Introduction
Several neurodegenerative conditions, including
Alzheimer's disease (AD) and Parkinson's disease, are associated
with homocysteine (Hcy) (1–3).
However, the mechanism involved remains to be fully elucidated.
Hyperhomocysteinemia (HHcy) is a condition characterized by an
elevation in the concentration of L-Hcy in plasma above its
physiological level, which varies between 5 and 15 µM (mean 10 µM).
Even moderate-mild HHcy is considered to be a risk factor for
several neurodegenerative diseases. The mechanisms underlying its
toxicity have been reported, including NMDA receptor- and group I
metabotropic glutamate receptor-mediated neurotoxicity. In
addition, Hcy can induce caspase-dependent apoptosis in human
dopaminergic cells, and in rat hippocampal and mouse cortical
neurons (4).
Although the neurotoxic effects of Hcy are well
documented, few studies have investigated the effect of Hcy in
astrocytes (4–6). Maler et al reported that Hcy
exhibited a dose-dependent cytotoxic effect at doses ≤2 mM in
cortical astrocytes (7). In order
to determine the influence of Hcy on the viability of the cells, a
previous study cultured glioblastoma cells with Hcy (0.5, 2, and 5
mM) for 72 h; the results indicated that the extent of cell death
increased with the concentration of Hcy in the culture medium
(8). Astrocytes are an important
cell type in the central nervous system and are critical in the
glial-vascular interface as part of the blood-brain barrier.
Astrocytes have been identified as the support and housekeeping
cells of the nervous system and exert structural, metabolic and
functional effects on neurons, which are either neurotoxic or
neuroprotective (9). It has been
shown that non-neural cells, mainly astrocytes, are crucial in the
occurrence and development of degenerative diseases (10).
A previous study has shown that Hcy exerts an
excitotoxic effect on cells by promoting free radical formation and
inducing oxidative stress (11).
It has been reported that, under oxidative stress, GAPDH
translocates to the nucleus and induces p53-dependent apoptosis
(12); Hcy-induced cell apoptosis
is also involved in the nuclear translocation of GAPDH (13). GAPDH, as an oxidant stress sensor,
contributes to the early stage of apoptosis, during which cellular
signals initiate the translocation of GAPDH into the nucleus.
Siah-1 proteins are a conserved family of E3
ubiquitin ligases that have been implicated in a variety of
cellular processes, including mitosis, DNA damage, tumor
suppression and apoptotic cell death (14–17).
Siah-1 consists of an N-terminal RING domain that can bind to E2
proteins, two novel zinc finger motifs that are involved in
protein-protein interactions, and a C-terminal sequence that can
regulate oligomerization and bind to target proteins (18–20).
GAPDH lacks a nuclear location signal (NLS), whereas siah-1 carries
an NLS motif allowing its translocation into the nucleus (21). As a binding partner of GAPDH,
siah-1 may translocate GAPDH from the cytosol to the nucleus,
contributing to cell death (22).
Glial cells are the most numerous cellular
constituent of the brain parenchyma. They serve a major role in
sustaining the physiological function of this tissue. Therefore,
the present study was undertaken to evaluate the viability of rat
C6 cells exposed to Hcy, mimicking HHcy in vitro. The
involvement of GAPDH/siah-1 nuclear translocation in Hcy-induced
cell damage was investigated in rat C6 cells. The model of
Hcy-induced impairment of C6 cells showed that siah-1 was
significantly upregulated by Hcy. In addition, siah-1 knockdown
using siah-1 small interfering RNA (siRNA) significantly decreased
the Hcy-induced nuclear accumulation of GAPDH/siah-1. These lines
of evidence show that siah-1 is important in the Hcy-induced
inhibition of C6 cell survival.
