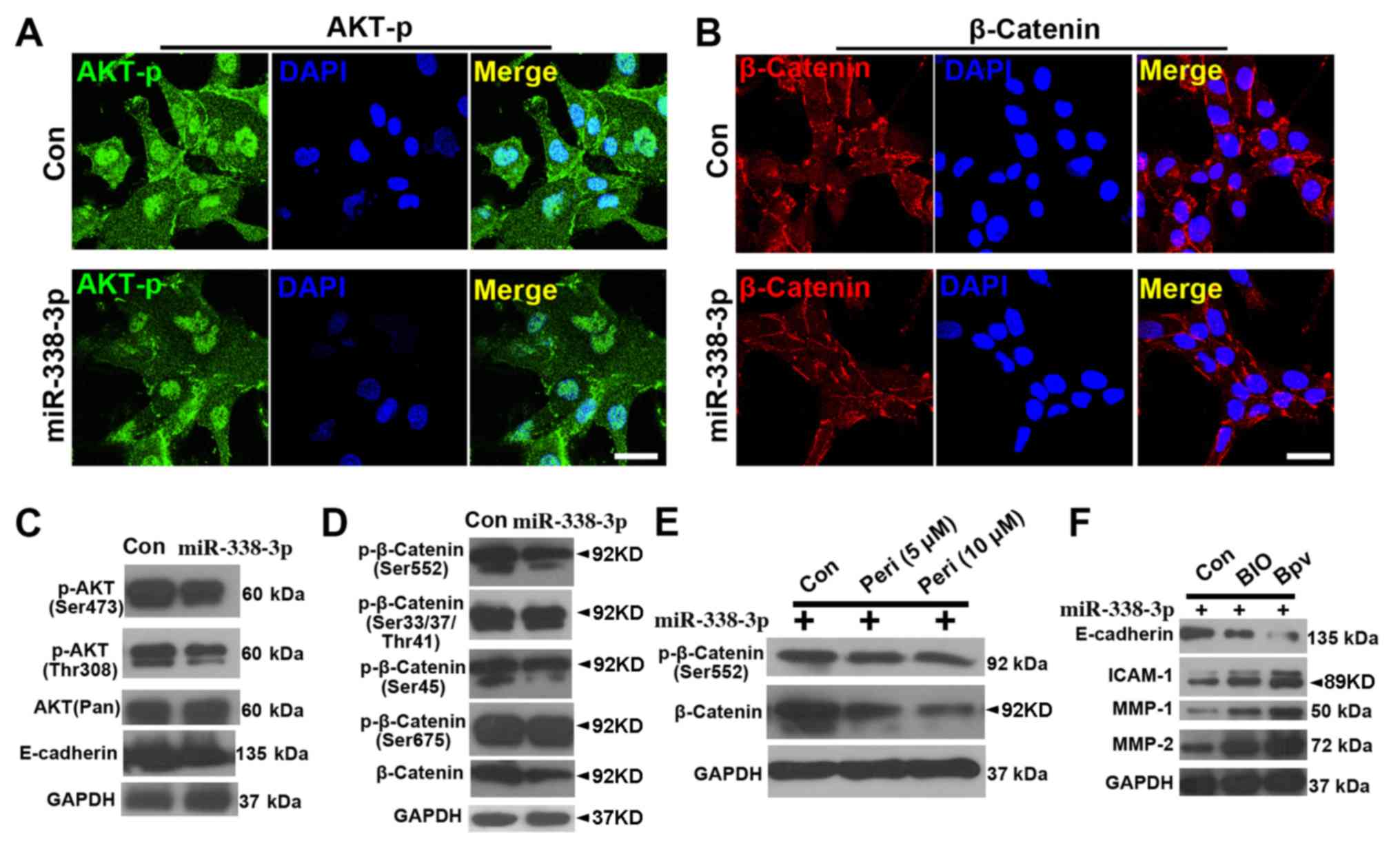 | Figure 3.miR-338-3p downregulates AKT and
β-catenin signaling pathway. Phosphorylated (Ser473) AKT (p-AKT) is
reduced in miR-338-3p-treated cells (A), and (B) similar result was
observed in the β-catenin staining. Western blotting results show
that (C) both p-AKT(Ser473) and p-AKT (Thr308) were downregulated
with miR-338-3p treatment, while (D) only p-β-catenin (Ser552), but
not p-β-catenin (Ser33/47/Thr41), p-β-catenin (Ser45) or
p-β-catenin (Ser675), was downregulated. (E) Inhibition of AKT
signaling pathway with perifosine attenuated the activity of both
p-β-catenin (Ser552) and β-catenin. (F) A decrease in E-cadherin
expression levels and an increase in ICAM-1, MMP-1 and MMP-2
