Introduction
Non-syndromic orofacial clefts (NSOC) are common
congenital birth defects in humans, with a worldwide incidence of
approximately 1 in 700 cases. The etiology of NSOC is complex, and
is generally believed to involve interactions between environmental
and genetic factors. NSOC can be divided into cleft lip with or
without cleft palate (CL/P) and cleft palate only (CPO) according
to different etiologies (1,2).
Previous research has focused on the etiological
mechanisms underlying NSOC, although much remains to be discovered
(3–5). A number of genome-wide association
studies (GWAS) have revealed that several gene variants can lead to
NSOC; however, these protein-coding genes can only partially
explain the causes of NSOC (6–8). The
Human Genome Project found that only 2% of genes in the genome code
for proteins. Moreover, previous GWAS data analyses have found that
approximately 80% of susceptible sites are not in protein coding
regions (9), illustrating that
genes that do not code for proteins also play important roles in
the incidence of NSOC.
Non-coding RNA (ncRNA) is a general term for a
variety of RNA types that do not encode proteins; nevertheless,
these RNAs regulate gene expression on multiple levels. ncRNAs can
be grouped according to their length. MicroRNAs (miRNAs or miRs)
are a kind of endogenous small RNAs with a length of approximately
19–25 nt that can regulate the expression of genes at the
transcriptional level (10).
miRNAs can degrade or inhibit the translation of target genes by
binding specifically to their 3′-UTRs. Mounting evidence indicates
that ncRNAs play important roles in various diseases, such as
cancer, cardiovascular diseases and nervous system diseases, as
well as others (11–13). In recent years, several researchers
have found that ncRNAs also affect the pathogenesis of NSOC. For
example, during lip development, the target genes of the miR-203
and miR-302 clusters are different subtypes of p63, and it has been
shown that the deletion of these genes can cause different degrees
of CL/P (14). In addition, a
functional analysis of zebrafish models found that the
overexpression of miR-23b led to the broadening of the ethmoid
plate and aberrant cartilage structures in the viscerocranium,
while the overexpression of miR-133b causes a reduction in ethmoid
plate size and a significant midfacial cleft (15).
Long non-coding RNAs (lncRNAs) are ncRNAs with a
length of >200 nt with no protein coding function. They regulate
the expression of genes at the transcriptional,
post-transcriptional and epigenetic levels (16). lncRNAs have drawn increasing public
attention in recent years; increasing evidence suggests that they
play crucial regulatory roles in various physiological and
pathological processes. However, reports of the functions and
mechanisms of action of lncRNAs as regards the development of the
lip and palate are limited. The lncRNA H19 gene was initially
identified as a CPO-related gene in transforming growth factor
(TGF) β3-knockout mice by RNA sequencing analysis (17). Based on this finding, the present
study investigated the expression of lncRNA H19 in mice with a
cleft palate induced by 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)
and retinoic acid (RA). The results revealed that the expression of
lncRNA H19 and its target gene, insulin like growth factor 2
(IGF2), changed from E13.5 to E15.5, exhibiting a negative
correlation. This finding indicated that lncRNA H19 may interact
with its target gene, IGF2, playing significant roles in palate
development (18,19). Although lncRNAs and miRNAs can
regulate gene expression, their roles are not independent, and they
form a complex regulatory network.
Although emerging data have revealed that ncRNAs and
mRNAs play important roles in NSOC, no studies of the potential
interactions between ncRNAs and mRNAs in the development of NSOC
have been reported to date, at least to the best of our knowledge.
Using high-throughput sequencing methods, this study identified
differentially expressed (DE) ncRNAs and mRNAs from the peripheral
blood of patients with CL/P and CPO. Using bioinformatics analyses,
we then predicted the function and potential regulatory
associations of the ncRNAs, with a goal of discovering novel
etiological molecular mechanisms of NSOC.
Materials and methods
Sample collection
This study was approved by the Ethics Committee of
Harbin Medical University (Harbin, China), and informed consent was
obtained from the patients' families before sampling. The
participants were divided into 3 groups as follows: A CL/P group, a
CPO group and a healthy control group. The selection criteria were
the following: Patients with a cleft lip and palate, without other
congenital malformations and genetic diseases, who were
hospitalized at the First Affiliated Hospital of Harbin Medical
University were selected for this study. All 36 participants
included in this study were aged between 1 month and 40 months and
enrolled from January 2017 to May 2018. On the morning of the 2nd
day of admission, 5 ml of peripheral blood were collected using
tubes containing EDTA. In this study, 9 samples (3 controls, 3 CL/P
and 3 CPO) were used for RNA-Seq analysis, and another 27 samples
(10 controls, 10 CL/P and 7 CPO) were used for reverse
transcription-quantitative polymerase chain reaction (RT-qPCR).
Participant information is presented in Table I.
 | Table I.Information regarding the
participants. |
Table I.
Information regarding the
participants.
| Group | Age, months | Sex | Sample use |
|---|
| CL/P | 3 | Male | RNA-seq |
| CL/P | 10 | Male | RNA-seq |
| CL/P | 11 | Female | RNA-seq |
| CL/P | 12 | Male | RT-qPCR |
| CL/P | 9 | Female | RT-qPCR |
| CL/P | 5 | Male | RT-qPCR |
| CL/P | 12 | Male | RT-qPCR |
| CL/P | 27 | Male | RT-qPCR |
| CL/P | 18 | Male | RT-qPCR |
| CL/P | 10 | Female | RT-qPCR |
| CL/P | 8 | Male | RT-qPCR |
| CL/P | 6 | Male | RT-qPCR |
| CL/P | 4 | Male | RT-qPCR |
| CPO | 24 | Male | RNA-seq |
| CPO | 18 | Female | RNA-seq |
| CPO | 14 | Male | RNA-seq |
| CPO | 15 | Female | RT-qPCR |
| CPO | 8 | Female | RT-qPCR |
| CPO | 11 | Male | RT-qPCR |
| CPO | 35 | Female | RT-qPCR |
| CPO | 37 | Female | RT-qPCR |
| CPO | 16 | Female | RT-qPCR |
| CPO | 15 | Female | RT-qPCR |
| Control | 9 | Male | RNA-seq |
| Control | 7 | Male | RNA-seq |
| Control | 5 | Male | RNA-seq |
| Control | 6 | Male | RT-qPCR |
| Control | 4 | Male | RT-qPCR |
| Control | 1 | Female | RT-qPCR |
| Control | 4 | Female | RT-qPCR |
| Control | 5 | Female | RT-qPCR |
| Control | 11 | Female | RT-qPCR |
| Control | 4 | Female | RT-qPCR |
| Control | 23 | Female | RT-qPCR |
| Control | 5 | Female | RT-qPCR |
| Control | 5 | Female | RT-qPCR |
RNA extraction and quality
control
Total RNA was extracted from the samples using
TRIzol® reagent (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) according to the manufacturer's instructions. RNA
degradation and contamination were monitored using 1% agarose gels.
RNA purity was examined using a NanoPhotometer®
spectrophotometer (Implen, Inc., Westlake Village, CA, USA). The
RNA concentration was measured using the Qubit® RNA
Assay kit in a Qubit® 2.0 Fluorometer (Thermo Fisher
Scientific, Inc.). RNA integrity was assessed using the RNA Nano
6000 Assay kit for the Bioanalyzer 2100 system (Agilent
Technologies, Santa Clara, CA, USA).
Construction of the RNA sequencing
library
A total of 3 µg of RNA per sample was used as input
material for the RNA sample preparations. First, ribosomal RNA was
removed using an Epicentre Ribo-zero™ rRNA Removal kit (Epicentre,
Charlotte, NC, USA) and rRNA-free residue was cleaned by ethanol
precipitation. Subsequently, sequencing libraries were generated
from the rRNA-depleted RNA using a NEBNext® Ultra™
Directional RNA Library Prep kit for Illumina® (NEB,
Ipswich, MA, USA), following the manufacturer's recommendations.
Briefly, fragmentation was carried out using divalent cations under
elevated temperature in NEBNext First Strand Synthesis Reaction
Buffer (5X). First-strand cDNA was synthesized using a random
hexamer primer and M-MuLV Reverse Transcriptase (RNase H-). Second
strand cDNA synthesis was subsequently performed using DNA
Polymerase I and RNase H. In the reaction buffer, dNTPs with dTTP
were replaced by dUTP. Remaining overhangs were converted into
blunt ends via exonuclease/polymerase activity. Following the
adenylation of the 3′ ends of DNA fragments, NEBNext Adaptors with
hairpin loop structures were ligated to prepare for hybridization.
To select cDNA fragments that were preferentially 150–200 bp in
length, the library fragments were purified with the AMPure XP
System (Beckman Coulter, Beverly, MA, USA). Subsequently, 3 µl of
USER Enzyme (NEB) was used with the size-selected, adaptor-ligated
cDNA at 37°C for 15 min followed by 5 min at 95°C before PCR. PCR
was performed with Phusion High-Fidelity DNA polymerase, universal
PCR primers and the Index (X) Primer. Finally, products were
purified (AMPure XP System), and library quality was assessed on
the Agilent Bioanalyzer 2100 system. (Agilent Technologies).
Construction and sequencing of small
RNA libraries
A total of 3 µg of total RNA per sample was used as
the input material for the small RNA libraries. Sequencing
libraries were generated using the NEBNext® Multiplex
Small RNA Library Prep Set for Illumina® (NEB) following
the manufacturer's recommendations and index codes were added to
attribute sequences for each sample. Briefly, NEB 3′ SR Adaptors
were directly and specifically ligated to the 3′ end of the miRNA.
Following the 3′ ligation reaction, the SR RT Primer hybridized to
the excess 3′ SR Adaptor (that remained free after the 3′ ligation
reaction) and transformed the single-stranded DNA adaptor into a
double-stranded DNA molecule. This step is important to prevent
adaptor-dimer formation. Furthermore, dsDNAs are not substrates for
ligation mediated by T4 RNA Ligase 1 and therefore would not ligate
to the 5′ SR Adaptor in the subsequent ligation step. The 5′ ends
adapter was ligated to the 5′ ends of the miRNAs. First-strand cDNA
was then synthesized using M-MuLV Reverse Transcriptase (RNase H-).
PCR amplification was performed using LongAmp Taq 2X Master Mix, SR
Primer for Illumina and the Index (X) primer (NEB). PCR products
were purified on an 8% polyacrylamide gel (100 V, 80 min). DNA
fragments corresponding to 140–160 bp (the length of the small
ncRNA plus the 3′ and 5′ adaptors) were recovered and dissolved in
8 µl of elution buffer. Finally, library quality was assessed on an
Agilent Bioanalyzer 2100 system using DNA High Sensitivity Chips.
Clustering of the index-coded samples was performed on a cBot
Cluster Generation System using a TruSeq SR Cluster kit v3-cBot-HS
(Illumina) according to the manufacturer's instructions. Following
cluster generation, the library preparations were sequenced on an
Illumina HiSeq 2500 platform (Illumina, Inc., San Diego, CA, USA),
and 125-bp paired-end and 50-bp single-end reads were
generated.