Materials and methods
Reagents
The following antibodies were used in the present
study: Goat anti-siah-1 antibody (cat. no. sc-5505), mouse
anti-lamin B antibody (cat. no. sc-374015; both Santa Cruz
Biotechnology, Inc., Dallas, TX, USA), mouse anti-GAPDH antibody
(cat. no. ab8245), mouse anti-β-actin antibody (cat. no. ab6276;
both Abcam, Cambridge, MA, USA), rabbit anti-cleaved caspase 3
antibody (cat. no. 9661), rabbit anti-phospho (p)-p53 antibody
(cat. no. 9284), mouse anti-p53 (cat. no. 2524; all Cell Signaling
Technology, Inc., Danvers, MA, USA), IRDye® 800CW goat
anti-mouse immunoglobulin G (IgG) heavy and light chain (H+L; cat.
no. P/N 925–32210), IRDye® 800CW goat anti-rabbit IgG
(H+L; cat. no. P/N 925-32211), IRDye® 800CW donkey
anti-goat IgG (H+L; cat. no. P/N 925-32214; all LI-COR Biosciences,
Lincoln, NE, USA), Alexa Fluor 488-AffiniPure goat anti-mouse IgG
(H+L; cat. no. 115-545-003) and Cy3 AffiniPure rabbit anti-goat IgG
(H+L; cat. no. 305-165-003; both Jackson ImmunoResearch
Laboratories, Inc., West Grove, PA, USA). Other materials used
included D,L-homocysteine (Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany), Cell Counting kit-8 (CCK-8; Dojindo Molecular
Technologies, Inc., Kumamoto, Japan), and TRIzol (Invitrogen;
Thermo Fisher Scientific, Inc., Waltham, MA, USA). All experimental
procedures were performed in accordance with the experimental
standards of Tongji University (Shanghai, China).
Cell culture
The rat C6 astroglioma cells were purchased from the
Cell Bank of Shanghai Institute of Cell Biology, Chinese Academy of
Sciences (Shanghai, China). The cells were maintained in Dulbecco's
modified Eagle's medium (DMEM; Gibco; Thermo Fisher Scientific,
Inc.), supplemented with 10% fetal bovine serum (FBS) and 1%
penicillin/streptomycin (both Gibco; Thermo Fisher Scientific,
Inc.) at 37°C and 5% CO2 in a humidified atmosphere. The
medium was refreshed once every 2–3 days, and cells were passaged
when the cell confluence reached 100%.
Cell proliferation assay
The proliferation of the C6 cells was detected using
a CCK-8 assay. The cells were seeded into 96-well plates (3,000
cells/well). After 24 h, the medium was refreshed and the cells
were treated with Hcy at various concentrations (0, 2, 4, 6, 8 and
10 mM) for 48 h. Following rinsing twice with PBS, 10 µl of CCK-8
solution was added to each well (100 µl). The absorbance was
measured at 450 nm using a microplate reader (Synergy 2; BioTek
Instruments, Inc., Winooski, VT, USA) after 2 h incubation at
37°C.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
Total RNA was isolated from the cells in each group
using TRIzol reagent according to the manufacturer's protocol. The
RNA (2 µg) was reverse transcribed into cDNA with the PrimeScript
RT reagent kit (Takara Biotechnology Co., Ltd., Dalian, China). The
qPCR protocol was used in conjunction with SYBR FAST qPCR Master
mix (KAPA; Roche Diagnostics, Basel, Switzerland). Reaction mix
included: 1 µl cDNA, 10 µl SYBR FAST qPCR master mix, 0.4 µl ROX
High, 0.4 µl primer forward (10 µM) and 0.4 µl primer reverse (10
µM) in a total Volume of 20 µl. The primers were as follows: Siah-1
forward, 5′-CAAAGTGTCCACCATCCCAGAG-3′ and reverse,
5′-GGTGGCAATACATAGTCAAAGCAG-3′; GAPDH forward,
5′-TTCCTACCCCCAATGTATCCG-3′ and reverse,
5′-CATGAGGTCCACCACCCTGTT-3′; β-actin forward,
5′-TGCTATGTTGCCCTAGACTTCG-3′ and reverse,
5′-GTTGGCATAGAGGTCTTTACGG-3′. Quantitation of mRNA expression was
performed on an ABI Prism 7900 Sequence Detection system (Applied
Biosystems; Thermo Fisher Scientific, Inc.). The reaction
conditions were as follows: 95°C for 3 min, 95°C for 3 sec, and
60°C for 30 sec, for a total of 40 cycles; following melting curve
analysis, the reaction conditions were as follows: 95°C for 15 sec,
60°C for 15 sec, and 95°C for 15 sec. All mRNA expression was
normalized to that of β-actin, and the detection was performed in
triplicate. Data normalization was performed using β-actin, and the
2−ΔΔCq method was used to calculate the relative gene
expression level (23).