expression levels, in miR-338-3p-treated A549 cells after treatment
with BIO and bpV. Scale bar, 20 µm. Con, control; ICAM,
intercellular adhesion molecule; miR, microRNA; MMP, matrix
metalloproteinase; Peri, perifosine. |
AKT and β-catenin signaling serve
crucial roles in miR-338-3p-inhibited A549 cell invasion
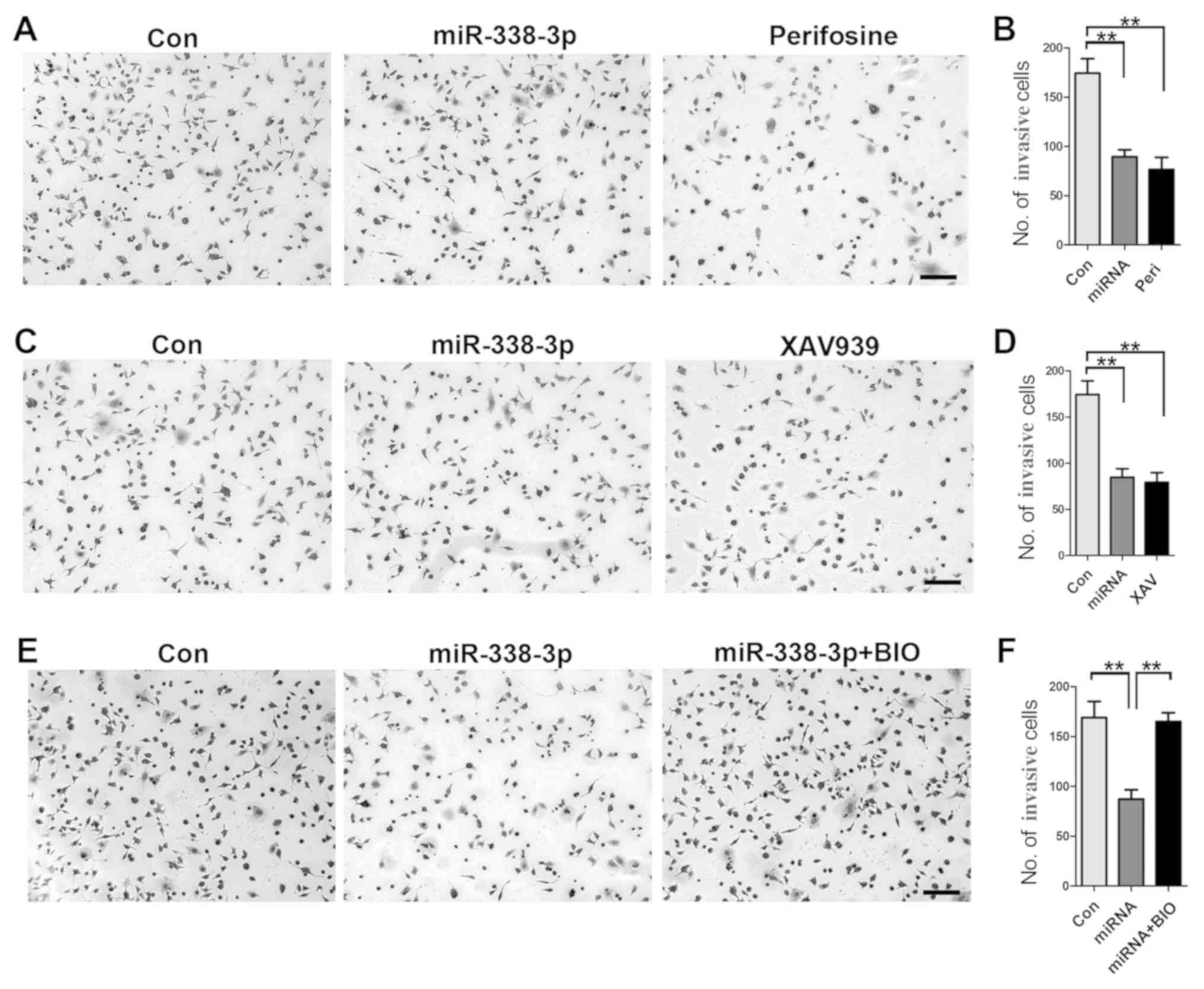
To gain further insight into the role of AKT and
β-catenin signaling pathways in how miR-338-3p inhibits A549 cell
invasion, a cell invasion assay was performed. First, inhibition of
AKT signaling pathway with perifosine resulted in a significant
reduction of invasive cells (Fig. 4A
and B); similarly, inhibition of β-catenin signaling with
