Introduction
Colorectal cancer (CRC) is one of the most common
types of malignancy with a high mortality rate in the world
(1,2). Although conventional anticancer
therapies, including surgery, chemotherapy and radiotherapy, have
improved, the long-term survival rate of patients with CRC treated
with conventional therapies remains low. Substantial efforts have
made in terms of targeted therapies for CRC (3–5).
However, there is limited information on valuable therapeutic
targets for CRC intervention. Therefore, the identification of
novel therapeutic targets for the development of therapies is of
significance in the management of patients with CRC.
CD24 is a small, heavily-glycosylated, mucin-like
glycosylphosphatidylinositol-anchored surface protein (6). It contains a small protein core of
only 27 amino acids with varying levels of potential O- and
N-linked glycosylation, which leads to a molecular weight range
between 38 and 70 kDa (6). Its
expression is upregulated in CRC (7–10).
Previous studies have shown that upregulated expression of CD24 is
associated with the promotion of angiogenesis and lymph node
metastasis, and poor prognosis in CRC (11–13).
Studies have also indicated that targeting CD24 can markedly
inhibit the growth of CRC (14,15),
and that CD24 may be a valuable therapeutic target for CRC
intervention (7). By contrast,
another study revealed that the inhibitory efficacy of targeting
CD24 in inhibiting human HCT116 and HT29 CRC cell growth was
limited (16). Currently, the
mechanism underlying the effect of targeting CD24 on the growth of
CRC has not been clarified.
Autophagy is a dynamic process involving the
intracellular degradation and recycling of cellular components to
eliminate non-essential proteins and damaged organelles for the
maintenance of cellular homeostasis (15,17–22).
During the process of autophagy, initiators stimulate the
expression of autophagy-related genes, including Beclin-1,
autophagy-related 3 (Atg3), autophagy-related 5 (Atg5) and
microtubule-associated protein-1 light chain-3 (LC3), to form
autophagosomes, which migrate and fuse with lysosomes to degrade
the carried intracellular cargos (17,23).
Simultaneously, LC3 on the outside of the vesicles is cleaved into
LC3A and LC3B, and this dynamic process of autophagic flux
modulates the expression of sequestosome-1/p62 (24,25).
Previous studies have shown that Atg5 is a key independent factor
involved in autophagy and apoptosis (20,26).
Accordingly, Atg5 was selected here as the key and representative
gene to investigate the crosstalk of autophagy and apoptosis.
Simultaneously, the expression of Beclin1, also known as Atg6
(20), was also detected in the
present study. The inhibition of autophagy by 3-methyladenine
(3-MA) can suppress the activity of PI3-kinase and the formation of
autophagosomes and autophagic vacuoles (25,27).
Autophagy is associated physiologically with supporting cell
survival, and aberrant autophagy can induce its apoptosis. However,
although targeting CD24 or inhibiting autophagy has shown potential
in anticancer therapy in recent years (7,14,15,26,28,29),
whether CD24 modulates the process of autophagy and how the
combination of targeting CD24 and inhibiting autophagy affects the
survival and apoptosis of human CRC cells remains unclear.
Elucidating the role of CD24 in the crosstalk of autophagy and
apoptosis may aid in designing CD24-based therapies for CRC.
Transcription factors in the nuclear factor (NF)-κB
family can regulate the expression of a broad range of genes
involved in the development, proliferation, survival,
differentiation and senescence of cancer cells (14,30–33).
The NF-κB family consists of heterodimers of five members,
including RelA (p65), c-Rel, RelB, NF-κB1 (p50 and its precursor
p105) and NF-κB2 (p52 and its precursor p100) (34). In physiological conditions, NF-κB
is sequestered in the cytoplasm by interacting with the specific
inhibitory factor, inhibitor of NF-κBα (IκBα). IκBα can be
phosphorylated by the IκB kinases (IKKs), leading to the
phosphorylation of NF-κB (35–39).
The activated NF-κB is translocated to the nucleus and regulates
the transcription of its target genes (34,40).
In our previous study, it was shown that CD24 can interact with
heat shock protein 90 to regulate the stability and degradation of
CD24 in CRC cells (41). Another
study demonstrated that heat shock activates NF-κB signaling, which
enhances autophagy and cell survival (42). In addition, the expression of CD24
potentiates DNA damage reagent-induced apoptosis by suppressing
NF-κB signaling in CD44+ breast cancer cells (43). Other studies have indicated that
autophagy is regulated by the IKK complex or NF-κB signaling
(44–48). By contrast, autophagy can regulate
the activation of NF-κB signaling (49). Therefore, the association between
autophagy and NF-κB signaling is of interest and has not been fully
clarified, particularly in CRC cells (50). In addition, whether autophagy
modulated by CD24 can be regulated by NF-κB signaling in CRC
remains to be elucidated. Accordingly, the present study
investigated whether NF-κB signaling was involved in the crosstalk
between autophagy and apoptosis modulated by CD24.
The present study investigated the effect of altered
expression of CD24 on the autophagy of human HCT116 and HT29 CRC
cells, which express low and high levels of CD24, respectively, in
our preliminary experiments. Furthermore, the consequences of
targeting CD24 and inhibiting autophagy on cell survival and
apoptosis were determined in vitro. The resulting data
indicated that there were varying expression levels of CD24 in
human CRC cells and that CD24 appeared to inhibit autophagy in CRC
cells. The combination of targeting CD24 and inhibiting autophagy
promoted the apoptosis of human CRC cells. Autophagy modulated by
CD24 was partially regulated by NF-κB signaling. In conclusion, the
present study is the first, to the best of our knowledge, to
demonstrate the association between autophagy and CD24, and
describe the mechanisms underlying resistance to CD24 targeted
therapy. These findings may provide insights into the regulation of
CD24 on autophagy and aid in the design of novel therapies for CRC
intervention.
Materials and methods
Specific reagents
Bay11-7082, cell counting kit-8 (CCK-8) and Hoechst
33342 were purchased from Beyotime Institute of Biotechnology
(Guangzhou, China). Antibodies against GAPDH (cat. no. TA-08) and
β-actin (cat. no. TA-09) were purchased from ZSGB-BIO; OriGene
Technologies, Inc. (Beijing, China). Antibodies against p62 (cat.
no. 5114), Beclin-1 (cat. no. 4445), LC3A (cat. no. 4445), LC3B
(cat. no. 4445), Atg5 (cat. no. 4445), Atg3 (cat. no. 4445),
cleaved caspase-3 (cat. no. 9664), cleaved poly (ADP-ribose)
polymerase (PARP; cat. no. 5625), IκBα (cat. no. 4812), NF-κBp65
(cat. no. 8242) and phosphorylated NF-κBp65 (cat. no. 3033) were
purchased from Cell Signaling Technology, Inc. (New York, NY, USA).
Antibody against CD24 was provided by Professor Peter Altevogt
(German Cancer Research Center, Germany).
Cell culture and transfection
Human HCT116 and HT29 CRC cells were obtained from
American Type Culture Collection (Manassas, VA, USA) and were
cultured in RPMI-1640 supplemented with 10% fetal bovine serum
(FBS) (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) in
a humidified atmosphere of 5% CO2 at 37°C. The HCT116
cells were transiently transfected with the control plasmid or a
plasmid for the overexpression of CD24, as described previously
(51), using Lipofectamine 2000
(Invitrogen; Thermo Fisher Scientific, Inc.), to generate control
(CD24VEC:HCT116) and CD24-overexpressing (CD24OE:HCT116) cells,
respectively. The HT29 cells were transiently transfected with the
control plasmid for scramble short hairpin (sh)RNA
(5′-UUCUCCGAACGUGUCACGUTT-3′) or the plasmid for CD24-specific
shRNA (5′-UCUCUCUUCUGCAUCUUUAdTdT-3′) using Lipofectamine 2000
(Invitrogen; Thermo Fisher Scientific, Inc.) to generate control
(CD24NC:HT29) and CD24-knockdown (CD24KD:HT29) cells, respectively.