Western blotting
The cells were placed into radioimmunoprecipitation
assay buffer and total protein was extracted. The nucleic fractions
were prepared using Nuclear and Cytoplasmic Protein Extraction kit
(Beyotime Institute of Biotechnology, Haimen, China). The bovine
serum albumin (BSA; Gibco; Thermo Fisher Scientific, Inc.) was
diluted to make a standard solution. Subsequently, the protein
samples, the standard solution and the bicinchoninic acid assay kit
(Beyotime Institute of Biotechnology) were separately added to the
96-well plate. The samples were incubated at 37°C for 30 min in the
dark. The optical density at 562 nm was measured using an ELISA kit
(ELX-800; BioTek Instruments, Inc.) and the protein concentration
was calculated. A total of 50 µg protein was loaded per lane.
Proteins were separated by 10% SDS-PAGE. Following electrophoretic
separation for 2 h at 80 V, the gel was subsequently transferred
onto a PVDF membrane at 20 mA for 1 h. The membranes were blocked
in 5% BSA (Gibco; Thermo Fisher Scientific, Inc.) at room
temperature for 1 h and subsequently incubated overnight at 4°C
with primary antibodies (anti-β-actin, 1:5,000; anti-lamin B,
1:400; anti-GAPDH, 1:1,000; anti-siah-1, 1:200; anti-p-p53,
1:1,000; anti-p53, 1:1,000; and anti-cleaved caspase 3, 1:1,000).
The free antibodies were washed away with 0.1 M Tris-buffered
saline with 0.1% Tween-20 buffer. The membrane was then incubated
with the appropriate secondary antibody (peroxidase-conjugated goat
antibody anti-rabbit or mouse immunoglobulin G and
peroxidase-conjugated donkey antibody anti-goat immunoglobulin G;
each secondary antibody was diluted 1:2,000) for 1 h at room
temperature. The IRDye-conjugated antibody was scanned with the
Odyssey imaging system (LI-COR Biosciences), analyzed with the
ImageJ software (version 1.8.0; National Institutes of Health,
Bethesda, MD, USA). Lamin B served as the nuclear fractionation
control, β-actin was used to determine total protein
concentration.
Immunofluorescence staining
The cells were treated with Hcy for 48 h. Following
fixation in 4% paraformaldehyde for 30 min at room temperature, the
cells were washed three times with PBS, blocked with 1% BSA and
treated with 0.1% Triton X-100 for 30 min at room temperature.
Then, cells were incubated overnight at 4°C with mouse anti-GAPDH
(1:500) and anti-siah-1 (1:200) prior to the treatment with
secondary antibody (Dylight 488 AffiniPure Goat anti-mouse IgG,
1:200; Cy3 AffiniPure rabbit anti-goat IgG, 1:200) and DAPI (0.2
µg/ml) for 30 min at room temperature. The fluorescence intensity
was measured under a ZEISS LSM710 confocal microscope (Zeiss GmbH,
Jena, Germany; magnification, ×100).
Co-immunoprecipitation assay
The cells were lysed in immunoprecipitation buffer
(protease inhibitor and phosphatase inhibitor mixture) and equal
quantities of protein were obtained from each sample (800 µg). The
Agarose Protein A+G beads were mixed and divided into two groups,
one for removing non-specific binding and one for binding to
antibodies. The sample was pretreated with Agarose A+G, shaken
slowly for 2 h at 4°C, and the pretreated sample was added to a new
EP tube, following which the protein A+G beads were removed. The
anti-siah-1 antibody (3 µg) was added to the total protein to react
with the target protein, with slow agitation of the
antigen-antibody mixture overnight at 4°C. Following washing with
pre-cooled PBS, the immune complexes were detected by western
blotting. GAPDH was used a positive control, eliminating false
positive interference.
siRNA transfection
The cells were transfected with 40 nM siRNA
targeting rat siah-1 (GenePharma, Shanghai, China) in the presence
of Lipofectamine™ 2000 reagent (Invitrogen; Thermo
Fisher Scientific, Inc.) according to the manufacturer's protocol.