Analysis of the sequencing
results
Low-quality raw reads containing adapters were
filtered to get clean reads. For lncRNAs, the clean reads were
aligned to the reference genome using HISAT2 (version 2.0.4)
(20), and then the mapped reads
of each sample were assembled by StringTie software (version 1.3.1)
(20,21). A series of strict screening
conditions was then set according to the structure and functional
characteristics to identify lncRNAs for the following analysis. The
screening conditions included: Number of exons ≥2, length >200
bp, fragments per kilobase million (FPKM) ≥0.5, and removing
transcripts that overlap the exon region of the database annotation
and with coding potential by either/all of the four tools: CNCI
version 2 (22), CPC version
cpc-0.9-r2 (23), Pfam-sca version
1.3 (24) and phyloCSF version
20121028 (25). FPKMs of both
lncRNAs and coding genes in each sample were calculated for
differential expression analysis using Ballgown software with a
P-adjust <0.05 (20). For
miRNAs, the clean reads were screened to include those with a
length of 18–25 nt and were aligned to the reference sequence using
Bowtie software (version bowtie-0.12.9) (26), which was combined with miREvo
software (version miREvo_v1.1) (27) and miRDeep2 software (version
mirdeep2_0_0_5) (28) to analyze
the functions of new miRNAs and adopt DESeq2 (version 1.12.0)
(29) with a negative binomial
distribution to analyze DE miRNAs. All sequencing programs were
carried out by Novogene Company (China, Beijing).
RT-qPCR validation
To validate the reliability of the RNAseq results, 2
miRNAs (miR-483-3p and miR-92b-5p), 2 lncRNAs (lncRNA RP11-731F5.2
and lncRNA XIST) and 2 mRNAs [BCL2 associated athanogene 5 (BAG5)
and zinc finger E-box binding homeobox 2 (ZEB2)] were selected for
further validation using the RT-qPCR method with another 27 samples
(10 controls, 10 CL/P and 7 CPO). The selection criteria were based
on the fold changes and P-values of DE genes, and the selection was
also based on the DE genes that have been associated with NSOC. The
total RNA of each sample was extracted, and first-strand cDNA was
synthesized using cDNA Synthesis kit (GK8030; Generay Biotech Co.,
Ltd., Shanghai, China) and RevertAid First Strand cDNA Synthesis
kit (K1621 and EN0521; Thermo Fisher Scientific, Inc.) according to
the manufacturer's instructions. The temperature protocol used for
reverse transcription reaction was as follows: Incubation for 5 min
at 25°C, followed by 60 min at 42°C and terminating the reaction by
heating at 70°C for 5 min. The cDNA was then used as the template
for a RT-qPCR reaction with Power SYBR® Green PCR Master
Mix (cat. no. 4367659; Applied Biosystems, Thermo Fisher
Scientific, Inc.) and Power qPCR PreMix (GK8020; Generay Biotech
Co., Ltd.). Reactions were performed in 3 independent wells. The
thermocycling conditions for qPCR were as follows: Initial
denaturation at 95°C for 10 min, followed by 40 cycles at 95°C for
10 sec and annealing at 60°C for 34 sec. The sequences of the
primers are presented in Table
II. The relative expression levels of miRNAs, mRNAs and lncRNAs
were calculated using miR-16-5p and β-actin as internal references.
The RT-qPCR results were quantified using the 2−ΔΔCq
method (30).
| Table II.Primers designed for reverse
transcription-quantitative polymerase chain reaction validation of
candidate noncoding RNAs and mRNAs. |
Table II.
Primers designed for reverse
transcription-quantitative polymerase chain reaction validation of
candidate noncoding RNAs and mRNAs.
| Gene | Primer sequence
(5′-3′) |
|---|
| ZEB2 | F: |
CAAGCACCACCTTATCGAGC |
|
| R: |
TGTGATTCATGTGCTGCGAG |
| BAG5 | F: |
CACATCCTTCCGTTGCCAAA |
|
| R: |
CCCGAGAGCACACAGGATAA |
| XIST | F: |
GACTACCCAAAGCCCCTTCT |
|
| R: |
AGTCAACACTGCACCAACAC |
| RP11-731F5.2 | F: |
CAGTCAAGACCATCGCCAAG |
|
| R: |
CCACTGGTCCCATCACTTCT |
| β-actin | F: |
GTCCACCTTCCAGCAGATGT |
|
| R: |
CTCAGTAACAGTCCGCCTAGAA |
| hsa-miR-483-3p | F: |
GCGCTCACTCCTCTCCTC |
|
| R: |
TGGGTTCATTTCTGGGTCTT |
| hsa-miR-92b-5p | F: |
GCGCAGGGACGGGACGCGG |
|
| R: |
TGGGTTCATTTCTGGGTCTT |
| hsa-miR-16-5p | F: |
GCGCTAGCAGCACGTAAAT |
|
| R: |
TGGGTTCATTTCTGGGTCTT |
Analysis of the miRNA-mRNA-lncRNA
regulatory network
lncRNAs have extensive regulatory functions. Not
only can they directly regulate the structure of DNA and the
transcription and translation of RNA, but they can also regulate
gene expression indirectly by acting as miRNA sponges by
competitively binding to the same miRNA binding sites with mRNAs.
Based on the theory of competing endogenous RNA (ceRNA), we
attempted to look for lncRNA-gene pairs with the same binding sites
as miRNAs to construct lncRNA-miRNA-mRNA regulatory networks with
lncRNA as a decoy, miRNA as a center and mRNA as a target, and we
visualized these networks using Cytoscape software (http://www.cytoscape.org/download.php).
Prediction and functional analysis of
lncRNA target genes
lncRNAs can regulate the expression of target genes