Subsequently, both types of transfected cells were transiently
transfected with control small interfering (si)RNA
(5′-UUCUCCGAACGUGUCACGUTT-3′) or Atg5-specific siRNA
(5′-GUCCAUCUAAGGAUGCAAUTT-3′) using Lipofectamine 2000 (Invitrogen;
Thermo Fisher Scientific, Inc.), to generate CD24OE +
Atg5KD:HCT116, CD24VEC + Atg5KD:HCT116, CD24NC + Atg5KD:HT29 and
CD24KD + Atg5KD:HT29 cells, respectively. After 2 days, the cells
were harvested for western blotting, cell viability analysis,
Hoechst 33258 staining and LC3 staining.
Extraction of cytoplasmic and nuclear
proteins
The cytoplasmic and nuclear proteins were extracted
from cells using nuclear and cytoplasmic extraction reagents
(Beyotime Institute of Biotechnology), respectively, according to
the manufacturer's protocols. The protein concentrations were
determined using a BCA Protein Assay kit (Beyotime Institute of
Biotechnology).
Western blot analysis
The harvested cells were lysed at 0°C for 30 min in
radioimmunoprecipitation assay buffer (Beyotime Institute of
Biotechnology) containing protease inhibitors and centrifuged at
4°C and 14,000 × g for 20 min. Following the determination of
protein concentrations using the BCA assay, the cell lysate samples
(20–30 µg/lane) were separated by sodium dodecyl
sulfate-polyacrylamide gel electrophoresis on 8–15% gels, and
transferred onto polyvinyldifluoride membranes. The membranes were
blocked with 5% non-fat dry milk in Tris-buffered saline-Tween 20,
and the proteins were probed with primary antibodies against p62
(1:1,000), Beclin1 (1:3,000), LC3A (1:2,000), LC3B (1:2,000), Atg5
(1:1,000), Atg3 (1:1,000), cleaved caspase-3 (1:3,000) and cleaved
PARP (1:1,000) (Cell Signaling Technology, Inc.), CD24 (1:500)
(German Cancer Research Center), GAPDH (1:2,000) and β-actin
(1:500) (ZSGB-BIO; OriGene Technologies, Inc.) overnight at 4°C.
After being washed, the bound antibodies were detected with
horseradish peroxidase-conjugated secondary antibodies (1:2,000;
cat. no. ZB-2301 or cat. no. ZB2305, ZSGB-BIO; OriGene
Technologies, Inc.) at room temperature for 1 h and visualized
using enhanced chemiluminescent reagents (Gibco; Thermo Fisher
Scientific, Inc.).
Expression of EGFP-LC3
The HCT116 and HT29 cells were cultured in 12-well
plates and co-transfected with the p-EGFP-LC3B plasmid for the
expression of EGFP-LC3B (cat. no. 11546, Addgene, Inc., Watertown,
MA, USA) and either the CD24-overexpression plasmid or control
plasmid. The HT29 cells were co-transfected with the p-EGFP-LC3B
plasmid and CD24-specific siRNA or control siRNA. After 2 days, the
cells were examined under a fluorescent microscope and images were
captured.
Cell proliferation assay
Cell proliferation was measured using CCK-8
(Beyotime Institute of Biotechnology). Briefly, individual groups
of cells (1×104/well) were pre-treated with, or without,
3-MA (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) in six
replicates in 96-well plates for 24 h, and cultured at 37°C for 48
and 72 h. During the last 3 h of culture, the cells were exposed to
10% CCK-8 and the absorbance of individual wells was measured at
450 nm using a microplate reader.
Hoechst 33258 staining
Cell apoptosis was examined by Hoechst 33258
staining (Beyotime Institute of Biotechnology). The different
groups of HCT116 and HT29 cells were pretreated with, or without,
3-MA for 24 h and cultured on glass coverslips in culture dishes
for 24 h. The cells were fixed in 4.0% paraformaldehyde, and
stained with Hoechst 33258. The cells were examined under a
fluorescence microscope or laser confocal microscope. The numbers
of cells with nuclear condensation and fragmentation in 200 cells
selected randomly from each group were calculated.
Inhibition of autophagy and NF-κB
signaling
The CD24OE:HCT116, CD24VEC:HCT116, CD24NC:HT29 and
CD24KD:HT29 cells were treated with vehicle (DMSO) or 5 µM
Bay11-7082, an inhibitor of NF-κB signaling for 24 h. The relative
levels of NF-κBp65 phosphorylation, and expression of Beclin-1,
Atg5 and LC3B were determined by western blot analysis. The
punctate cells in individual groups were examined by
immunofluorescence.
Statistical analysis
All experiments were repeated at least three times.
Data are expressed as the mean ± SD. Statistical analysis was
performed using SPSS 13.0 for windows (SPSS, Inc., Chicago, IL,
USA). Differences between groups were analyzed using (two-tailed)
Student's t-test, whereas differences among groups were analyzed by
one-way ANOVA multiple comparison and the Bonferroni post hoc test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
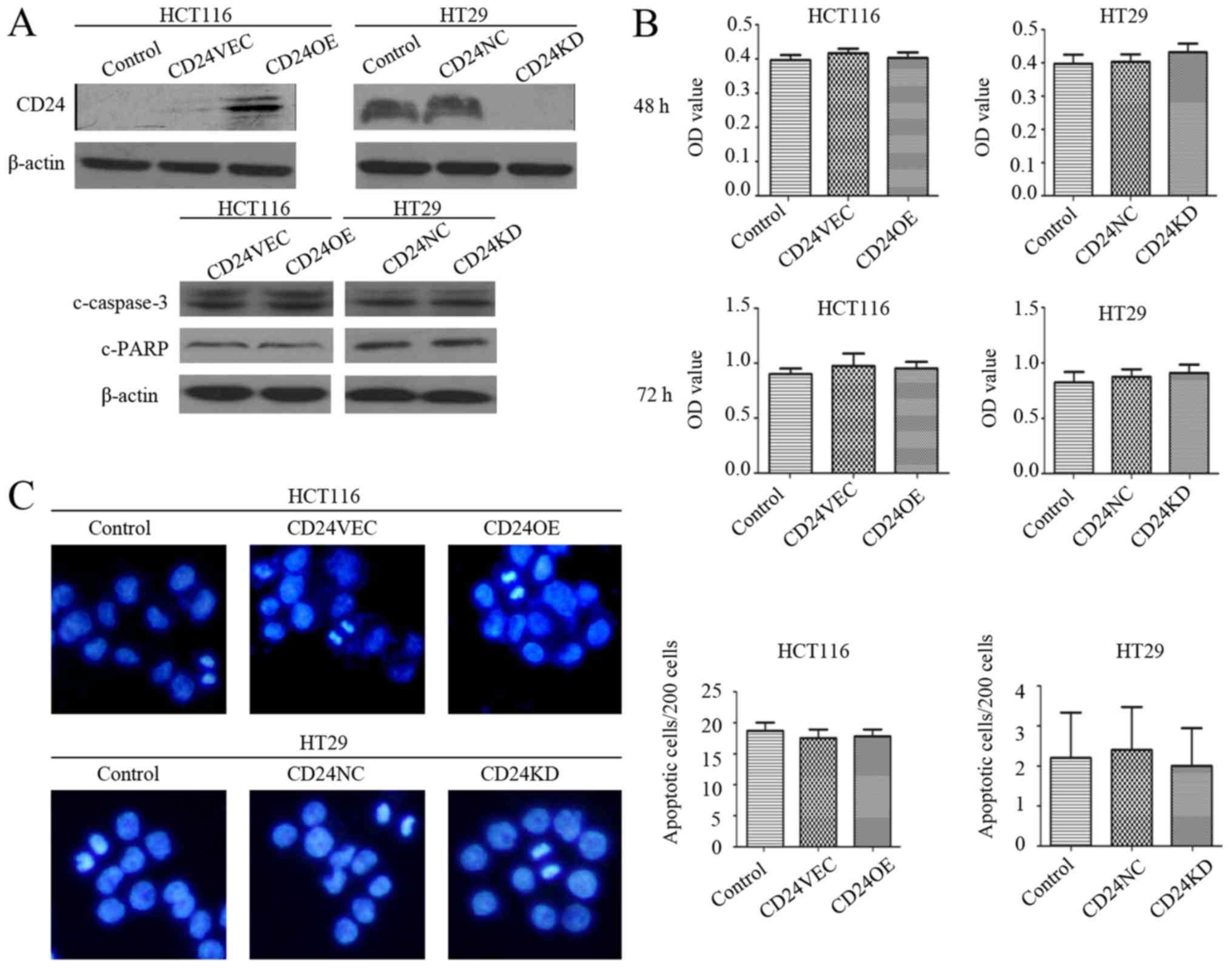
CD24 does not alter the proliferation
or apoptosis of HCT116 or HT29 CRC cells
The preliminary findings revealed lower expression
levels of CD24 in HCT116 cells and higher levels in HT29 cells.