The two RNAi oligos targeting siah-1 were 1#-siRNA siah-1:
5′-GCAACAGCCAUCAUGAAUATT-3′ and 2#-siRNA siah-1:
5′-UAUUCAUGAUGGCUGUUGCTT-3′. The medium was refreshed 6 h later and
the cells were maintained for another 24 h. The transfection
efficiency was detected by RT-qPCR and western blot analyses.
Statistical analysis
The data were analyzed with either one-way or
two-way analysis of variance, followed by the Newman-Keuls post hoc
test for pairwise comparisons, using SPSS 16.0 software (SPSS,
Inc., Chicago, IL, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
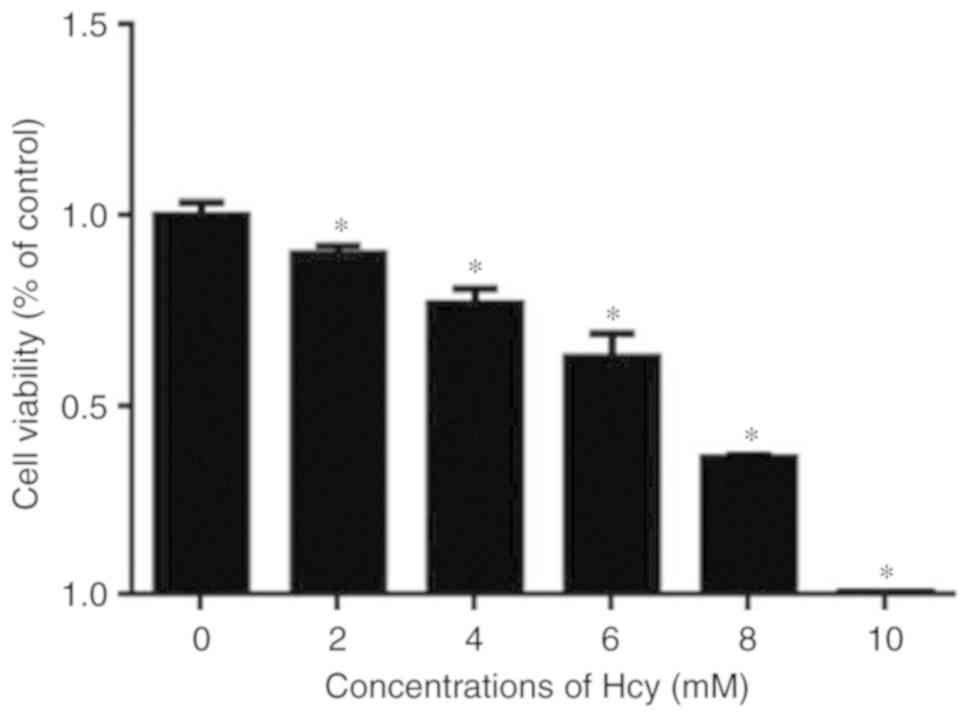
Hcy affects the viability of C6
cells
The viability of C6 cells was determined following
incubation with Hcy at various concentrations for 48 h. The
viability of the Hcy-treated cells was normalized to that of cells
without Hcy treatment. As shown in Fig. 1, Hcy treatment reduced cell
viability in a dose-dependent manner, as assessed using a CCK-8
assay.