by colocalization and co-expression. We set the threshold of
colocalization as 100 kb upstream and downstream of the lncRNAs.
Pearson correlation coefficients with absolute values >0.95 were
used to concurrently analyze the correlation between lncRNAs and
mRNAs. The target genes of DE miRNAs were predicted using the
intersection of the miRanda (http://miranda.org.uk/), PITA (https://genie.weizmann.ac.il/pubs/mir07/mir07_prediction.html)
and RNAhybrid databases (https://bibiserv.cebitec.uni-bielefeld.de/rnahybrid/).
Subsequently, the candidate target genes obtained from the above
methods were analyzed by Gene Ontology (GO, http://www.geneontology.org/) and Kyoto Encyclopedia
of Genes and Genomes (KEGG, http://www.genome.jp/kegg/) pathway enrichment
analysis using GOseq (version 2.12) (31) and KOBAS (version 2.0) (32), respectively. GO is an international
standard classification system for gene function. According to the
distribution of predicted genes, which are identified in each group
in GO, the functions of DE ncRNAs were predicted and classified.
The groups include biological process (BP), cellular component (CC)
and molecular function (MF). Different genes coordinate with each
other to perform their biological functions in vivo.
Significant pathway enrichment can determine the main biochemical
metabolic pathways and signal transduction pathways involving
target genes. KEGG is the main public database of pathways and uses
the KEGG pathway as a unit, applying hypergeometric tests to define
significantly enriched pathways involving target genes in
comparison to the whole genome background.
Statistical analysis
All measurement data are presented as the means ±
SD. The relative expression levels of miR-92b-5p and lncRNA XIST
between the CPO and control groups were analyzed by Student's
t-test using GraphPad Prism® version 7.04 software
(GraphPad Software, Inc., La Jolla, CA, USA). The relative
expression levels of miR-483-3p, lncRNA RP11-731F5.2, BAG5 and ZEB2
in the CPO, CL/P and control groups were analyzed by one-way ANOVA
with Dunnett's multiple comparisons test using GraphPad Prism
Version 7.04 software (GraphPad Software, Inc.). P<0.05 was
considered to indicate a statistically significant difference.
Results
DE ncRNAs and mRNAs
A total of 3,450 annotated lncRNAs, 1,447 annotated
miRNAs, 3,990 novel lncRNAs and 97 novel miRNAs were obtained after
mapping to the reference genome and carrying out prediction
according to the structure and functional characteristics of
lncRNAs and miRNAs. We analyzed ncRNAs and mRNAs according to fold
change values and significance levels to identify DE lncRNAs,
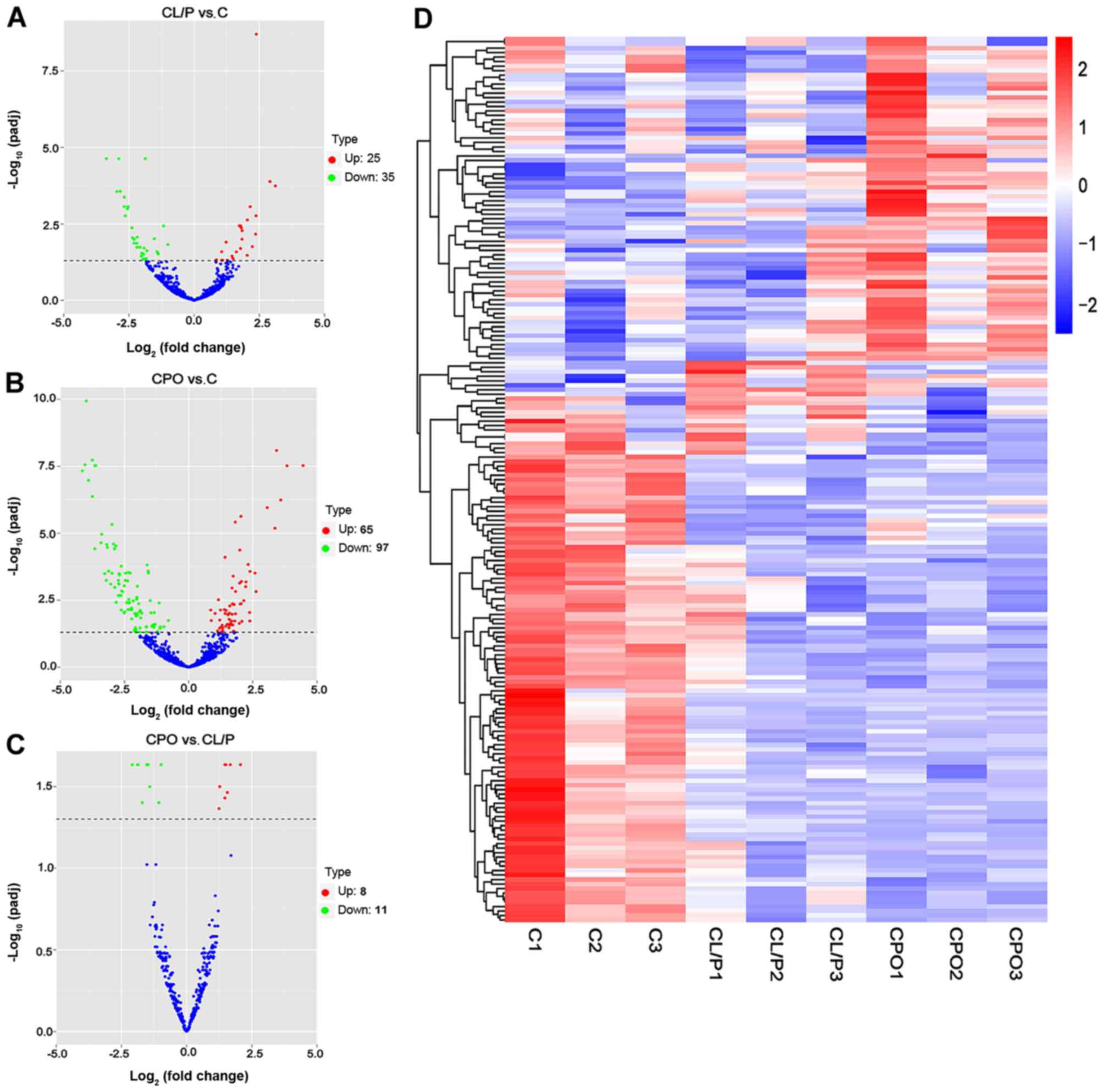
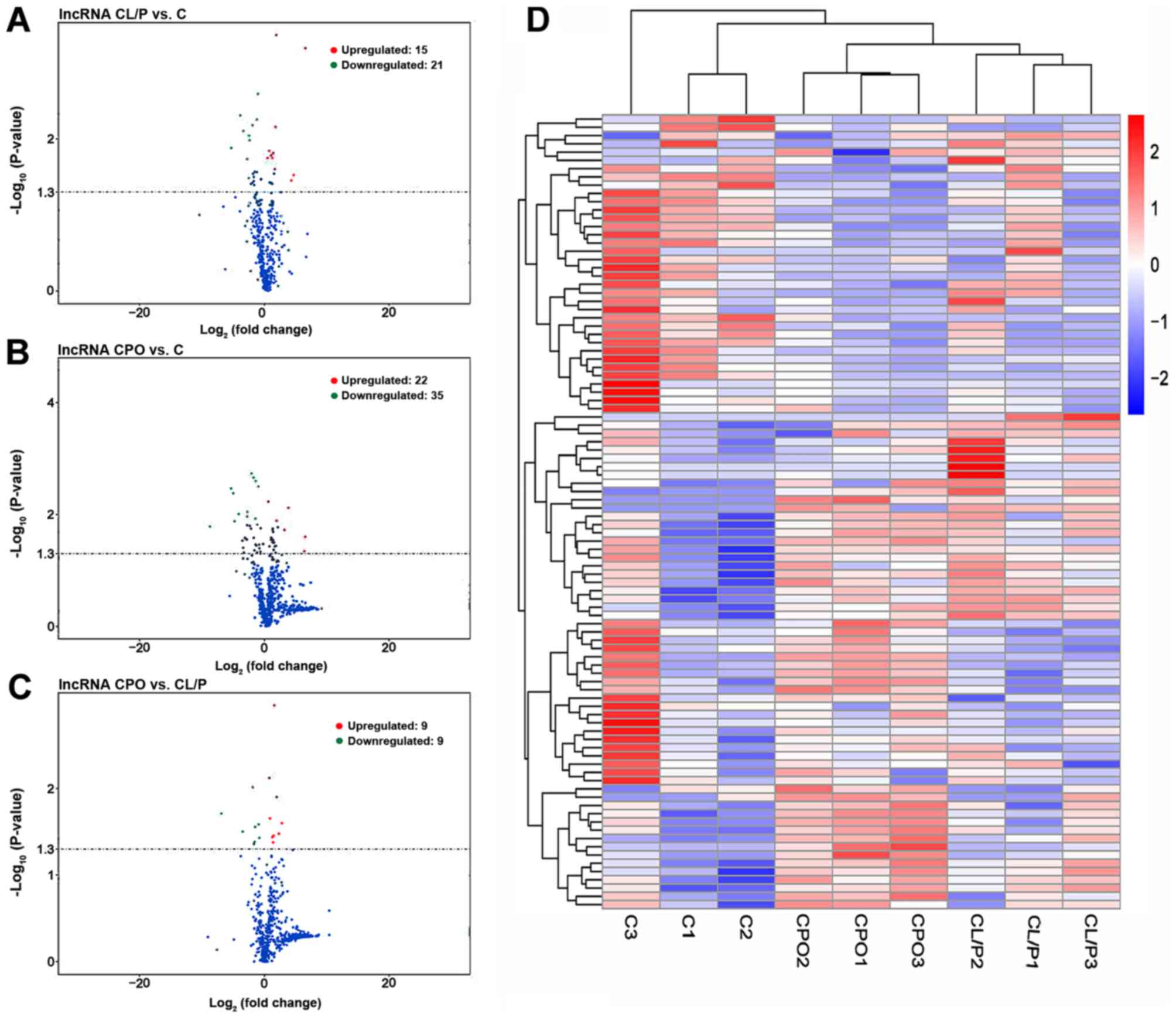
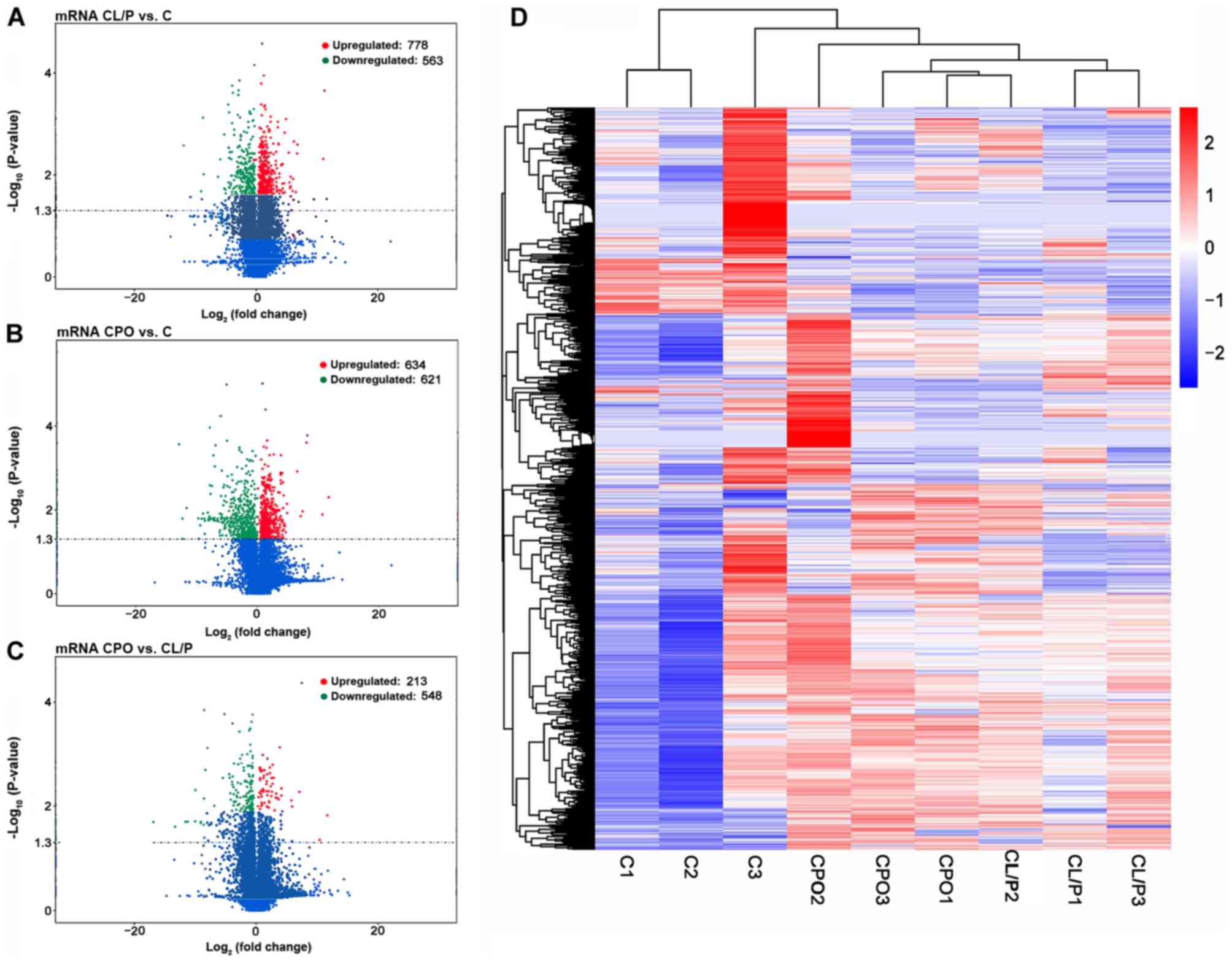
miRNAs and mRNAs. The distribution of DE miRNAs (Fig. 1A-C), DE lncRNAs (Fig. 2A-C) and DE mRNAs (Fig. 3A-C) was directly visualized using
volcano plots. The results were as follows: There were 36 DE
lncRNAs (15 upregulated and 21 downregulated), 1,341 DE mRNAs (778
upregulated and 563 downregulated), and 60 DE miRNAs (25
upregulated and 35 downregulated) in the CL/P group compared to the
control group. There were 57 DE lncRNAs (22 upregulated and 35
downregulated), 1,255 DE mRNAs (634 upregulated and 621
downregulated), and 162 DE miRNAs (65 upregulated and 97
downregulated) in the CPO group compared to the control group.
There were 18 DE lncRNAs (9 upregulated and 9 downregulated), 761
DE mRNAs (213 upregulated and 548 downregulated) and 19 DE miRNAs
(8 upregulated and 11 downregulated) in the CPO group compared to
the CL/P group. Since there were a number of DE genes obtained from
RNA-seq in the study, detailed information of the top 10 DE miRNAs,
lncRNAs and mRNAs in the CL/P group compared to the control group
and in the CPO group compared to the control group is presented in
Tables III–VIII. Hierarchical clustering analysis
was used to reveal the expression profiles of DE miRNAs (Fig. 1D), DE lncRNAs (Fig. 2D) and DE mRNAs (Fig. 3D) in the control, CL/P and CPO
groups.
| Table III.Detailed information of the top 10
differentially expressed microRNAs in the cleft lip with or without
cleft palate group compared to the control group. |
Table III.
Detailed information of the top 10
differentially expressed microRNAs in the cleft lip with or without
cleft palate group compared to the control group.
| microRNA | Log fold
change | P-value |
|---|
| hsa-let-7b-5p | 3.119 |
1.42×10−6 |
|
hsa-miR-3200-5p | 2.913 |
8.58×10−7 |
| hsa-miR-150-3p | 2.387 |
2.60×10−12 |
|
hsa-miR-3200-3p | 2.378 |
3.40×10−5 |
| hsa-miR-511-5p | 2.362 |
2.07×10−4 |
|
hsa-miR-376c-3p | −3.365 |
7.32×10−8 |
| hsa-miR-655-3p | −2.964 |
2.91×10−6 |
| hsa-miR-483-3p | −2.889 |
1.08×10−7 |
| hsa-miR-4467 | −2.836 |
2.51×10−6 |
| hsa-miR-561-5p | −2.680 |
1.29×10−5 |
| Table VIII.Detailed information of the top 10
differentially expressed mRNAs in the cleft palate only group
compared to the control group. |
Table VIII.