Accordingly, the effect of altered expression of CD24 on
-proliferation and apoptosis in HCT116 and HT29 cells was examined
by inducing the overexpression of CD24 in HCT116 cells and
silencing CD24 in HT29 cells, respectively (Fig. 1A). Subsequently, the impact of the
altered expression of CD24 on cell proliferation and apoptosis was
determined using a CCK-8 assay and Hoechst 33258 staining. It was
found that neither the overexpression or silencing of CD24
significantly altered the viability of HCT116 and HT29 cells
following culture for 48 and 72 h (Fig. 1B). Similarly, the altered
expression of CD24 did not change the frequency of spontaneous
apoptosis of HCT116 and HT29 cells (Fig. 1C). Western blot analysis revealed
that there were similar levels of cleaved PARP and caspase-3 among
the groups of cells, regardless of the overexpression and silencing
of CD24 (Fig. 1A). Therefore, the
altered expression of CD24 did not significantly affect the
proliferation or spontaneous apoptosis of the CRC cells.
CD24 inhibits autophagy in CRC
cells
The impact of altered expression of CD24 on the
autophagy of CRC cells was then assessed. It was found that
induction of the overexpression of CD24 decreased the relative
expression levels of Beclin-1, Atg3, Atg5, LC3A and LC3B in HCT116
cells, and CD24 silencing increased the relative expression levels
of Beclin-1, Atg3, Atg5, LC3A and LC3B in HT29 cells (Fig. 2A and B). Compared with the control,
increased expression levels of p62 were detected in the
CD24-overexpressing CD24OE:HCT116 cells, whereas decreased levels
were observed in the CD24-knockdown CD24KD:HT29 cells at 24–72 h
post-culture (Fig. 2C). The
different groups of cells were also transfected with the
p-EGFP-LC3B plasmid to monitor the process of autophagy. The
punctuated expression of LC3 was significantly decreased in the
CD24OE:HCT116 cells, but increased in the CD24KD:HT29 cells
(P<0.01 for both, Fig. 2D and
E). These data indicated that CD24 inhibited autophagy and
autophagic flux in CRC cells.
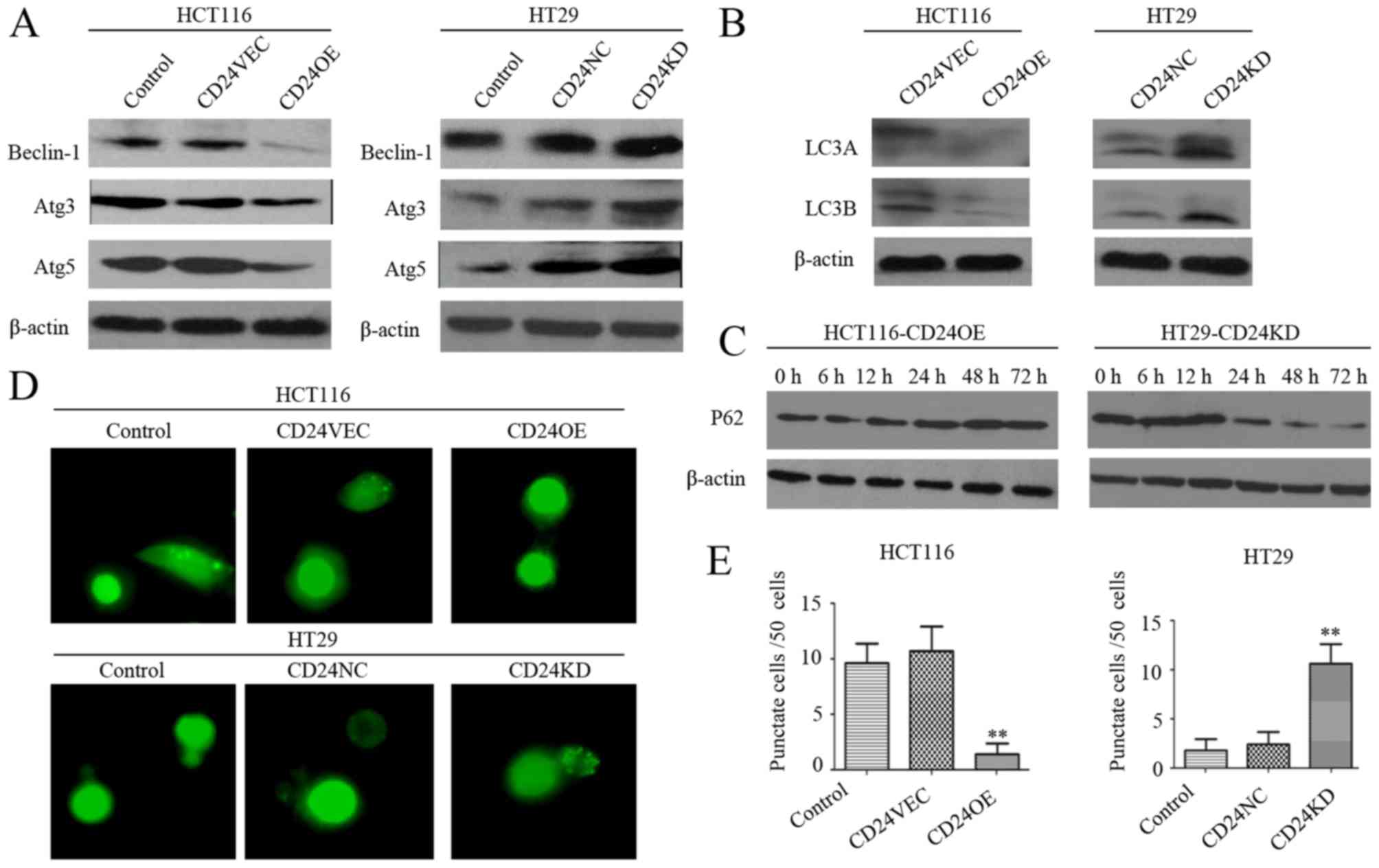 | Figure 2.Altered expression of CD24 modulates
autophagy in CRC cells. (A) Western blot analysis of the relative
levels of Beclin-1, Atg5 and Atg3 in individual groups of cells.
(B) Western blot analysis of the expression of LC3A and LC3B in
individual groups of cells. (C) Western blot analysis of the
dynamic changes in the relative expression levels of p62 in
CD24OE:HCT116 and CD24KD:HT29 cells. (D) Immunofluorescent analysis
of punctate LC3B expression in CRC cells. (E) Quantitative analysis
of punctate cells. Data are representative images (magnification,
×200) or are expressed as the mean ± SD of each group of cells from
three separate experiments. **P<0.01 vs. control. CRC,
colorectal cancer; VEC, vector control; NC, negative control; OE,
overexpression; KD, knockdown; LC3, microtubule-associated
protein-1 light chain-3; Atg3, autophagy-related 3; Atg5,
autophagy-related 5; 3-MA, 3-methyladenine. |
Inhibition of autophagy by 3-MA
modulates the apoptosis of CRC cells
The present study also examined the association
between the inhibition of autophagy and expression of CD24 in CRC
cell apoptosis. First, treatment with 3-MA significantly reduced
the relative expression levels of Beclin-1 and Atg5 in the HCT116
and HT29 cells, regardless of the overexpression and knockdown of
CD24, relative to the levels in the untreated cells (Fig. 3A). Furthermore, treatment with 3-MA
did not change the viability of control CD24VEC:HCT116 and
CD24NC:HT29 cells (Fig. 3B). By
contrast, treatment with the same dose of 3-MA significantly
enhanced the viability of the CD24OE:HCT116 cells, but reduced the
viability of the CD24KD:HT29 cells (P<0.01 for CD24OE:HCT116
cells, P<0.05 for CD24KD:HT29 cells, Fig. 3B), relative to their corresponding
controls. Compared with that of the untreated control cells,
treatment with 3-MA significantly decreased the frequency of
apoptotic CD24OE:HCT116 cells and increased the percentages of
apoptotic CD24KD:HT29 cells (P<0.01 for both), although
treatment with 3-MA did not alter the frequency of apoptotic cells
in the control CD24VEC:HCT116 and CD24NC:HT29 groups (Fig. 3C and D). Evidently, treatment with
3-MA reduced the relative expression levels of cleaved PARP and
caspase-3 in CD24OE:HCT116 cells and enhanced their levels in
CD24KD:HT29 cells, compared with those in the untreated control
cells (Fig. 3A). Therefore, the
inhibition of autophagy modulated the apoptosis of CRC cells.