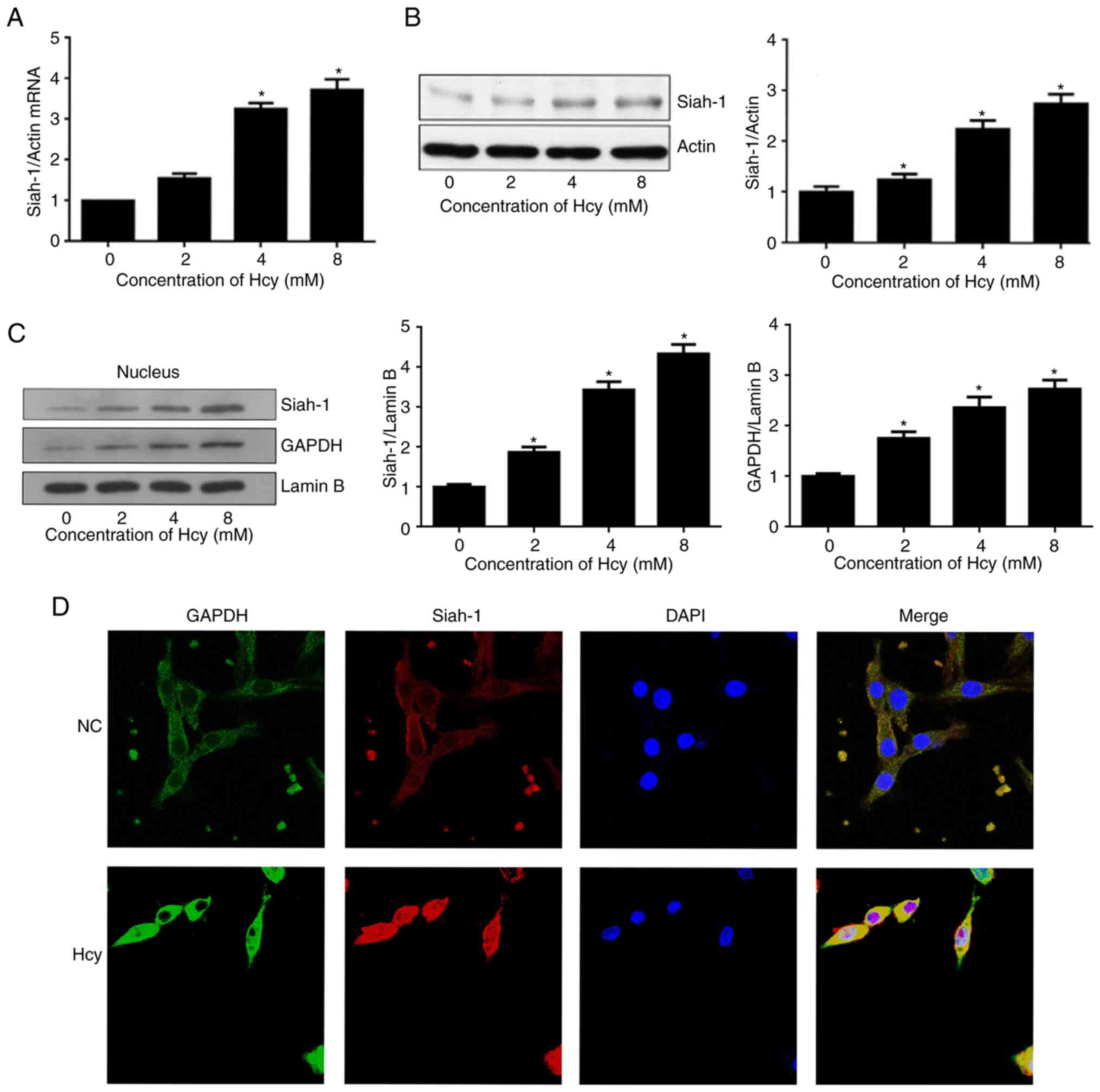
Hcy increases the expression of
siah-1
Siah-1 is expressed in several tissues, including
the brain (24). A previous study
showed that apoptosis-inducing stimuli may markedly elevate
cellular expression of siah-1 (15). Therefore, the expression of siah-1
was also measured following Hcy treatment. The cells were treated
with Hcy at various concentrations for 48 h. The results showed
that Hcy significantly increased the mRNA expression of siah-1
within 48 h compared with the untreated cells (Fig. 2A). In addition, a significant
increase in the protein expression of siah-1 was observed following
Hcy treatment, as shown by the western blotting (Fig. 2B). The increases in the mRNA and
protein expression levels of siah-1 were dependent on the
concentrations of Hcy.