Detailed information of the top 10
differentially expressed mRNAs in the cleft palate only group
compared to the control group.
| Transcript_id | Gene name | Log fold
change | P-value |
|---|
|
ENST00000299204 | BAG5 | 11.721 | 0.0050 |
|
ENST00000376619 | HDAC6 | 10.693 | 0.0128 |
|
ENST00000439744 | HVCN1 | 8.190 | 0.0002 |
|
ENST00000473745 | IRF5 | 8.080 | 0.0002 |
|
ENST00000554215 | DCAF5 | 7.436 | 0.0108 |
|
ENST00000302779 | PXK | −12.868 | 0.0003 |
|
ENST00000564243 | EIF3C | −12.316 | 0.0161 |
|
ENST00000428575 | XRCC6 | −12.309 | 0.0491 |
|
ENST00000636026 | ZEB2 | −11.635 | 0.0090 |
|
ENST00000519501 | FAXDC2 | −9.719 | 0.0159 |
RT-qPCR validation of ncRNA and mRNA
expression
To validate the reliability of the sequencing
results and to provide evidence for further functional experiments
with the DE ncRNAs we identified, the expression levels of 6 genes,
including miR-483-3p, miR-92b-5p, BAG5, ZEB2, lncRNA RP11-731F5.2
and lncRNA XIST, were analyzed by RT-qPCR along with another 27
samples (10 CL/P, 7 CPO and 10 healthy controls). The expression
levels of each gene according to RT-qPCR and RNAseq are presented
in Fig. 4. With the exception of
ZEB2, the expression trends of these genes according to RT-qPCR
were almost the same as those obtained with RNAseq, indicating that
the sequencing results were reliable.
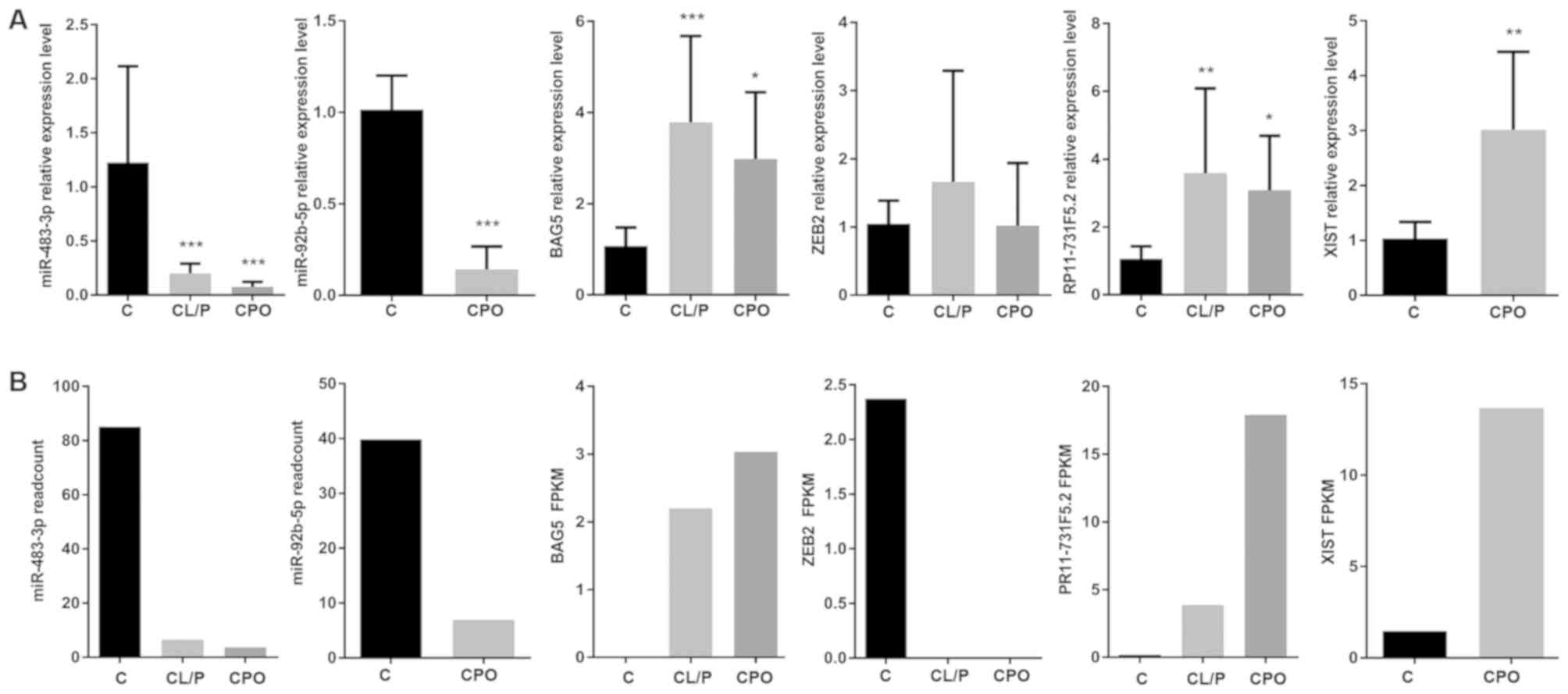 | Figure 4.(A) The RT-qPCR validations of DE
ncRNAs and mRNAs in the control, CL/P and CPO groups. The vertical
axis shows the relative expression level of each RNA measured by
RT-qPCR, *P<0.05, **P<0.01 and ***P<0.001, compared to the
control. (B) The sequencing results for corresponding mRNAs, miRNAs
and lncRNAs with qPCR verification. CL/P, cleft lip with or without
cleft palate; CPO, cleft palate only; C, healthy control group;
RT-qPCR, quantitative real-time polymerase chain reaction; FPKM,
fragments per kilobase million. |
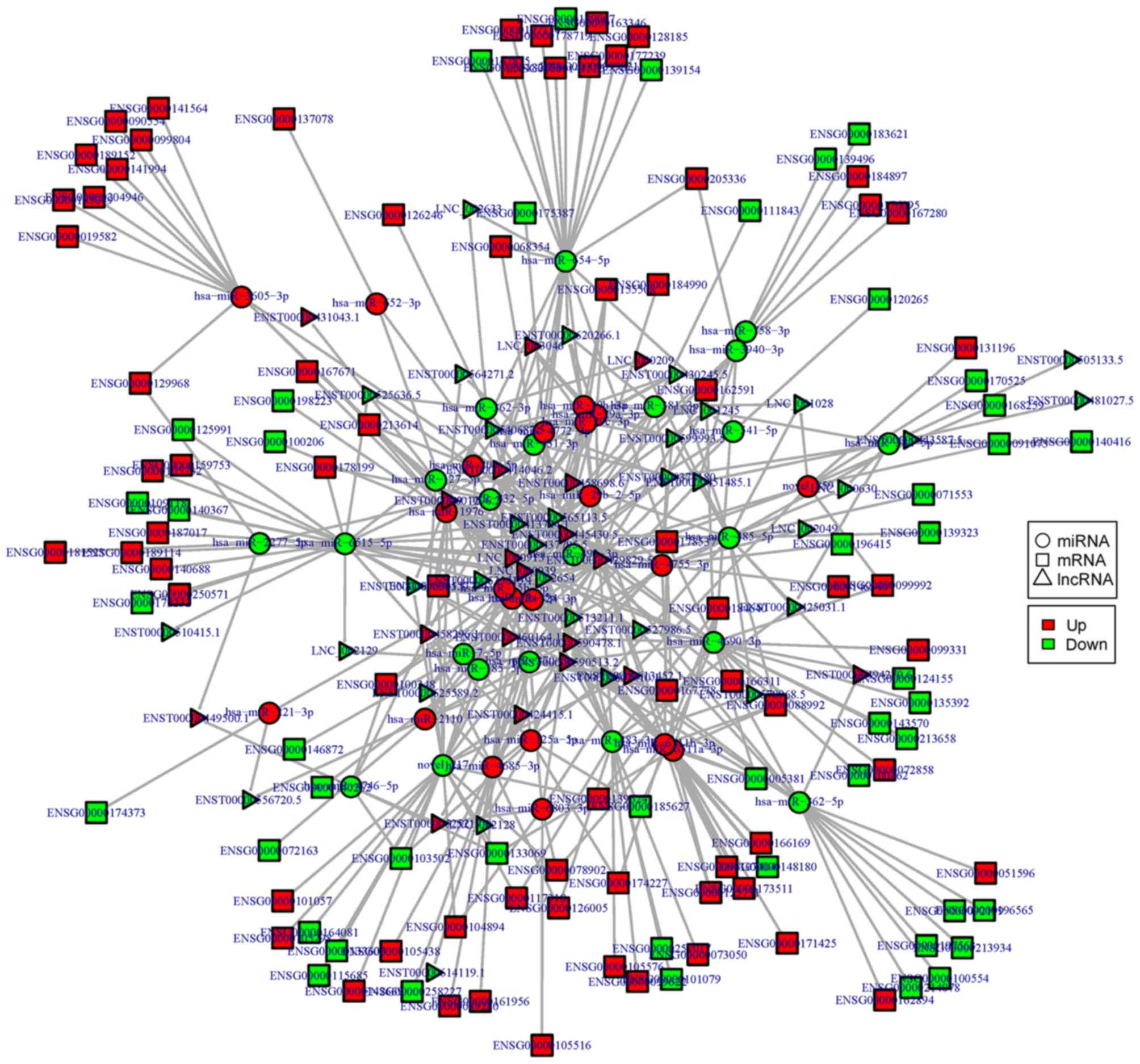
Analysis of miRNA-lncRNA-mRNA
regulatory networks
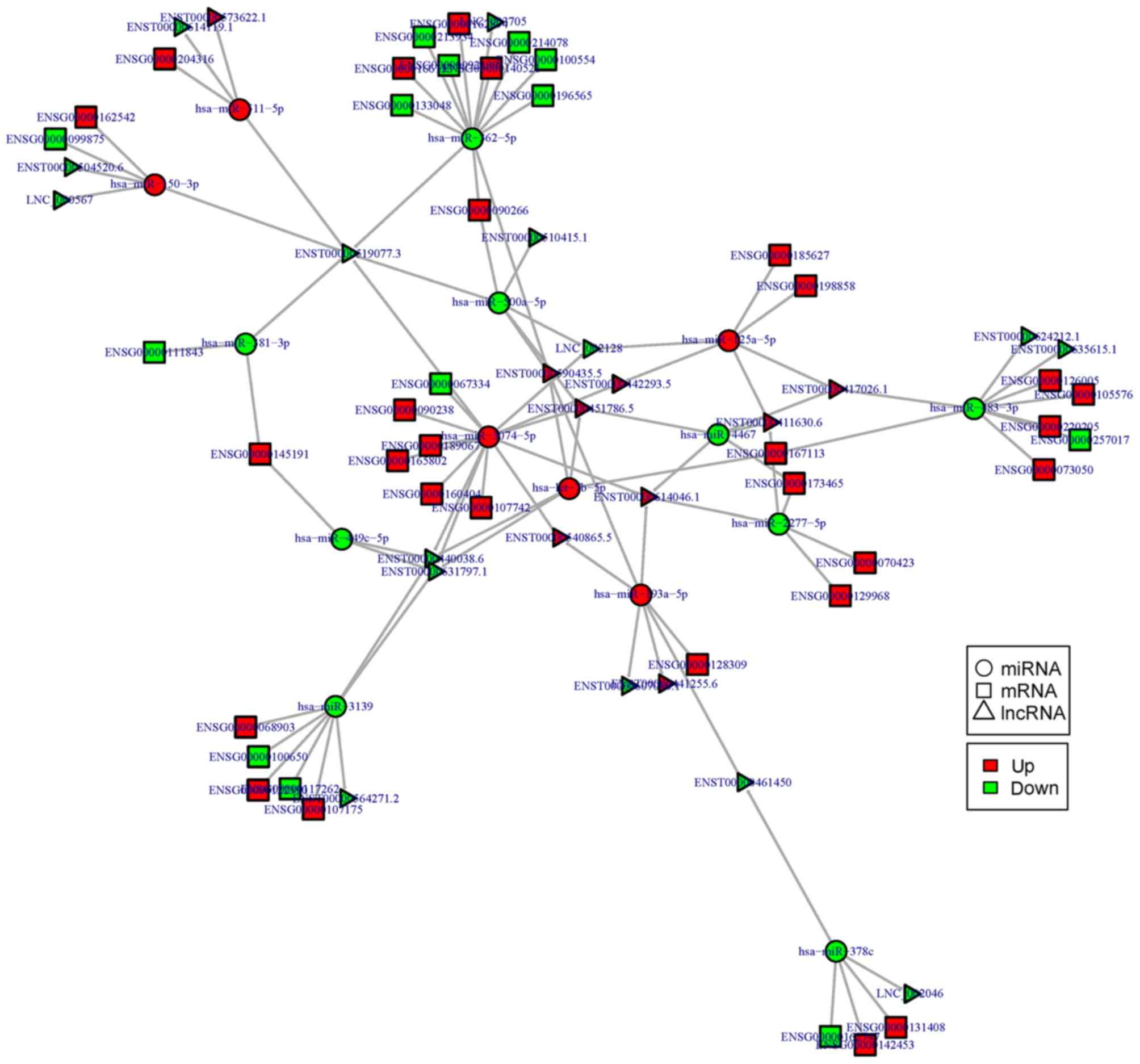
Based on the theory of ceRNA, miRNA-lncRNA-mRNA
regulatory networks were constructed and visualized using Cytoscape
software in the CL/P (Fig. 5) and
CPO groups (Fig. 6). Thus, at the
whole-transcriptome level, the possible regulatory mechanism
between ncRNAs and mRNAs was revealed in the CL/P and CPO groups.