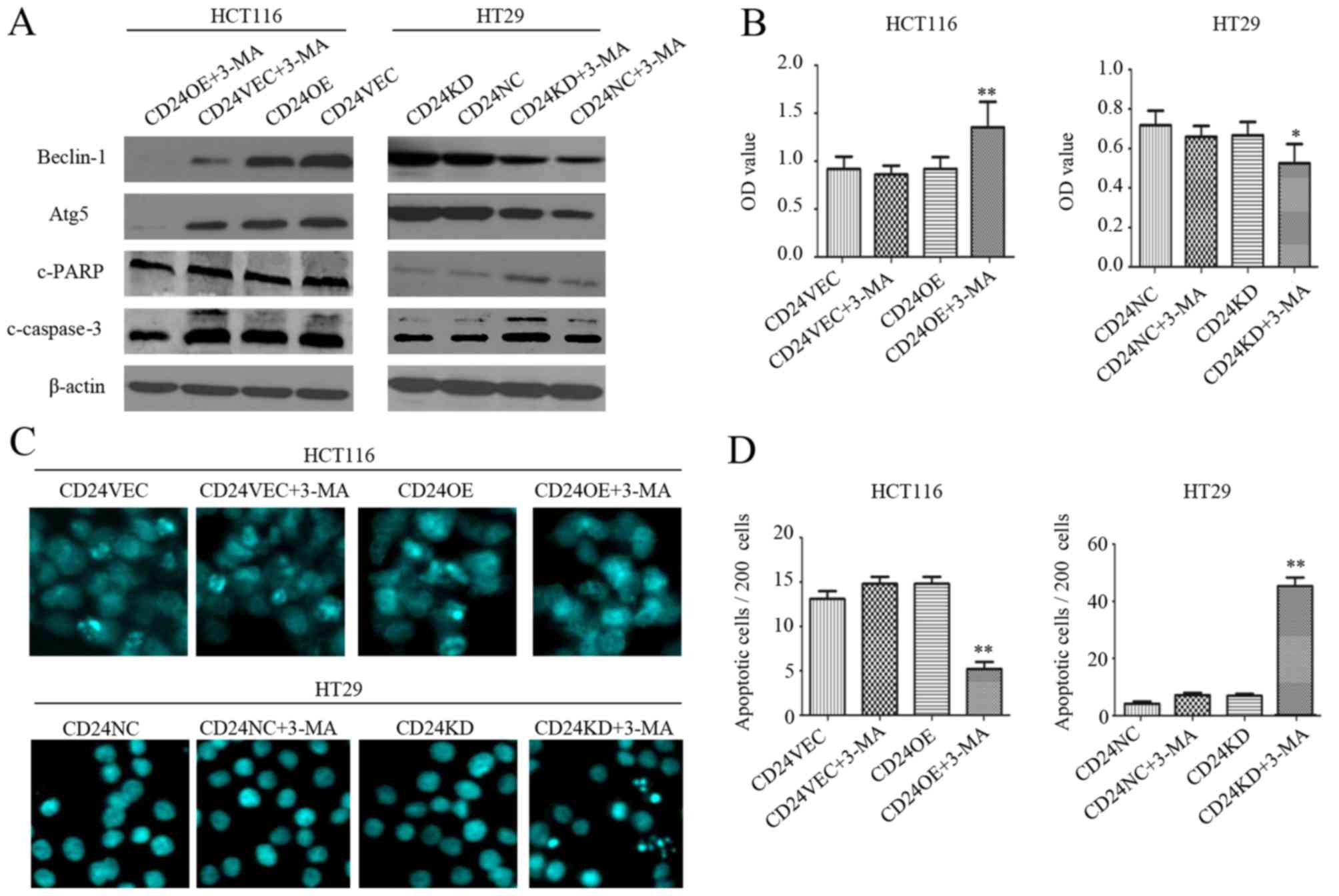 | Figure 3.Inhibition of autophagy by 3-MA
promotes apoptosis of CRC cells. CD24VEC:HCT116, CD24OE:HCT116,
CD24NC:HT29 and CD24KD:HT29 cells were treated with, or without,
3-MA for 24 h. (A) Western blot analysis of the relative expression
levels of Beclin-1, Atg5, c-PARP and c-caspase-3 in individual
groups of cells. (B) Cell counting kit-8 examination of cell
viability. (C) Hoechst 33258 staining for the detection of
apoptotic cells. (D) Quantitative analysis of apoptotic cells. Data
are representative images (magnification, ×200) or are expressed as
the mean ± SD of each group of cells from three separate
experiments. *P<0.05 and **P<0.01 vs. corresponding control
cells. CRC, colorectal cancer; VEC, vector control; NC, negative
control; OE, overexpression; KD, knockdown; 3-MA, 3-methyladenine;
PARP, poly(ADP-ribose) polymerase; c-, cleaved; Atg,
autophagy-related 5. |
Knockdown of Atg5 promotes the
apoptosis of CD24KD:HT29 cells
To obtain additional evidence for the inhibition of
autophagy modulating CRC apoptosis, the HCT116 and HT29 cells were
transfected Atg5-specific or control siRNA (Atg5NC). It was found
that transfection with Atg5-specific siRNA markedly reduced the
relative expression levels of Atg5 in the HCT116 and HT29 cells,
demonstrating the efficacy of Atg5 silencing (Fig. 4A). Compared with the Atg5NC
controls, Atg5 silencing also decreased the relative expression
levels of Beclin-1, cleaved PARP and cleaved caspase-3 in the
CD24OE:HCT116 cells, but increased their expression in CD24KD:HT29
cells (Fig. 4B). Functionally,
Atg5 silencing significantly increased the viability of
CD24OE:HCT116 cells and decreased the viability of CD24KD:HT29
cells (P<0.05 for CD24OE:HCT116 cells, P<0.01 for CD24KD:HT29
cells, Fig. 4C). Atg5 silencing
significantly decreased the percentage of apoptotic CD24OE:HCT116
cells and increased the frequency of apoptotic CD24KD:HT29 cells
(P<0.01 for both, Fig. 4D and
E). Therefore, the inhibition of autophagy modulated the
apoptosis of CRC cells in vitro.