XAV939 resulted in decreased number of invasive cells (Fig. 4C and D), indicating the important
role of both AKT and β-catenin signaling pathways in
miR-338-3p-mediated inhibition of A549 cell invasion. Furthermore,
increased activity of β-catenin with BIO rescued the inhibitory
effect of miR-338-3p on A549 cell invasion (Fig. 4E and F), validating the crucial
role of AKT/β-catenin signaling pathway in miR-338-3p-induced
inhibition of A549 cell invasion.
miR-338-3p inhibits lung cancer
development in vivo
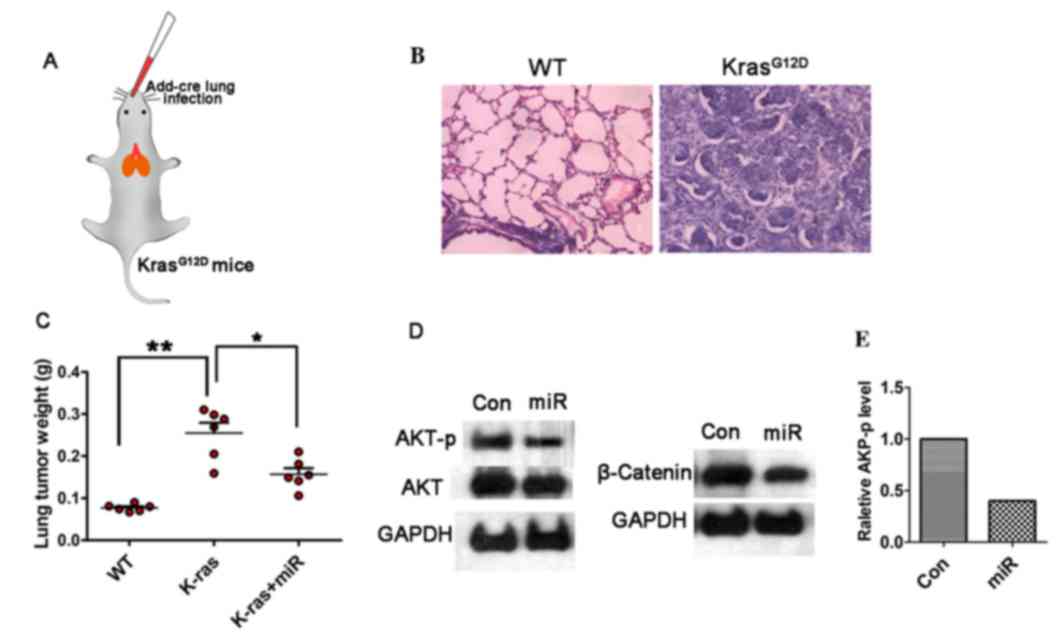
To investigate the efficiency of miR-338-3p on tumor
inhibition, in vivo, a lung cancer model in
KrasG12D mice was established, in which the
cre-advenovirus (AdCre) was administrated via nasal cavity
KrasG12D mice (Fig.