As shown in the figures, crucial ncRNAs and mRNAs with more edges
may play important roles in the pathology of CPO and CL/P; these
ncRNAs and mRNAs include miR-483-3p, miR-4690-3p, miR-654-3p,
miR-6515-5p, lncRNA RP11-731F5.2, lncRNA XIST, lncRNA
RP11-591C20.9, RARA and SMPD1, as well as others. Through the
analysis of the regulatory networks, we found that there may be a
complex regulatory association between ncRNAs and mRNAs in the CL/P
and CPO groups. Some of these key nodes and regulatory pairs need
to be further validated in future studies.
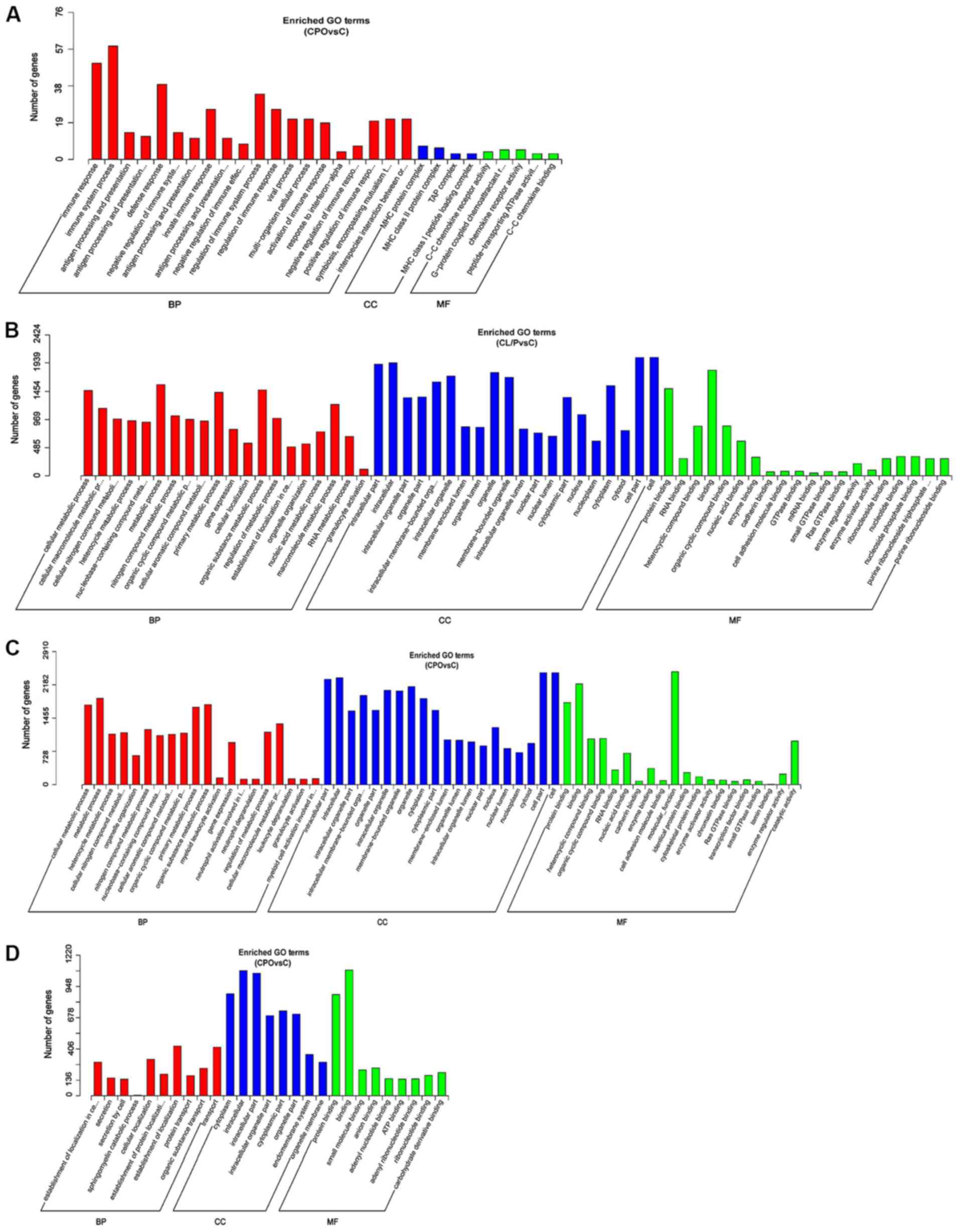
Functional prediction of ncRNAs
It has been reported that lncRNAs can regulate the
expression of target genes on multiple levels; however, the
specific underlying mechanism remain unclear (33–36).
In this study, the biological functions of lncRNAs were predicted
by colocalization and co-expression. GO enrichment analysis was
performed for the colocalization of target genes of DE lncRNAs
between the CPO and control groups (Fig. 7A), the co-expression of target
genes of DE lncRNAs between the CL/P and control groups (Fig. 7B), the co-expression target genes
of DE lncRNAs between the CPO and control groups (Fig. 7C), and the target genes of DE
miRNAs between the CPO and control groups (Fig. 7D). However, no significant
enrichment (P>0.05) was found in the GO analysis of the target
genes of DE miRNAs (Table SI) and
in the colocalization of target genes of DE lncRNAs (Table SII) between the CL/P and control
groups; thus, GO enrichment analysis figures could not be obtained.
The genes with significant enrichment for each GO term were
statistically analyzed and are displayed as a histogram. The
results of the analysis indicated that the functions of
dysregulated ncRNAs in the CPO group compared to the control group
were mainly associated with ‘immune response’, ‘immune system
process’, ‘antigen processing and presentation’, ‘MHC protein
complex’ (Fig. 7A),
‘intracellular’, ‘protein binding’, ‘cellular metabolic process’,
‘metabolic process’ (Fig. 7C),
‘cytoplasm’, ‘protein transport’, ‘secretion by cell’, ‘cellular
localization’ (Fig. 7D), as well
as others. Additionally, the functions of dysregulated ncRNAs in
the CL/P group compared to the control group were mainly associated
with ‘intracellular part’, ‘cytoplasmic part’, ‘protein binding’,
‘cellular metabolic process’, ‘metabolic process’, ‘gene
expression’, ‘cellular localization’, ‘organic substance metabolic
process’, ‘regulation of metabolic process’, and others (Fig. 7B).
In this study, KEGG pathway analysis was performed
for the target genes of DE lncRNAs and the target genes of DE
miRNAs between the CL/P and control group and between the CPO and
control group. The KEGG pathway enrichment was analyzed with an
enrichment criterion of P-value <0.05. However, only the
analysis of the colocalization of target genes of DE lncRNAs
between the CPO group and control group presented significant
enrichment (Table IX). The rich
factor refers to the ratio of the number of DE genes in the pathway
to the total number of genes in the annotated gene list of the
pathway. When the rich factor is greater and the P-value is closer
to zero, the enrichment is more significant. The results revealed
that the most significantly involved pathways in the CPO group were
‘antigen processing and presentation’, ‘phagosome’, ‘allograft
rejection’, ‘type I diabetes mellitus’, ‘intestinal immune network
for IgA production’, ‘viral myocarditis’, ‘rheumatoid arthritis’
and ‘ABC transporters’.
| Table IX.Kyoto Encyclopedia of Genes and
Genomes pathway analysis of colocalization target genes of
differentially expressed long noncoding RNAs in the cleft palate
only group compared to the control group. |
Table IX.
Kyoto Encyclopedia of Genes and
Genomes pathway analysis of colocalization target genes of
differentially expressed long noncoding RNAs in the cleft palate
only group compared to the control group.
| Pathway_term | Rich_factor | P-value | Gene_number |
|---|
| Antigen processing
and presentation | 0.101 | 0.0019 | 8 |
| Phagosome | 0.065 | 0.0032 | 10 |
| Allograft
rejection | 0.128 | 0.0097 | 5 |
| Graft-versus-host
disease | 0.116 | 0.0107 | 5 |
| Type I diabetes
mellitus | 0.111 | 0.0107 | 5 |
| Intestinal immune
network for IgA production | 0.102 | 0.0127 | 5 |
| Autoimmune thyroid
disease | 0.093 | 0.0160 | 5 |
| Asthma | 0.125 | 0.0160 | 4 |
| Staphylococcus
aureus infection | 0.088 | 0.0160 | 5 |
| Viral
myocarditis | 0.083 | 0.0171 | 5 |
| Rheumatoid
arthritis | 0.066 | 0.0171 | 6 |
| ABC
transporters | 0.091 | 0.0310 | 4 |
| Leishmaniasis | 0.068 | 0.0310 | 5 |
| Herpes simplex
infection | 0.043 | 0.0310 | 8 |
Discussion
This study first identified transcriptome profiles,
including mRNAs, lncRNAs and miRNAs, from the peripheral blood of
patients with CL/P and CPO using a high-throughput sequencing
method. Subsequently, the potential functions of the DE ncRNAs were
predicted using bioinformatics tools and databases, and based on
the theory of ceRNAs, we constructed lncRNA-miRNA-mRNA regulatory
networks to identify the possible regulatory mechanisms between
ncRNAs and mRNAs in NSOC.