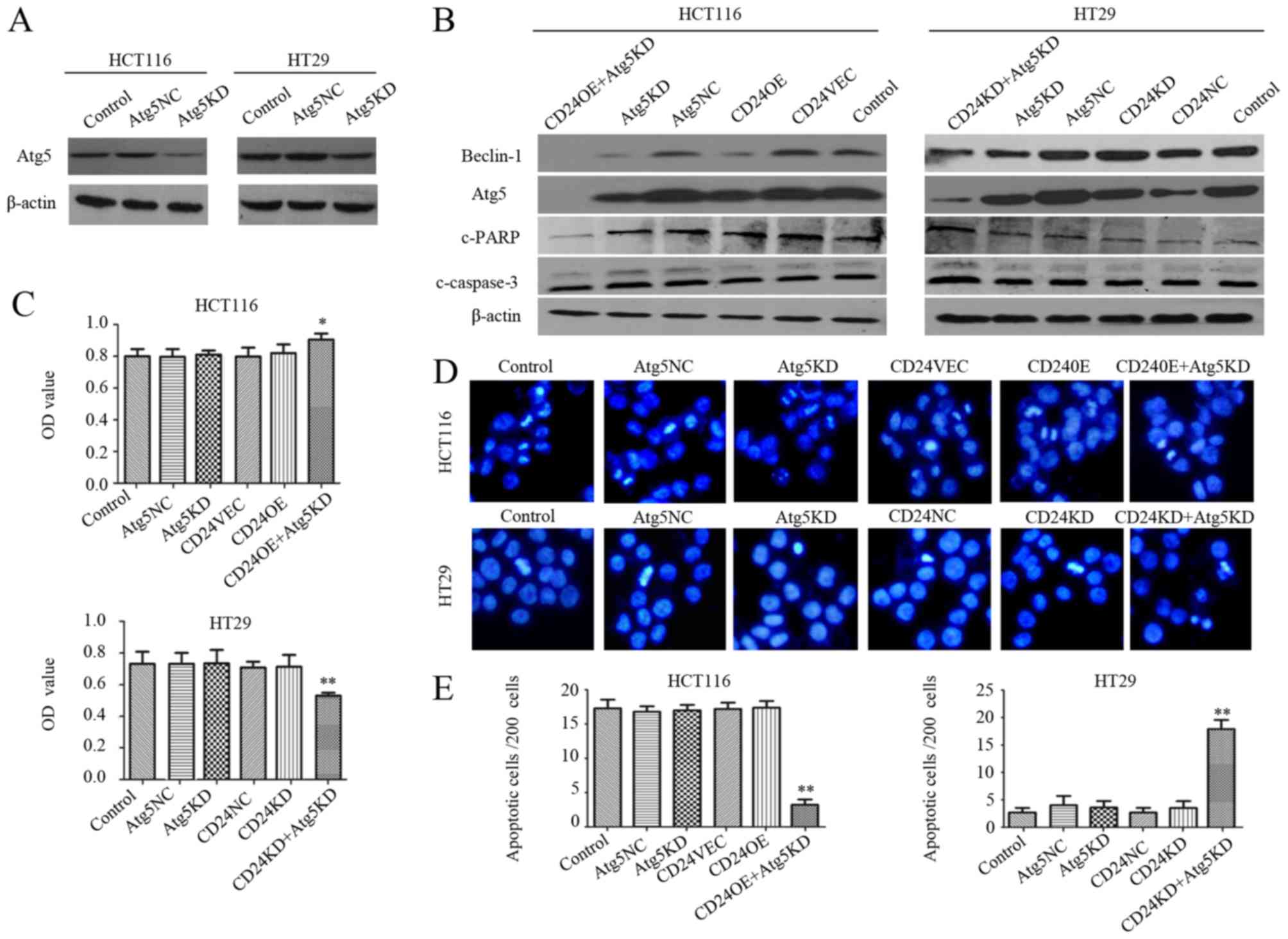 | Figure 4.Inhibition of autophagy by Atg5
silencing modulates the altered expression of CD24-regulated cell
viability and apoptosis in colorectal cancer cells. (A) HCT116 and
HT29 cells were transfected Atg-specific siRNA or control siRNA and
the relative expression levels of Atg5 were determined by a western
blot assay. (B) CD24VEC:HCT116, CD24OE:HCT116, CD24NC:HT29 and
CD24KD:HT29 cells were transfected Atg5-specific siRNA or control
siRNA and the relative expression levels of Beclin-1, Atg5, cleaved
PARP and cleaved caspase-3 were determined by western blot
analysis. (C) Viabilities of individual groups of cells were
determined using cell counting kit-8 assays. (D) Apoptotic cells
were examined by Hoechst 33258 staining. (E) Quantitative analysis
of apoptotic cells. Data are representative images (magnification,
×200) or are expressed as the mean ± SD of each group of cells from
three separate experiments. *P<0.05 and **P<0.01 vs.
corresponding control cells. VEC, vector control; NC, negative
control; OE, overexpression; KD, knockdown; siRNA, small
interfering RNA; PARP, poly (ADP-ribose) polymerase; c-, cleaved;
Atg, autophagy-related 5. |
Autophagy inhibited by CD24 is partly
regulated by NF-κB signaling
Increasing evidence suggests that NF-κB signaling
positively regulates the process of autophagy (30,42,48,52).
Finally, the present study investigated the effect of the NF-κB
signaling inhibitor on the CD24-induced reduction of autophagy.
First, inducting the overexpression of CD24 marginally increased
the phosphorylation of NF-κBp65 and decreased the expression of
IκBα in HCT116 cells, whereas CD24 silencing decreased the
phosphorylation of NF-κBp65 and increased the expression of IκBα in
HT29 cells (Fig. 5A). Compared
with the control, treatment with Bay11-7082 marginally increased
the phosphorylation of NF-κBp65, but did not alter the relative
expression levels of Beclin-1, Atg5 or LC3B in the CD24OE:HCT116
cells (Fig. 5B). Furthermore,
treatment with Bay11-7082 significantly reduced the frequency of
LC3 punctate cells in the HCT116 cells, but enhanced their
frequency in HT29 cells, regardless of the altered expression of
CD24 (P<0.01 for all, Fig. 5C and
D). Therefore, autophagy regulated by the altered expression of
CD24 was partially modulated by activating NF-κB signaling in CRC
cells.
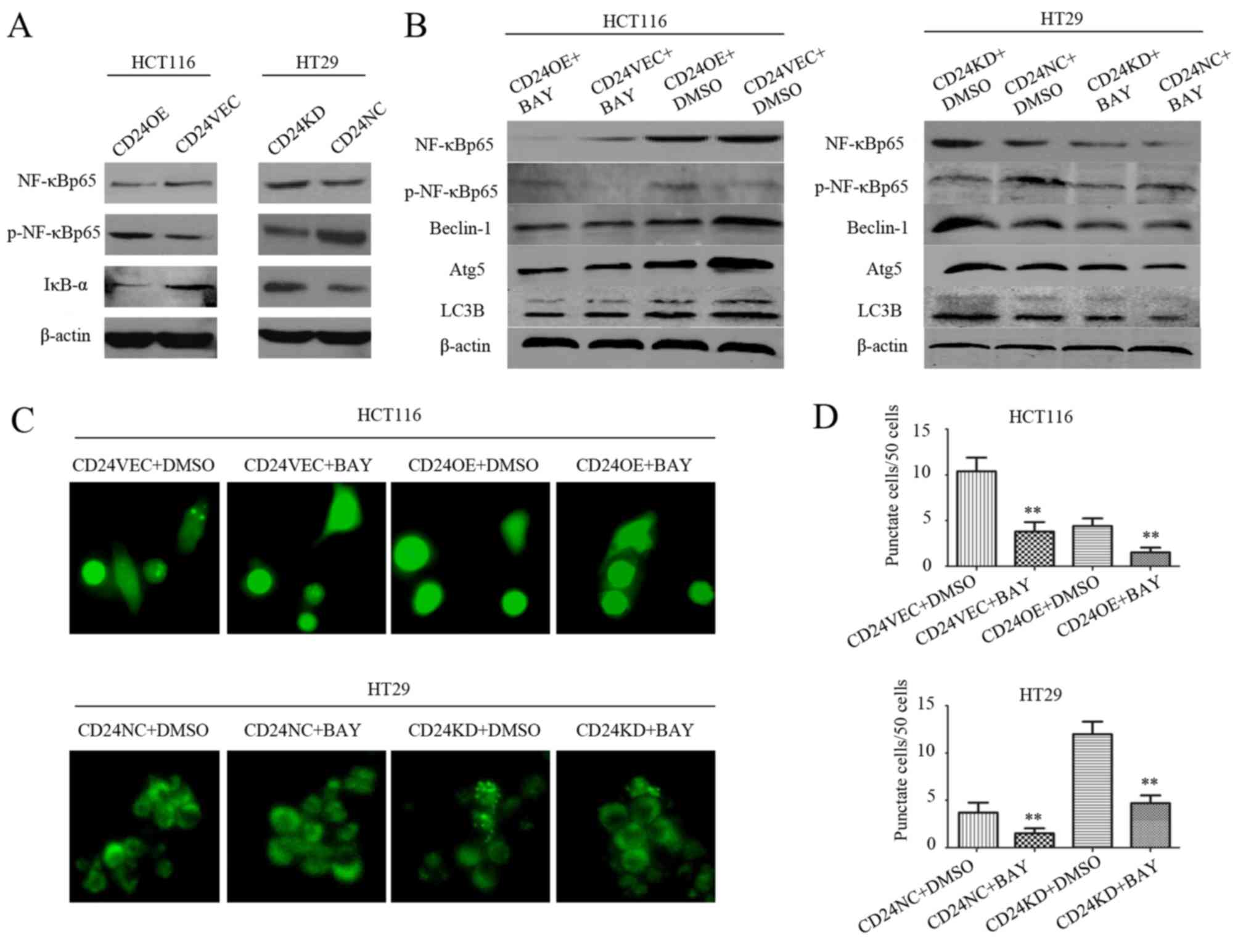 | Figure 5.Autophagy regulated by altered
expression of CD24 is partially regulated by activating NF-κBp65
signaling in colorectal cancer cells. (A) Western blot analysis of
the relative levels of NF-κBp65 and IκB-α and p-NF-κBp65 in
individual groups of cells. (B) Western blot analysis of
p-NF-κBp65, Beclin-1, Atg5 and LC3B in individual groups of cells
treated with vehicle DMSO or 5 µM BAY for 24 h. (C) Punctate LC3B
expression was determined by immunofluorescence and (D)
quantitatively analyzed for cells with punctate LC3B. Data are
representative images (magnification, ×200) or are expressed as the
mean ± SD of each group of cells from three separate experiments.