5A). Lung cancer was developed within five weeks after AdCre
treatment, H&E staining showed malignant cell
hyperproliferation in the AdCre treated tissue (Fig. 5B). To test the efficiency of
miR-338-3p in this primary lung cancer model, the in vivo
miR-338-3p delivery system was applied via mPEG-PLGA-PLL-LA
nanoparticles. The result showed that following the continuous
treatment of miR-338-3p for four weeks, the tumor weight on the
35th day after the primary tumor initiation was decreased in the
miR-338-3p treated group compared with the control groups (Fig. 5C). To further evaluate the role of
miR-338-3p on modulating AKT and β-catenin signaling pathways,
western blotting on the lung tissue in different treated group was
performed. Phosphorylation of β-catenin was analyzed to determine
the underlying mechanism of the upregulation of β-catenin, and it
was identified that β-catenin (ser552) was the phosphorylation
peptide. For this experiment, the total expression level of
β-catenin was assessed to evaluate the change of Wnt/β-catenin. The
result showed that both the AKT-p and β-catenin expression levels
were reduced in the miR-338-3p treated group (Fig. 5D and E). Although the total AKT
level was slightly decreased in the miR-338-3p treated group, the
AKT-p was significantly reduced, thus miR-338-3p may efficiently
reduce AKT signaling; however, this requires further investigation.
These data support that miR-338-3p is capable to suppress AKT and
β-catenin signaling pathway in lung cancer in vivo. Taken
together, the present data indicate that miR-338-3p may inhibit
A549 lung cancer cell proliferation and invasion by targeting AKT
and β-catenin signaling, in vitro and in vivo.
Discussion
The most common clinical metastases are lung cancer
(21). The recurrence and
metastasis of lung cancer are complicated processes with multiple
steps involving the degradation of tumor extracellular matrix, the
enhancement of the invasive ability of tumor cells, tumor
neovascularization and other cell biological behaviors. Some
contributors to these processes include changes in gene expression
and associated gene regulation, molecular signal pathway
abnormalities (22,23).
A previous study has shown that EMT is associated
with tumor invasion, tumor metastasis and drug resistance (24). Therefore, a novel tumor treatment
strategy target for EMT was established in a previous study
(25). MicroRNAs serve a
regulatory role in human gene expression, and abnormal tumor
expression and microRNA regulation extensively affect the
development of the tumor. Inhibition of EMT-associated miRNAs in
the tumor results in the abatement of tumor invasion and
metastasis; however, promoting the expression of EMT-associated
miRNAs accelerate tumor development. Although a small number of
miRNAs have been identified by biochemical or bioinformatics
methods, more studies were needed to investigate the biological
functions of them and to develop a systematic understanding of
their roles in tumor development and metastasis.
Accumulating data support Akt promote the growth and
proliferation of tumor cells, inhibit cell apoptosis, increase
hypoxia tolerance of cells via phosphorylating various downstream
substrates (26,27). Invasion, metastasis, and promotion
of resistance to chemotherapy and radiotherapy of cancer cells are
linked with overexpression and activation of AKT in a variety of
tumor tissues (28). p-AKT occurs
during AKT activation and AKT1 is one of the important subtypes of
AKT, and its expression is also increased in human gastric cancer
(29). Abnormal expression and
activity of AKT1 are also found in breast cancer, serving an
important role in its metastasis (30). The data of the present study
indicated that miR-338-3p is capable to inhibit the metastasis of
lung cancer cells, in vitro, in which the AKT signaling was
a crucial mediator. Additionally, the data of the present study
supported that β-catenin acts downstream of AKT signaling, as
inhibition of AKT signaling attenuates the β-catenin expression
level. And inhibition of both AKT and β-catenin attenuated A549
cells invasion. Furthermore, in the present study it was also found
that AKT and β-catenin were downregulated in the miR-338-3p treated
lung tumor. However, the mechanism underlying miR-338-3p modulating
AKT and β-catenin signaling in vivo still needs to be
investigated.