Hcy induces the nuclear accumulation
of siah-1 and GAPDH in C6 cells
To confirm that Hcy induces the nuclear accumulation
of siah-1 and GAPDH, western blotting was used to determine the
expression levels of siah-1 and GAPDH in the nucleus following
treatment with Hcy at 8 mM for 48 h. As shown in Fig. 2C, the nuclear protein expression
levels of siah-1 and GAPDH were significantly increased in a
dose-dependent manner following Hcy treatment. The localization of
siah-1 and GAPDH expressed in the C6 cells was also determined by
immunofluorescence staining. As shown in Fig. 2D, compared with the control cells,
a high proportion of cells showed nuclear expression of siah-1 and
GAPDH following Hcy treatment. Therefore, it was hypothesized that
Hcy promoted the translocation of siah-1 and GAPDH into the
nucleus.
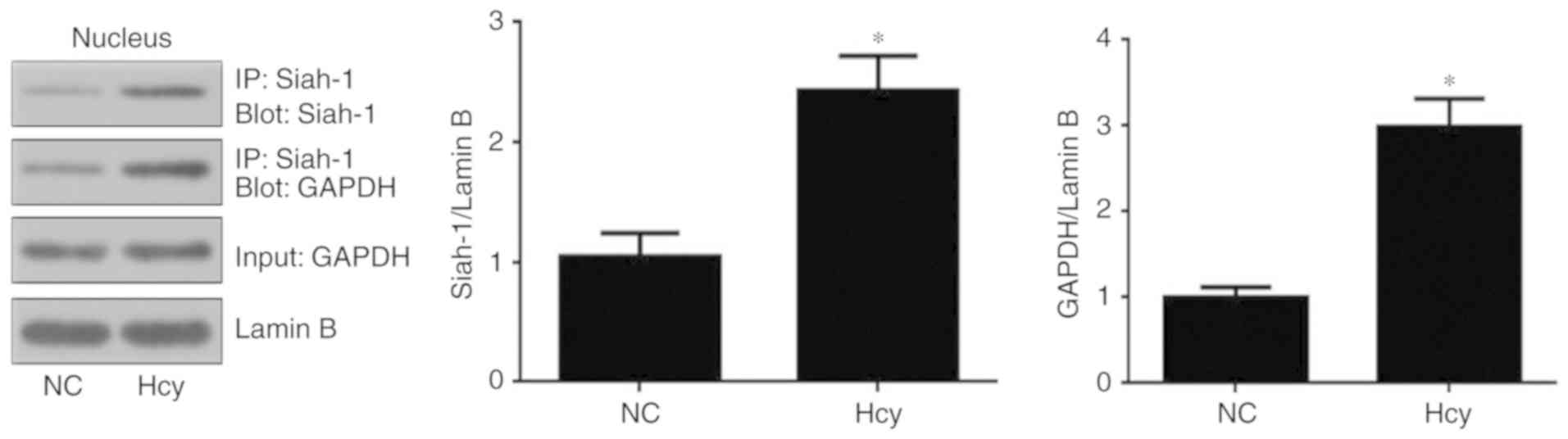
Hcy increases the interaction between
siah-1 and GAPDH
To determine whether Hcy induces the formation of
siah-1-GAPDH complexes, co-immunoprecipitation was performed using
equal quantities of nuclear fractions from cells following
treatment with 8 mM Hcy for 48 h. The results showed that siah-1
and GAPDH formed complexes in response to Hcy treatment. As shown
in Fig. 3, Hcy significantly
induced the formation of GAPDH/siah-1 complexes compared with the
control group.