Peripheral blood is the only tissue in the body that
has contact with all organs. It carries RNA, DNA, vesicles and
other biological substances that can aid clinicians in the
diagnosis and identification of a variety of diseases, and its use
is also more convenient and less invasive than other tissues
(37,38). Although the pathogenesis of NSOC is
related to a number of factors during embryonic development, it has
been found that a number of these differences persist after birth
(39). In this study, the
peripheral blood of patients with CL/P and CPO was used for
high-throughput sequencing in an attempt to find DE ncRNAs compared
to healthy individuals and to explore the possible regulatory
mechanisms, although there were some limitations and differences in
using peripheral blood instead of tissue samples due to ethical
limitations. This study lays the foundation for future research,
and the key DE ncRNAs and regulatory associations in NSOC need to
be investigated in future studies.
Due to ethical limitations, there have been no
studies to date (at least to the best of our knowledge) on the
expression profiles of ncRNAs of lip or palate tissues of humans.
In a previous study, a research group used microarrays to screen
the expression profiles of NSOC, and the results revealed that
there were 305, 221 and 132 DE miRNAs in the plasma of patients
with CLO, CL/P and CPO, respectively (40,41).
In those studies, miRNA-483-3p was significantly downregulated in
both the CL/P and CPO groups, exhibiting a similar trend as that
observed in this study. Although there have been no reports to date
on miR-483-3p in NSOC (to the best of our knowledge), matrix
metalloproteinase (MMP)9, the target gene of miR-483-3p, was found
to be related to palate development. MMPs comprise a group of
likely candidate proteins involved in the etiology of CL/P due to
their role in modeling craniofacial tissues. The temporospatial
expression of MMPs 2, 3, 7, 9 and 13 has been observed during
murine palatal fusion (42–44).
MMP9 is known for its ability to degrade type IV collagen, a main
component of the extracellular matrix (ECM), and to facilitate cell
migration (45). In a recent
study, researchers analyzed miRNA expression in cultured palate
fibroblasts from patients with CL/P and CPO using high-throughput
sequencing methods. The results revealed that there were 9 DE
miRNAs in the CPO group, including miR-93-5p, miR-18a-5p,
miR-92a-3p, miR-29c-5p, miR-549a, miR-3182, miR-181a-5p, miR-451a
and miR-92b-5p. Patients with CL/P had only one DE miRNA
(miR-505-3p) compared to the control group (46). In this study, the expression of
miRNA-92b-5p was found to be also significantly downregulated in
the CPO group compared to the control group. At present, miR-92b-5p
has been studied mainly in tumors, cardiovascular and
cerebrovascular diseases (47–50);
however, its mechanism of action has rarely been reported. Based on
the analysis of the results of this study with a target gene
prediction software program, it should be noted that the expression
of lncRNA XIST, which is the target gene of miR-92b-5p, was
significantly upregulated in the CPO group. Although the regulatory
association between lncRNA XIST and miR-92b-5p in NSOC has not yet
been reported, at leas to the best of our knowledge, other studies
have found that lncRNA XIST and miR-92b directly interact with and
repress each other, and lncRNA XIST inhibits hepatocellular
carcinoma cell proliferation and metastasis by targeting miR-92b
(50). Since the same regulatory
relationship can be involved in multiple pathways, we speculated
whether the lncRNA XIST-miR-92b relationship functions in the
pathogenesis of CPO. These genes need to be verified in future
experiments. Additionally, we found that some of the DE ncRNAs (or
their target genes) and some DE mRNAs, including miR-96-5p,
miR-92b-5p, miR-200b-3p, CD44, ZEB2, runt related transcription
factor 3 (RUNX3), SMAD2, lncRNA RP11-731F5.2, and others, were
reported in previous studies on NSOC or in related experiments. The
present study found that T-Box 1 (TBX1) regulated the proliferation
of dental progenitor cells and craniofacial development through
miR-96-5p and paired-like homeodomain transcription factor 2
(PITX2). Additionally, TBX1, as a candidate gene of NSOC, can
maintain the normal growth and development of palatal shelves,
mediated through the regulation of genes involved in muscle cell
differentiation, nervous system development and biomineral tissue
development (51). The present
study hypothesized that the dysregulated expression of miR-96-5p
may affect the development of palatal shelves by inhibiting the
expression of its target gene. CD44 is an integral cell membrane
glycoprotein that acts as a receptor for ECM molecules involved in
cell-cell interactions and cell adhesion and migration, and plays a
postulated role in the proliferation of mesenchymal cells (52). Park et al (53) also found that CD44, as a candidate
gene for CL/P, exhibited significant evidence of linkage in the
presence of disequilibrium in 58 case-parent trios from Maryland
and it was also found to be expressed in the developing palate and
lip. RUNX3/PEBP2αC has multiple functions and was first found to be
associated with the pathogenesis and progression of human gastric
cancer as a tumor suppressor. The expression of RUNX3 was also
observed in mouse embryonic palate and palatal shelf epithelium,
which has been reported to express TGFβ3, bone morphogenetic
protein (BMP)2 and BMP4 (54).
SMAD2, a crucial regulator during palatal fusion, has been reported
in a number of studies on NSOC. SMAD2 is necessary for the
induction of Snail in TGFβ-mediated epithelial-mesenchymal
transition (EMT) at the cellular level (55,56),
and the cleft palates of TGFβ3-knockout mice were shown to be
‘rescued’ by the overexpression of SMAD2 in medial edge epithelia
(MEE) cells (57). Another study
found that miR-200b was expressed in the development of palatal
shelves in mice and played a crucial role in regulating SMAD2 in
apoptosis during palatogenesis by acting as a direct negative
regulator (58). In addition, Shin
et al (59) found that the
ZEB family may be involved in cell migration during palate
development and that excessive levels of miR-200b can lead to
non-fused medial edge epithelium seam (MES) through the inhibition
of ZEB1 and ZEB2 during palatogenesis. In this study, the
expression of miR-200b-3p was upregulated, that of SMAD2 was
downregulated and that of ZEB2 was also downregulated in the CPO
group. It was thus hypothesized that miR-200b-3p may interact with
ZEB2 or SMAD2, which play significant roles in the development of
the palate.
Based on previous studies on the expression profiles
of miRNAs in NSOC, this study adopted the high-throughput
sequencing of peripheral blood of patients with CL/P and CPO to
identify DE ncRNAs and mRNAs. The results of this study differ from
those of previous studies, although there are still a few overlaps.
It was hypothesized that this was due to the following reasons:
First, we used different sample sources, resulting in different
expression profiles. Second, the high-throughput sequencing method
is more full-scale than the microarray method, which is more
responsive to low-abundance transcripts. Finally, the
false-positive results from high-throughput sequencing technology
and analysis methods vary with each individual due to the different
choices of software, databases and thresholds (60–62).
According to the fold changes, P-values and previous
studies on NSOC, we selected 6 genes for further validation by
RT-qPCR method with another 27 cases (10 controls, 10 CL/P and 7
CPO). The results revealed that the expression levels of 5 of these
genes differed significantly and were consistent with the trends
displayed by the sequencing results, apart from ZEB2, illustrating
that the sequencing results are reliable and can provide a
reference for further study. In addition, it was hypothesized that
the following reasons account for the different trends in ZEB2
expression in the RT-qPCR and sequencing results. First, the
samples were derived from human peripheral blood, which carries a
large amount of genetic information and may lead to large
differences between individuals. Second, the number of cases for
sequencing was small, and false-positive results obtained with
next-generation sequencing technology cannot be ruled out.
Therefore, further in vitro or in vivo experiments
are warranted in order to validate the present findings.
The primary and secondary palates have different
embryonic sources; thus, orofacial clefts are generally divided
into CL/P and CPO. The results of this study demonstrated that
there were 19, 18 and 761 DE miRNAs, lncRNAs and mRNAs,
respectively, in the CPO group compared to the CL/P group,
reflecting the separate etiologies of these 2 groups. Among the
identified miRNAs, Warner et al (63) previously found that miR-199a-3p was
expressed in the murine medial nasal and maxillary processes, which
may be involved in the development of the lip and palate.
Additionally, RUNX1 and ZEB1, which are the target genes of
miR-199a-3p, have been reported to be associated with cleft lip and
palate. Runx1 previously has been shown to be important for the
fusion of the primary and secondary palates (64). ZEB1 has been shown to regulate EMT
in a number of systems, including the fusion of the secondary
palate; however, there are no studies available to date examining
its potential role in the fusion of the facial prominences
(65). Thus, the potential role of
miR-199a-3p in CL/P or CPO warrants further investigation in
vivo or in vitro in the future. It worth noting that
there were 5 overlapping genes between the CL/P and control groups
and between the CPO and control groups, including miR-7b-5p,
miR-3200-5p, miR-3200-3p, miR-483-3p and lncRNA RP11-731F5.2. Yu
et al (8) conducted a NSCLP
GWAS using case-control samples from China and replicated 14 novel
and 12 reported NSCLP risk loci in a total of 23,463 samples from
sub-phenotypes of NSOFC. They found that 1 novel and 2 reported
NSCLP risk loci exhibited significant associations with CPO
(8). This result indicated that
the 2 sub-groups appeared to share distinct etiologies, although
the overlapping risk factors between the 2 groups cannot be ruled
out. Therefore, the overlapping genes need to be further analyzed,
for example, miR-483-3p and lncRNA RP11-731F5.2, mentioned
above.
Salmena proposed the ceRNA hypothesis, which
suggests that RNA molecules with the same miRNA response elements
(MREs), including mRNAs, lncRNAs, pseudogenes and circRNAs, can
regulate the expression abundance and translation activity of
target genes by competitively binding to the same miRNA (66). The ceRNA causes protein-coding
genes and ncRNAs to form complex and elaborate regulatory networks
within the transcriptome and has been confirmed in diverse
biological processes, including the differentiation of embryonic
stem cells, the growth of the midbrain, cancer metastasis and
others (67,68). In this study, based on the ceRNA
hypothesis, lncRNA-miRNA-mRNA regulatory networks were constructed
to explore the potential regulatory associations between ncRNAs and
mRNAs in NSOC. Crucial ncRNAs and mRNAs with more edges may play
important roles in the pathology of CPO and CL/P. miR-483-3p and
lncRNA XIST, mentioned above, were also the key nodes of the
network in the CPO group. Additionally, lncRNA RP11-731F5.2 and its
target gene, RARA, were found to have more edges in the network of
the CPO group. Previous studies have reported that retinoic acid
receptor α (RARA) may be a candidate gene for NSOC, and retinoic
acid also may bind to RARA to inhibit apoptosis, resulting in the
failure of palatal shelves fusion, whereas others have proposed
that retinoic acid disrupts elevation or retards the growth of
palatal shelves (69–71).