In conclusion, in the present study, miR-338-3p was
validated as an inhibitor of lung cancer cell invasion via
downregulation of AKT/β-catenin signaling. However, the regulatory
role of miR-338-3p in other signaling pathways and their underlying
mechanism requires further investigation.
Acknowledgements
Not applicable.
Funding
The present study was funded by Heilongjiang
Province Scientific Research Institution Innovation Grants (grant
no. YC2016D002) and Harbin Scientific Research and Innovation
Grants (grant no. 2015DB3AS010).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JL and TL designed the present study, performed the
experiments, analyzed the data and wrote the manuscript. LC, NZ,
YF, ZY, YL, CT and JH performed the experiments and analyzed the
data.
Ethics approval and consent to
participate
All procedures were performed with the approval of
the Animal Ethics Committee of Shenzhen Jiake Biotechnology Co.,
Ltd. (Shenzhen, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics for Hispanics/Latinos, 2012. CA Cancer J Clin.
62:283–298. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel R, DeSantis C, Virgo K, Stein K,
Mariotto A, Smith T, Cooper D, Gansler T, Lerro C, Fedewa S, et al:
Cancer treatment and survivorship statistics, 2012. CA Cancer J
Clin. 62:220–241. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2012. CA Cancer J Clin. 62:10–29. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wang X and Adjei AA: Lung cancer and
metastasis: New opportunities and challenges. Cancer Metastasis
Rev. 34:169–171. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Catela Ivkovic T, Voss G, Cornelia H and
Ceder Y: microRNAs as cancer therapeutics: A step closer to
clinical application. Cancer Lett. 407:113–122. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chen X, Pan M, Han LL, Lu HT, Hao XW and
Dong Q: miR-338-3p suppresses neuroblastoma proliferation, invasion
and migration through targeting PREX2a. FEBS Lett. 587:3729–3737.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Baek SH, Ko JH, Lee JH, Kim C, Lee H, Nam
D, Lee J, Lee SG, Yang WM, Um JY, et al: Ginkgolic acid inhibits
invasion and migration and TGF-β-induced EMT of lung cancer cells
through PI3K/Akt/mTOR inactivation. J Cell Physiol. 232:346–354.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jiao D, Wang J, Lu W, Tang X, Chen J, Mou
H and Chen QY: Curcumin inhibited HGF-induced EMT and angiogenesis
through regulating c-Met dependent PI3K/Akt/mTOR signaling pathways
in lung cancer. Mol Ther Oncolytics. 3:160182016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Meng J, Zhang XT, Liu XL, Fan L, Li C, Sun
Y, Liang XH, Wang JB, Mei QB, Zhang F and Zhang T: WSTF promotes
proliferation and invasion of lung cancer cells by inducing EMT via
PI3K/Akt and IL-6/STAT3 signaling pathways. Cell Signal.