**P<0.01 vs. untreated corresponding cells. VEC, vector control;
NC, negative control; OE, overexpression; KD, knockdown; BAY,
Bay11-7082; NF-κB, nuclear factor-κB; IκB-α, inhibitor of NF-κBα;
p-, phosphorylated; Atg5, autophagy-related 5; LC3B,
microtubule-associated protein-1 light chain-3B. |
Discussion
The upregulated expression of CD24 is inversely
associated with the poor outcomes for patients with CRC (10–13,53),
and CD24 has been considered as a valuable target for the treatment
of CRC (7,8,15,51).
Targeting CD24 using anti-CD24 antibody or siRNA can inhibit the
growth of CD24-expressing CRC cells, and the combination of
anti-CD24 and chemotherapy markedly reduces the growth of CRC
(15,53). However, how CD24 affects the growth
of CRC remains controversial. In the present study, varying levels
of CD24 were found in human CRC cells, and altering the expression
of CD24 by inducing its overexpression or silencing did not alter
viability or spontaneous apoptosis in HCT116 and HT29 cells. Such
data were consistent with a previous observation (16), but were in disagreement with
another study (51). These
findings suggest that CRC with different expression levels of CD24
may have varying sensitivity to anti-CD24 therapy.
Autophagy is crucial for cell survival and
apoptosis, including cancer cells (17,18),
and aberrant autophagy can lead to the apoptosis of cancer cells
(21,54,55).
The induction of autophagy can promote drug resistance and cancer
cell survival (26,56,57),
while the inhibition of autophagy sensitizes cancer cells to
anticancer therapies (26,58–60).
Accordingly, inhibiting autophagy may be a promising strategy to
improve the efficacy of conventional anticancer chemotherapies
(28,29). In the present study, it was found
that CD24 inhibited autophagy in human CRC cells. Evidentially,
induction of the overexpression of CD24 decreased the levels of
autophagy in HCT116 cells, whereas CD24 silencing increased
autophagy in HT29 cells. Of note, although inhibiting autophagy by
3-MA treatment did not alter the spontaneous apoptosis of wild-type
HCT116 or HT29 cells, the same treatment significantly decreased
CD24-overexpressing HCT116 cell apoptosis, but increased
CD24-silenced HT29 cell apoptosis. Similar patterns of cell
survival and apoptosis were observed among different groups of
HCT116 and HT29 cells following Atg5 silencing. Such findings
indicated that the combination of targeting CD24 and inhibiting
autophagy effectively triggered the apoptosis of CRC cells with a
low expression of CD24, but not those with a high expression of
CD24. Therefore, these novel findings may provide insights into the
regulation of CD24 in autophagy in CRC and assist in the design of
novel therapies for CRC intervention.
The present study also examined the underlying
mechanisms of the CD24-induced reduction of autophagy. CD24
silencing reduced the activation of NF-κB signaling, whereas the
inhibition the NF-κB activation inhibited autophagy, indicating
that the CD24-induced reduction of autophagy was at least partly
regulated by NF-κB signaling. Therefore, the CD24-induced reduction
of autophagy may inhibit the activation of NF-κB signaling in CRC
cells. Thus, enhanced autophagy may be a potential way of
overcoming the resistance of CRC cells to chemotherapy, and
targeting CD24 may involve the activation of NF-κB signaling.
The association between the regulation of autophagy
and NF-κB signaling appears complex. NF-κB has emerged as a
negative mediator of tumor necrosis factor-α-induced autophagy
(48). By contrast, NF-κB is
positively regulated by inhibition of TSC2/mammalian target of
rapamycin activity, a key inhibitor of autophagy (61). In addition, the NF-κB family member
p65/RelA can upregulate the expression of Beclin-1 in different
types of cell, and the inhibition of NF-κBp65 signaling reduces the
expression of Beclin-1, suggesting that the regulation of autophagy
requires NF-κB activation (62).
These findings support our hypothesis that CD24-mediated autophagy
may require the activation of NF-κB signaling and our data
confirmed that the inhibition of NF-κB signaling attenuated CD24
silencing-induced autophagy in CRC cells. Accordingly, it is
possible that the activation or inactivation of NF-κB signaling
regulates autophagy, dependent on the cell genetic background, cell
context or experimental conditions. However, whether the
interactions of altered expression of CD24, NF-κB activation and
autophagy are regulated by the NF-κBp65-induced expression of
Beclin-1 in CRC cells remains to be elucidated. Further
investigations are required to determine how the altered expression
of CD24 regulates NF-κB signaling during the development and
progression of CRC.
In conclusion, the data obtained in the present
study indicated the presence of varying levels of CD24 in different
human CRC cells and that altered expression of CD24 failed to alter
the viability or spontaneous apoptosis of CRC cells. Furthermore,
CD24 inhibited autophagy in CRC cells and the combination of
targeting CD24 and inhibiting autophagy effectively triggered the
apoptosis of CRC cells, particularly in cells with low expression
levels of CD24. In addition, the overexpression of CD24 increased
NF-κBp65 activation, which was decreased following CD24 silencing,
in CRC cells. The inhibition of NF-κB activation enhanced autophagy
in HCT116:CD24OE cells, whereas the same treatment led to reduced
autophagy in HT29:CD24KD cells. These data suggest that autophagy
regulated by the altered expression of CD24 may be partially
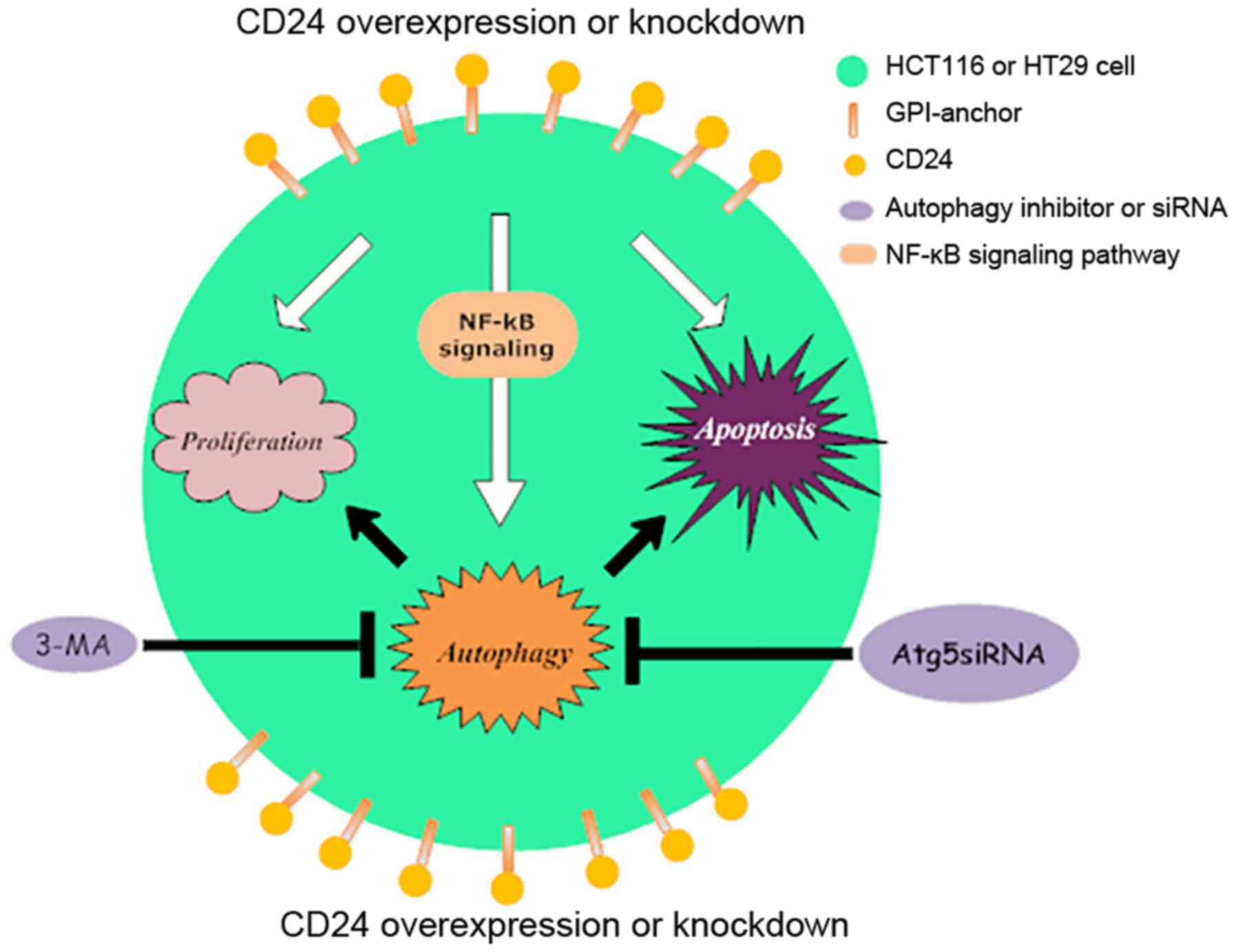
regulated by activating NF-κBp65 signaling in CRC cells (Fig. 6). These novel findings may provide
further explanation as to why targeting CD24 therapies are
unsatisfactory in certain patients with CRC, and may assist in the
design of novel therapeutic strategies for CRC intervention. The
present study had limitations, including all mechanistic findings
being from in vitro cellular experiments. Further
investigations are warranted on the molecular mechanisms underlying
the therapeutic effect of combined autophagy inhibition and CD24
targeting CRC apoptosis in vivo.