In this study, GO and KEGG pathway enrichment
analysis was used to evaluate the biological functions and pathways
of the target genes of DE ncRNAs. Through GO enrichment analysis of
the target genes, we found that the function of DE ncRNAs in the
CPO and CL/P groups was mainly related to cellular behaviors,
including ‘intracellular part’, ‘cell adhesion molecule binding’,
‘cytoplasmic part’, ‘cellular metabolic process’, ‘cellular
localization’ and others. These cellular functions are coordinated
by numerous genes encoding various growth factors, signaling
mediators, transcriptional factors, cytokines, and extracellular
matrix proteins (72). As is
widely known, various cellular and molecular events are related to
palatogenesis, including apoptosis, EMT, cell proliferation and
cell migration, and any perturbation of these programs may cause
NSOC. Therefore, it is crucial to explore genes and molecules that
regulate the above-mentioned biofunctions in order to understand
cellular behavior during palatogenesis.
The KEGG enrichment analysis revealed that the most
significantly involved pathways in the CPO group were ‘antigen
processing and presentation’, ‘phagosome’, ‘allograft rejection’,
‘type I diabetes mellitus’, ‘intestinal immune network for IgA
production’, ‘viral myocarditis’, ‘rheumatoid arthritis’, and ‘ABC
transporters’. The etiology of NSOC is complex and related to
multiple factors, suggesting that its development may involve other
physiological systems and biological processes. According to a
long-term follow-up survey, patients with cleft lip and palate have
a higher risk of cancer, cardiovascular disease, central nervous
system disease, and even suicide later in life compared to healthy
individuals, although the underlying mechanisms have not been
studied in depth (73). It was
hypothesized that patients with cleft lip and palate may carry
variations that do not cause other abnormalities at birth, but may
increase the susceptibility to other diseases later on in life.
Based on the data from the analysis of this study, an association
between the risk of NSOC and the target genes enriched in other
functions and signaling pathways cannot be ruled out.
Next-generation RNA sequencing was performed to
identify the transcriptome profiles, including mRNAs, lncRNAs and
miRNAs, in patients with CL/P and CPO. Subsequently, GO and KEGG
enrichment analysis was performed to predict the functions of the
DE ncRNAs; however, there are still some limitations. First,
high-throughput sequencing technology itself has errors, and
false-positive results can also be obtained from the analysis
methods and prediction software. In addition, the exact mechanisms
of action of the ncRNAs should be verified in vitro or in
vivo. Second, when data was analyzed, WGCNA analysis was
tentatively used to determine the correlation of lncRNA with mRNA.
WGCNA analysis is a method based on a correlation coefficient,
which is suitable for multi-sample analysis. According to previous
studies, it is recommended that the sample size should be no less
than 25, and the more samples there are, the more reliable the
results will be (74,75). In this study, 9 samples were used
for RNA-Seq (3 controls, 3 CL/P and 3 CPO). Given the
above-mentioned considerations, WGCNA analysis was not performed in
this study. Instead, a threshold of colocalization of 100 kb
upstream and downstream of the lncRNAs was used. Pearson
correlation coefficients with absolute values >0.95 were used to
concurrently analyze the correlation between lncRNAs and mRNAs.
However, if samples are sufficient in subsequent studies, the WGCNA
analysis will be used to obtain more thorough results. Finally, the
use of peripheral blood as a sample for sequencing is different
than using tissue samples due to ethical limitations. Thus, this
study provides only initial results regarding the pathogenesis of
NSOC in epigenetics and lays the foundation for future verification
using tissues. Additional studies may lead to the identification of
novel diagnostic markers and therapeutic targets for NSOC.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by the Innovation
Research Project of Harbin Medical University (grant no.
YJSCX2017-51HYD), and the National Key Research and Development
Program of China (grant no. 2016YFC1000504).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
XJ, SF and WS were responsible for the design of the
study. XW, QZ and YL diagnosed the patients with NSOC and collected
the patients' peripheral blood. HS and WZ analyzed the data. YG
performed the experiments and drafted the manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of Harbin Medical University, Heilongjiang, China and
informed consent was obtained from the patients' families before
sampling.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
NSOC
|
non-syndromic orofacial clefts
|
|
CL/P
|
cleft lip with or without cleft
palate
|
|
CPO
|
cleft palate only
|
|
lncRNA
|
long non-coding RNA
|
|
miRNA
|
microRNA
|
|
ncRNA
|
non-coding RNA
|
|
ceRNA
|
competing endogenous RNA
|
|
RNA-seq
|
next-generation RNA sequencing
|
|
GWAS
|
genome-wide association study
|
|
RT-qPCR
|
reverse transcription-quantitative
polymerase chain reaction
|
|
GO
|
gene ontology
|
|
KEGG
|
Kyoto Encyclopedia of Genes and
Genomes
|
|
BP
|
biological process
|
|
CC
|
cellular component
|
|
MF
|
molecular function
|
|
MRE
|
miRNA response elements
|
|
FPKM
|
fragments per kilobase million
|
|
DE
|
differentially expressed
|
References
|
1
|
Mossey PA, Little J, Munger RG, Dixon MJ
and Shaw WC: Cleft lip and palate. Lancet. 374:1773–1785. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Leslie EJ and Marizita ML: Genetics of
cleft lip and cleft palate. Am J Med Genet C Semin Med Genet 163C.
246–258. 2013. View Article : Google Scholar
|
|
3
|
Ladd-Acosta C and Beaty TH: Integrating
RNA expression identifies candidate gene for orofacial clefts. J
Dent Res. 97:31–32. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chiquet BT, Hashmi SS, Henry R, Burt A,
Mulliken JB, Stal S, Bray M, Blanton SH and Hecht JT: Genomic
screening identifies novel linkages and provides further evidence
for a role of MYH9 in nonsyndromic cleft lip and palate. Eur J Hum
Genet. 17:195–204. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chiquet BT, Yuan Q, Swindell EC, Maili L,
Plant R, Dyke J, Boyer R, Teichgraeber JF, Greives MR, Mulliken JB,
et al: Knockdown of Crispld2 in zebrafish identifies a novel
network for nonsyndromic cleft lip with or without cleft palate
candidate genes. Eur J Hum Genet. 26:1441–1450. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ludwig KU, Mangold E, Herms S, Nowak S,
Reutter H, Paul A, Becker J, Herberz R, AlChawa T, Nasser E, et al:
Genome-wide meta-analyses of nonsyndromic cleft lip with or without
cleft palate identify six new risk loci. Nat Genet. 44:968–971.
2012. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Beaty TH, Murray JC, Marazita ML, Munger
RG, Ruczinski I, Hetmanski JB, Liang KY, Wu T, Murray T, Fallin, et
al: A genome-wide association study of cleft lip with and without
cleft palate identifies risk variants near MAFB and ABCA4. Nat
Genet. 42:525–529. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yu Y, Zuo X, He M, Gao J, Fu Y, Qin C,
Meng L, Wang W, Song Y, Cheng Y, et al: Genome-wide analyses of
non-syndromic cleft lip with palate identify 14 novel loci and
genetic heterogeneity. Nat Commun. 8:143642017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Schoen C, Aschrafi A, Thonissen M,
Poelmans G, Von den Hoff JW and Carels CEL: MicroRNAs in
palatogenesis and cleft palate. Front Physiol. 8:1652017.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Moreno-Moya JM, Vilella F and Simón C:
MicroRNA: Key gene expression regulators. Fertil Steril.
101:1516–1523. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhou J, Xiong Q, Chen H, Yang C and Fan Y:
Identification of the spinal expression profile of Non-coding RNAs
involved in neuropathic pain following spared nerve injury by
sequence analysis. Front Mol Neurosci. 10:912017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Huang YK and Yu JC: Circulating microRNAs
and long non-coding RNAs in gastric cancer diagnosis: An update and
review. World J Gastroenterol. 21:9863–9886. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhao B, Lu M, Wang D, Li H and He X:
Genome-wide identification of long noncoding RNAs in human
intervertebral disc degeneration by RNA sequencing. Biomed Res Int.
2016:36848752016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mukhopadhyay P, Brock G, Pihur V, Webb C,
Pisano MM and Greene RM: Developmental microRNA expression
profiling of murine embryonic orofacial tissue. Birth Defects Res A
Clin Mol Teratol. 88:511–534. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ding HL, Hooper JE, Batzel P, Eames BF,
Postlethwait JH, Artinger KB and Clouthier DE: MicroRNA profiling
during craniofacial development: Potential roles for Mir23b and
Mir133b. Front Physiol. 7:2812016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dey BK, Mueller AC and Dutta A: Long
non-coding RNAs as emerging regulators of differentiation,
development, and disease. Transcription. 5:e9440142014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ozturk F, Li Y, Zhu X, Guda C and Naπwshad
A: Systematic analysis of palatal transcriptome to identify cleft
palate genes within TGFβ3-knockout mice alleles: RNA-Seq analysis
of TGFβ3 Mice. BMC Genomics. 14:1132013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gao L, Yin J and Wu W: Long non-coding RNA
H19-mediated mouse cleft palate induced by
2,3,7,8-tetrachlorodibenzo- p-dioxin. Exp Ther Med. 11:2355–2360.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gao L, Liu Y, Wen Y and Wu W: LncRNA
H19-mediated mouse cleft palate induced by all-trans retinoic acid.
Hum Exp Toxicol. 36:395–401. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pertea M, Kim D, Pertea GM, Leek JT and
Salzberg SL: Transcript-level expression analysis of RNA-seq
experiments with HISAT, StringTie and Ballgown. Nat Protoc.
11:1650–1667. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pertea M, Pertea GM, Antonescu CM, Chang
TC, Mendell JT and Salzberg SL: StringTie enables improved
reconstruction of a transcriptome from RNA-seq reads. Nat
Biotechnol. 33:290–295. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sun L, Luo H, Bu D, Zhao G, Yu K, Zhang C,
Liu Y, Chen R and Zhao Y: Utilizing sequence intrinsic composition
to classify protein-coding and long non-coding transcripts. Nucleic
Acids Res. 41:e1662013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kong L, Zhang Y, Ye ZQ, Liu XQ, Zhao SQ,
Wei L and Gao G: CPC: Assess the protein-coding potential of
transcripts using sequence features and support vector machine.
Nucleic Acids Res 35 (Web Server Issue). W345–W349. 2007.
View Article : Google Scholar
|
|
24
|
Mistry J, Bateman A and Finn RD:
Predicting active site residue annotations in the Pfam database.
BMC Bioinformatics. 8:2982007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lin MF, Jungreis I and Kellis M: PhyloCSF:
A comparative genomics method to distinguish protein coding and
non-coding regions. Bioinformatics. 27:i275–i282. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Langmead B, Trapnell C, Pop M and Salzberg
SL: Ultrafast and memory-efficient alignment of short DNA sequences
to the human genome. Genome Biol. 10:R252009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wen M, Shen Y, Shi S and Tang T: miREvo:
An integrative microRNA evolutionary analysis platform for
next-generation sequencing experiments. BMC Bioinformatics.