28:1673–1682. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pan H, Jiang T, Cheng N, Wang Q, Ren S, Li
X, Zhao C, Zhang L, Cai W and Zhou C: Long non-coding RNA BC087858
induces non-T790M mutation acquired resistance to EGFR-TKIs by
activating PI3K/AKT and MEK/ERK pathways and EMT in non-small-cell
lung cancer. Oncotarget. 7:49948–49960. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hong-Yuan W and Xiao-Ping C: miR-338-3p
suppresses epithelial-mesenchymal transition and metastasis in
human nonsmall cell lung cancer. Indian J Cancer. 52 (Suppl
3):E168–E171. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang G, Zheng H, Zhang G, Cheng R, Lu C,
Guo Y and Zhao G: MicroRNA-338-3p suppresses cell proliferation and
induces apoptosis of non-small-cell lung cancer by targeting
sphingosine kinase 2. Cancer Cell Int. 17:462017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bos JL: Ras oncogenes in human cancer: A
review. Cancer Res. 49:4682–4689. 1989.PubMed/NCBI
|
|
14
|
Avruch J, Zhang XF and Kyriakis JM: Raf
meets Ras: Completing the framework of a signal-transduction
pathway. Trends Biochem Sci. 19:279–283. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Khosravi-Far R and Der CJ: The Ras
signal-transduction pathway. Cancer Metastasis Rev. 13:67–89. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mills NE, Fishman CL, Rom WN, Dubin N and
Jacobson DR: Increased prevalence of K-Ras oncogene mutations in
lung adenocarcinoma. Cancer Res. 55:1444–1447. 1995.PubMed/NCBI
|
|
17
|
Pellegata NS, Sessa F, Renault B, Bonato
M, Leone BE, Solcia E and Ranzani GN: K-Ras and P53 gene-mutations
in pancreatic-cancer: Ductal and nonductal tumors progress through
different genetic lesions. Cancer Res. 54:1556–1560.
1994.PubMed/NCBI
|
|
18
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2 (-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jackson EL, Willis N, Mercer K, Bronson
RT, Crowley D, Montoya R, Jacks T and Tuveson DA: Analysis of lung
tumor initiation and progression using conditional expression of
oncogenic K-ras. Gene Dev. 15:3243–3248. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cai CL, Xie YX, Wu LL, Chen XJ, Liu HM,
Zhou Y, Zou H, Liu D, Zhao Y, Kong X and Liu P: PLGA-based dual
targeted nanoparticles enhance miRNA transfection efficiency in
hepatic carcinoma. Sci Rep. 7:462502017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tamura T, Kurishima K, Nakazawa K,
Kagohashi K, Ishikawa H, Satoh H and Hizawa N: Specific organ
metastases and survival in metastatic non-small-cell lung cancer.
Mol Clin Oncol. 3:217–221. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Blandin Knight S, Crosbie PA, Balata H,
Chudziak J, Hussell T and Dive C: Progress and prospects of early
detection in lung cancer. Open Biol. 7:1700702017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lemjabbar-Alaoui H, Hassan OU, Yang YW and
Buchanan P: Lung cancer: Biology and treatment options. Biochim
Biophys Acta. 1856:189–210. 2015.PubMed/NCBI
|
|
24
|
Tsai JH and Yang J: Epithelial-mesenchymal
plasticity in carcinoma metastasis. Genes Dev. 27:2192–2206. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shibue T and Weinberg RA: EMT, CSCs, and
drug resistance: The mechanistic link and clinical implications.
Nat Rev Clin Oncol. 14:611–629. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Linnerth-Petrik NM, Santry LA, Moorehead
R, Jücker M, Wootton SK and Petrik J: Akt isoform specific effects
in ovarian cancer progression. Oncotarget. 7:74820–74833. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mundi PS, Sachdev J, McCourt C and
Kalinsky K: AKT in cancer: New molecular insights and advances in
drug development. Br J Clin Pharmacol. 82:943–956. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tanno S, Tanno S, Mitsuuchi Y, Altomare
DA, Xiao GH and Testa JR: AKT activation up-regulates insulin-like
growth factor I receptor expression and promotes invasiveness of
human pancreatic cancer cells. Cancer Res. 61:589–593.
2001.PubMed/NCBI
|
|
29
|
Han J, Zhang L, Zhang J, Jiang Q, Tong D,
Wang X, Gao X, Zhao L and Huang C: CREBRF promotes the
proliferation of human gastric cancer cells via the AKT signaling
pathway. Cell Mol Biol (Noisy-le-grand). 64:40–45. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Enomoto A, Murakami H, Asai N, Morone N,
Watanabe T, Kawai K, Murakumo Y, Usukura J, Kaibuchi K and
Takahashi M: Akt/PKB regulates actin organization and cell motility
via Girdin/APE. Dev Cell. 9:389–402. 2005. View Article : Google Scholar : PubMed/NCBI
|