Acknowledgements
The authors thank Dr Liang Peng (Department of
Gastroenterology, Nanfang Hospital, Southern Medical University)
for his technical assistance and providing the CD24-overexpression
plasmid, and Professor Bo Jiang (Department of Gastroenterology,
Nanfang Hospital, Southern Medical University) for his support.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
XW and JZ conceived and designed the study, JZ
performed all experiments and wrote the manuscript. XW reviewed and
edited the manuscript. Both authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in China,
2015. CA Cancer J Clin. 66:115–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kuipers EJ, Grady WM, Lieberman D,
Seufferlein T, Sung JJ, Boelens PG, van de Velde CJ and Watanabe T:
Colorectal cancer. Nat Rev Dis Primers. 1:150652015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kerr D: Clinical development of gene
therapy for colorectal cancer. Nat Rev Cancer. 3:615–622. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kuipers EJ, Rösch T and Bretthauer M:
Colorectal cancer screening-optimizing current strategies and new
directions. Nat Rev Clin Oncol. 10:130–142. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Palta M, Czito BG and Willett CG:
Colorectal cancer: Adjuvant chemotherapy for rectal cancer-an
unresolved issue. Nat Rev Clin Oncol. 11:182–184. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kristiansen G, Sammar M and Altevogt P:
Tumour biological aspects of CD24, a mucin-like adhesion molecule.
J Mol Histol. 35:255–262. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Eyvazi S, Kazemi B, Dastmalchi S and
Bandehpour M: Involvement of CD24 in multiple cancer related
pathways makes it an interesting new target for cancer therapy.
Curr Cancer Drug Targets. 18:328–336. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sagiv E, Memeo L, Karin A, Kazanov D,
Jacob-Hirsch J, Mansukhani M, Rechavi G, Hibshoosh H and Arber N:
CD24 is a new oncogene, early at the multistep process of
colorectal cancer carcinogenesis. Gastroenterology. 131:630–639.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lee JH, Kim SH, Lee ES and Kim YS: CD24
overexpression in cancer development and progression: A
meta-analysis. Oncol Rep. 22:1149–1156. 2009.PubMed/NCBI
|
|
10
|
Lim SC: CD24 and human carcinoma: Tumor
biological aspects. Biomed Pharmacother. 59 (Suppl 2):S351–S354.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang JL, Guo CR, Su WY, Chen YX, Xu J and
Fang JY: CD24 overexpression related to lymph node invasion and
poor prognosis of colorectal cancer. Clin Lab. 64:497–505. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Weichert W, Denkert C, Burkhardt M,
Gansukh T, Bellach J, Altevogt P, Dietel M and Kristiansen G:
Cytoplasmic CD24 expression in colorectal cancer independently
correlates with shortened patient survival. Clin Cancer Res.
11:6574–6581. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Duex JE, Owens C, Chauca-Diaz A, Dancik
GM, Vanderlinden LA, Ghosh D, Leivo MZ, Hansel DE and Theodorescu
D: Nuclear CD24 drives tumor growth and is predictive of poor
patient prognosis. Cancer Res. 77:4858–4867. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shapira S, Shapira A, Starr A, Kazanov D,
Kraus S, Benhar I and Arber N: An immunoconjugate of anti-CD24 and
pseudomonas exotoxin selectively kills human colorectal tumors in
mice. Gastroenterology. 140:935–946. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sagiv E, Starr A, Rozovski U, Khosravi R,
Altevogt P, Wang T and Arber N: Targeting CD24 for treatment of
colorectal and pancreatic cancer by monoclonal antibodies or small
interfering RNA. Cancer Res. 68:2803–2812. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ahmed MA, Jackson D, Seth R, Robins A,
Lobo DN, Tomlinson IP and Ilyas M: CD24 is upregulated in
inflammatory bowel disease and stimulates cell motility and colony
formation. Inflamm Bowel Dis. 16:795–803. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mizushima N and Komatsu M: Autophagy:
Renovation of cells and tissues. Cell. 147:728–741. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lozy F and Karantza V: Autophagy and
cancer cell metabolism. Semin Cell Dev Biol. 23:395–401. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chen N and Karantza-Wadsworth V: Role and
regulation of autophagy in cancer. Biochim Biophys Acta.
1793:1516–1523. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Rikiishi H: Novel insights into the
interplay between apoptosis and autophagy. Int J Cell Biol.
2012:3176452012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tschan MP and Simon HU: The role of
autophagy in anticancer therapy: Promises and uncertainties. J
Intern Med. 268:410–418. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tsujimoto Y and Shimizu S: Another way to
die: Autophagic programmed cell death. Cell Death Differ. 12 (Suppl
2):S1528–S1534. 2005. View Article : Google Scholar
|
|
23
|
Cecconi F and Levine B: The role of
autophagy in mammalian development: Cell makeover rather than cell
death. Dev Cell. 15:344–357. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Paillas S, Causse A, Marzi L, de Medina P,
Poirot M, Denis V, Vezzio-Vie N, Espert L, Arzouk H, Coquelle A, et
al: MAPK14/p38α confers irinotecan resistance to TP53-defective
cells by inducing survival autophagy. Autophagy. 8:1098–1112. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Klionsky DJ, Abdalla FC, Abeliovich H,
Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M,
Agostinis P, Aguirre-Ghiso JA, et al: Guidelines for the use and
interpretation of assays for monitoring autophagy. Autophagy.
8:445–544. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Selvakumaran M, Amaravadi RK, Vasilevskaya
IA and O'Dwyer PJ: Autophagy inhibition sensitizes colon cancer
cells to antiangiogenic and cytotoxic therapy. Clin Cancer Res.