13:1402012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Friedländer MR, Mackowiak SD, Li N, Chen W
and Rajewsky N: miRDeep2 accurately identifies known and hundreds
of novel microRNA genes in seven animal clades. Nucleic Acids Res.
40:37–52. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Love MI, Huber W and Anders S: Moderated
estimation of fold change and dispersion for RNA-seq data with
DESeq2. Genome Biol. 15:5502014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Young MD, Wakefield MJ, Smyth GK and
Oshlack A: Gene ontology analysis for RNA-seq: Accounting for
selection bias. Genome Biol. 11:R142010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Mao X, Cai T, Olyarchuk JG and Wei L:
Automated genome annotation and pathway identification using the
KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics.
21:3787–3793. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ponting CP, OliPver PL and Reik W:
Evolution and functions of long noncoding RNAs. Cell. 136:629–641.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Nagano T and Fraser P: No-nonsense
functions for long noncoding RNAs. Cell. 145:178–181. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wilusz JE, Sunwoo H and Spector DL: Long
noncoding RNAs: Functional surprises from the RNA world. Genes Dev.
23:1494–1504. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kopp F and Mendell JT: Functional
classification and experimental dissection of long noncoding RNAs.
Cell. 172:393–407. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Huan T, Joehanes R, Schurmann C, Schramm
K, Pilling LC, Peters MJ, Mägi R, DeMeo D, O'Connor GT, Ferrucci L,
et al: A whole-blood transcriptome meta-analysis identifies gene
expression signatures of cigarette smoking. Hum Mol Genet.
25:4611–4623. 2016.PubMed/NCBI
|
|
38
|
Hardy JJ, Mooney SR, Pearson AN, McGuire
D, Correa DJ, Simon RP and Meller R: Assessing the accuracy of
blood RNA profiles to identify patients with post-concussion
syndrome: A pilot study in a military patient population. PLoS One.
12:e01831132017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhang J, Zhou S, Zhang Q, Feng S, Chen Y,
Zheng H, Wang X, Zhao W, Zhang T, Zhou Y, et al: Proteomic analysis
of RBP4/vitamin A in children with cleft lip and/or palate. J Dent
Res. 93:547–552. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Li J, Zou J, Li Q, Chen L, Gao Y, Yan H,
Zhou B and Li J: Assessment of differentially expressed plasma
microRNAs in nonsyndromic cleft palate and nonsyndromic cleft lip
with cleft palate. Oncotarget. 7:86266–86279. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zou J, Li J, Li J, Ji C, Li Q and Guo X:
Expression profile of plasma microRNAs in nonsyndromic cleft lip
and their clinical significance as biomarkers. Biomed Pharmacother.
82:459–466. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Iamaroon A, Wallon UM, Overall CM and
Diewert VM: Expression of 72-kDa gelatinase (matrix
metalloproteinase-2) in the developing mouse craniofacial complex.
Arch Oral Biol. 41:1109–1119. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Morris-Wiman J, Du Y and Brinkley L:
Occurrence and temporal variation in matrix metalloproteinases and
their inhibitors during murine secondary palatal morphogenesis. J
Craniofac Genet Dev Biol. 19:201–212. 1999.PubMed/NCBI
|
|
44
|
Morris-Wiman J, Burch H and Basco E:
Temporospatial distribution of matrix metalloproteinase and tissue
inhibitors of matrix metalloproteinases during murine secondary
palate morphogenesi. Anat Embryol (Berl). 202:129–141. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Letra A, da Silva RA, Menezes R, de Souza
AP, de Almeida AL, Sogayar MC and Granjeiro JM: Studies with MMP9
gene promoter polymorphism and nonsyndromic cleft lip and palate.
Am J Med Genet A 143A. 89–91. 2007. View Article : Google Scholar
|
|
46
|
Schoen C, Glennon JC, Abghari S, Bloemen
M, Aschrafi A, Carels CEL and Von den Hoff JW: Differential
microRNA expression in cultured palatal fibroblasts from infants
with cleft palate and controls. Eur J Orthod. 40:90–96. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wu T, Chen Y, Du Y, Tao J, Zhou Z and Yang
Z: Serum exosomal MiR-92b-5p as a potential biomarker for acute
heart failure caused by dilated cardiomyopathy. Cell Physiol
Biochem. 46:1939–1950. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Wu T, Chen Y, Du Y, Tao J, Li W, Zhou Z
and Yang Z: Circulating exosomal miR-92b-5p is a promising
diagnostic biomarker of heart failure with reduced ejection
fraction patients hospitalized for acute heart failure. J Thorac
Dis. 10:6211–6220. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zhao C, Zhao F, Feng H, Xu S and Qin G:
MicroRNA-92b inhibits epithelial-mesenchymal transition-induced
migration and invasion by targeting Smad3 in nasopharyngeal cancer.
Oncotarget. 8:91603–91613. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zhuang LK, Yang YT, Ma X, Han B, Wang ZS,
Zhao QY, Wu LQ and Qu ZQ: MicroRNA-92b promotes hepatocellular
carcinoma progression by targeting Smad7 and is mediated by long
non-coding RNA XIST. Cell Death Dis. 7:e22032016. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Gao S, Moreno M, Eliason S, Cao H, Li X,
Yu W, Bidlack FB, Margolis HC, Baldini A and Amendt BA: TBX1
protein interactions and microRNA-96-5p regulation controls cell
proliferation during craniofacial and dental development:
Implications for 22q11.2 deletion syndrome. Hum Mol Genet.
24:2330–2348. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Baroni T, Bellucci C, Lilli C, Pezzetti F,
Carinci F, Lumare E, Palmieri A, Stabellini G and Bodo M: Human
cleft lip and palate fibroblasts and normal nicotine-treated
fibroblasts show altered in vitro expressions of genes related to
molecular signaling pathways and extracellular matrix metabolism. J
Cell Physiol. 222:748–756. 2010.PubMed/NCBI
|
|
53
|
Park JW, Cai J, McIntosh I, Jabs EW,
Fallin MD, Ingersoll R, Hetmanski JB, Vekemans M, Attie-Bitach T,
Lovett M, et al: High throughput SNP and expression analyses of
candidate genes for non-syndromic oral clefts. J Med Genet.
43:598–608. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Yamamoto H, Ito K, Kawai M, Murakami Y,
Bessho K and Ito Y: Runx3 expression during mouse tongue and palate
development. Anat Rec A Discov Mol Cell Evol Biol. 288:695–699.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Nawshad A, Medici D and Liu CC: TGFbeta3
inhibits E-cadherin gene expression in palate medial-edge
epithelial cells through a Smad2-Smad4-LEF1 transcription complex.
J Cell Sci. 120:1646–1653. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Nawshad A, LaGamba D and Hay ED:
Transforming growth factor beta (TGFbeta) signalling in palatal
growth, apoptosis and epithelial mesenchymal transformation (EMT).
Arch Oral Biol. 49:675–689. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Cui XM, Shiomi N, Chen J, Saito T,
Yamamoto T, Ito Y, Bringas P, Chai Y and Shuler CF: Overexpression
of Smad2 in Tgf-beta3-null mutant mice rescues cleft palate. Dev
Biol. 278:193–202. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Shin JO, Lee JM, Cho KW, Kwak S, Kwon HJ,
Lee MJ, Cho SW, Kim KS and Jung HS: MiR-200b is involved in Tgf-β
signaling to regulate mammalian palate development. Histochem Cell
Biol. 137:67–78. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Shin JO, Nakagawa E, Kim EJ, Cho KW, Lee
JM, Cho SW and Jung HS: miR-200b regulates cell migration via Zeb
family during mouse palate development. Histochem Cell Biol.
137:459–470. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Mutz KO, Heilkenbrinker A, Lönne M, Walter
JG and Stahl F: Transcriptome analysis using next-generation
sequencing. Curr Opin Biotechnol. 24:22–30. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Mantione KJ, Kream RM, Kuzelova H, Ptacek
R, Raboch J, Samuel JM and Stefano GB: Comparing bioinformatic gene
expression profiling methods: Microarray and RNA-Seq. Med Sci Monit
Basic Res. 20:138–142. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Lowe R, Shirley N, Bleackley M, Dolan S
and Shafee T: Transcriptomics technologies. PLoS Comput Biol.
13:e10054572017. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Warner DR, Mukhopadhyay P, Brock G, Webb
CL, Michele Pisano M and Greene RM: MicroRNA expression profiling
of the developing murine upper lip. Dev Growth Differ. 56:434–447.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Charoenchaikorn K, Yokomizo T, Rice DP,
Honjo T, Matsuzaki K, Shintaku Y, Imai Y, Wakamatsu A, Takahashi S,
Ito Y, et al: Runx1 is involved in the fusion of the primary and
the secondary palatal shelves. Dev Biol. 326:392–402. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Liu Y, El-Naggar S, Darling DS, Higashi Y
and Dean DC: Zeb1 links epithelial-mesenchymal transition and
cellular senescence. Development. 135:579–588. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Salmena L, Poliseno L, Tay Y, Kats L and
Pandolfi PP: A ceRNA hypothesis: The Rosetta Stone of a hidden RNA
language? Cell. 146:353–358. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Lai K, Jia S, Yu S, Luo J and He Y:
Genome-wide analysis of aberrantly expressed lncRNAs and miRNAs
with associated co-expression and ceRNA networks in β-thalassemia
and hereditary persistence of fetal hemoglobin. Oncotarget.
5:49931–49943. 2017.
|
|
68
|
Tay Y, Rinn J and Pandolfi PP: The
multilayered complexity of ceRNA crosstalk and competition. Nature.
505:344–352. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Dai J, Yu H, Si J, Fang B and Shen SG:
Irf6-related gene regulatory network involved in palate and lip
development. J Craniofac Surg. 26:1600–1605. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Choi JW, Park HW, Kwon YJ and Park BY:
Role of apoptosis in retinoic acid-induced cleft palate. J
Craniofac Surg. 22:1567–1571. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Chenevix-Trench G, Jones K, Green AC,
Duffy DL and Martin NG: Cleft lip with or without cleft palate:
Associations with transforming growth factor alpha and retinoic
acid receptor loci. Am J Hum Genet. 51:1377–1385. 1992.PubMed/NCBI
|
|
72
|
Zhu X, Ozturk F, Pandey S, Guda CB and
Nawshad A: Implications of TGFβ on transcriptome and cellular
biofunctions of palatal mesenchyme. Front Physiol. 3:852012.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Christensen K, Juel K, Herskind AM and
Murray JC: Long term follow up study of survival associated with
cleft lip and palate at birth. BMJ. 328:14052004. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Langfelder P and Horvath S: WGCNA: An R
package for weighted correlation network analysis. BMC
Bioinformatics. 9:5592008. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
De Oliveira PSN, Coutinho LL, Tizioto PC,
Cesar ASM, de Oliveira GB, Diniz WJDS, De Lima AO, Reecy JM, Mourão
GB, Zerlotini A and Regitano LCA: An integrative transcriptome
analysis indicates regulatory mRNA-miRNA networks for residual feed
intake in Nelore cattle. Sci Rep. 8:170722018. View Article : Google Scholar : PubMed/NCBI
|