19:2995–3007. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Petiot A, Ogier-Denis E, Blommaart EF,
Meijer AJ and Codogno P: Distinct classes of phosphatidylinositol
3′-kinases are involved in signaling pathways that control
macroautophagy in HT-29 cells. J Biol Chem. 275:992–998. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li J, Hou N, Faried A, Tsutsumi S and
Kuwano H: Inhibition of autophagy augments 5-fluorouracil
chemotherapy in human colon cancer in vitro and in vivo model. Eur
J Cancer. 46:1900–1909. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li J, Hou N, Faried A, Tsutsumi S,
Takeuchi T and Kuwano H: Inhibition of autophagy by 3-MA enhances
the effect of 5-FU-induced apoptosis in colon cancer cells. Ann
Surg Oncol. 16:761–771. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Trocoli A and Djavaheri-Mergny M: The
complex interplay between autophagy and NF-κB signaling pathways in
cancer cells. Am J Cancer Res. 1:629–649. 2011.PubMed/NCBI
|
|
31
|
Bassères DS and Baldwin AS: Nuclear
factor-kappaB and inhibitor of kappaB kinase pathways in oncogenic
initiation and progression. Oncogene. 25:6817–6830. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Keeratichamroen S, Leelawat K, Thongtawee
T, Narong S, Aegem U, Tujinda S, Praditphol N and Tohtong R:
Expression of CD24 in cholangiocarcinoma cells is associated with
disease progression and reduced patient survival. Int J Oncol.
39:873–881. 2011.PubMed/NCBI
|
|
33
|
Luo JL, Kamata H and Karin M:
IKK/NF-kappaB signaling: Balancing life and death-a new approach to
cancer therapy. J Clin Invest. 115:2625–2632. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hoffmann A, Natoli G and Ghosh G:
Transcriptional regulation via the NF-kappaB signaling module.
Oncogene. 25:6706–6716. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Haskill S, Beg AA, Tompkins SM, Morris JS,
Yurochko AD, Sampson-Johannes A, Mondal K, Ralph P and Baldwin AS
Jr: Characterization of an immediate-early gene induced in adherent
monocytes that encodes I kappa B-like activity. Cell. 65:1281–1289.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Thompson JE, Phillips RJ,
Erdjument-Bromage H, Tempst P and Ghosh S: I kappa B-beta regulates
the persistent response in a biphasic activation of NF-kappa B.
Cell. 80:573–582. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Whiteside ST, Epinat JC, Rice NR and
Israël A: I kappa B epsilon, a novel member of the I kappa B
family, controls RelA and cRel NF-kappa B activity. EMBO J.
16:1413–1426. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Senftleben U, Cao Y, Xiao G, Greten FR,
Krähn G, Bonizzi G, Chen Y, Hu Y, Fong A, Sun SC and Karin M:
Activation by IKKalpha of a second, evolutionary conserved,
NF-kappa B signaling pathway. Science. 293:1495–1499. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Xiao G, Harhaj EW and Sun SC:
NF-kappaB-inducing kinase regulates the processing of NF-kappaB2
p100. Mol Cell. 7:401–409. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hayden MS and Ghosh S: Shared principles
in NF-kappaB signaling. Cell. 132:344–362. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wang X, Zhang Y, Zhao Y, Liang Y, Xiang C,
Zhou H, Zhang H, Zhang Q, Qing H, Jiang B, et al: CD24 promoted
cancer cell angiogenesis via Hsp90-mediated STAT3/VEGF signaling
pathway in colorectal cancer. Oncotarget. 7:55663–55676.
2016.PubMed/NCBI
|
|
42
|
Nivon M, Richet E, Codogno P, Arrigo AP
and Kretz-Remy C: Autophagy activation by NFkappaB is essential for
cell survival after heat shock. Autophagy. 5:766–783. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ju JH, Jang K, Lee KM, Kim M, Kim J, Yi
JY, Noh DY and Shin I: CD24 enhances DNA damage-induced apoptosis
by modulating NF-κB signaling in CD44-expressing breast cancer
cells. Carcinogenesis. 32:1474–1483. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kristiansen G, Pilarsky C, Pervan J,
Sturzebecher B, Stephan C, Jung K, Loening S, Rosenthal A and
Dietel M: CD24 expression is a significant predictor of PSA relapse
and poor prognosis in low grade or organ confined prostate cancer.
Prostate. 58:183–192. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Sagiv E, Kazanov D and Arber N: CD24 plays
an important role in the carcinogenesis process of the pancreas.
Biomed Pharmacother. 60:280–284. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Comb WC, Cogswell P, Sitcheran R and
Baldwin AS: IKK-dependent, NF-κB-independent control of autophagic
gene expression. Oncogene. 30:1727–1732. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Criollo A, Senovilla L, Authier H, Maiuri
MC, Morselli E, Vitale I, Kepp O, Tasdemir E, Galluzzi L, Shen S,
et al: The IKK complex contributes to the induction of autophagy.
EMBO J. 29:619–631. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Djavaheri-Mergny M, Amelotti M, Mathieu J,
Besançon F, Bauvy C, Souquère S, Pierron G and Codogno P: NF-kappaB
activation represses tumor necrosis factor-alpha-induced autophagy.
J Biol Chem. 281:30373–30382. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Djavaheri-Mergny M and Codogno P:
Autophagy joins the game to regulate NF-kappaB signaling pathways.
Cell Res. 17:576–577. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Huett A, Goel G and Xavier RJ: A systems
biology viewpoint on autophagy in health and disease. Curr Opin
Gastroenterol. 26:302–309. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Wang W, Wang X, Peng L, Deng Q, Liang Y,
Qing H and Jiang B: CD24-dependent MAPK pathway activation is
required for colorectal cancer cell proliferation. Cancer Sci.
101:112–119. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Xiao G: Autophagy and NF-kappaB: Fight for
fate. Cytokine Growth Factor Rev. 18:233–243. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Smith SC, Oxford G, Wu Z, Nitz MD, Conaway
M, Frierson HF, Hampton G and Theodorescu D: The
metastasis-associated gene CD24 is regulated by Ral GTPase and is a
mediator of cell proliferation and survival in human cancer. Cancer
Res. 66:1917–1922. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Su M, Mei Y and Sinha S: Role of the
crosstalk between autophagy and apoptosis in cancer. J Oncol.
2013:1027352013. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Yang SY and Winslet MC: Dual role of
autophagy in colon cancer cell survival. Ann Surg Oncol. 18 (Suppl
3):S2392011. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Zheng HY, Zhang XY, Wang XF and Sun BC:
Autophagy enhances the aggressiveness of human colorectal cancer
cells and their ability to adapt to apoptotic stimulus. Cancer Biol
Med. 9:105–110. 2012.PubMed/NCBI
|
|
57
|
Sato K, Tsuchihara K, Fujii S, Sugiyama M,
Goya T, Atomi Y, Ueno T, Ochiai A and Esumi H: Autophagy is
activated in colorectal cancer cells and contributes to the
tolerance to nutrient deprivation. Cancer Res. 67:9677–9684. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Sasaki K, Tsuno NH, Sunami E, Tsurita G,
Kawai K, Okaji Y, Nishikawa T, Shuno Y, Hongo K, Hiyoshi M, et al:
Chloroquine potentiates the anti-cancer effect of 5-fluorouracil on
colon cancer cells. BMC Cancer. 10:3702010. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Singh P, Godbole M, Rao G, Annarao S,
Mitra K, Roy R, Ingle A, Agarwal G and Tiwari S: Inhibition of
autophagy stimulate molecular iodine-induced apoptosis in hormone
independent breast tumors. Biochem Biophys Res Commun. 415:181–186.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Guo XL, Li D, Hu F, Song JR, Zhang SS,
Deng WJ, Sun K, Zhao QD, Xie XQ, Song YJ, et al: Targeting
autophagy potentiates chemotherapy-induced apoptosis and
proliferation inhibition in hepatocarcinoma cells. Cancer Lett.
320:171–179. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Ghosh S, Tergaonkar V, Rothlin CV, Correa
RG, Bottero V, Bist P, Verma IM and Hunter T: Essential role of
tuberous sclerosis genes TSC1 and TSC2 in NF-kappaB activation and
cell survival. Cancer Cell. 10:215–226. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Copetti T, Bertoli C, Dalla E, Demarchi F
and Schneider C: p65/RelA modulates BECN1 transcription and
autophagy. Mol Cell Biol. 29:2594–2608. 2009. View Article : Google Scholar : PubMed/NCBI
|