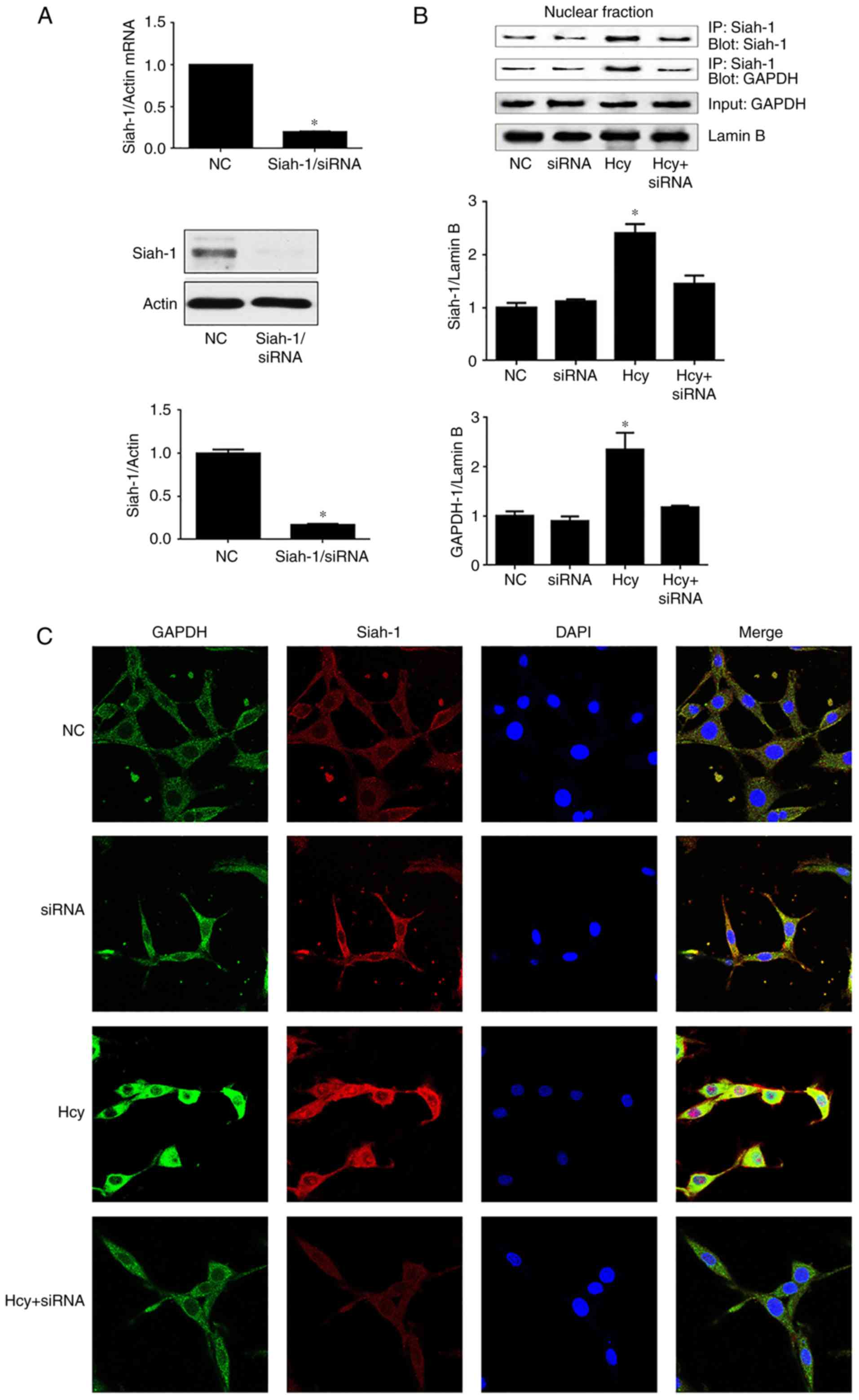
Siah-1 mediates the nuclear
accumulation of GAPDH
To assess whether a decrease in siah-1 is involved
in the nuclear accumulation of GAPDH, the expression of siah-1 was
silenced by siRNA-mediated knockdown in C6 cells, which were then
treated with Hcy at 8 mM for 48 h. Pull-down assays were performed,
as described above. The knockdown efficiency and other quality
control aspects of the siRNA experiments are shown in Fig. 4A. The results demonstrated that
siah-1 siRNA significantly reduced the mRNA and protein levels of
siah-1 by >80% in the C6 cells.
The nuclear interaction between GAPDH/siah-1
following treatment with Hcy was inhibited in C6 cells following
siah-1 silencing (Fig. 4B). In
addition, nuclear aggregation and nuclear translocation of siah-1
and GAPDH was decreased in cells transfected with siah-1 siRNA
compared with the group treated with Hcy (Fig. 4C).
Siah-1 silencing suppresses
Hcy-induced cell death
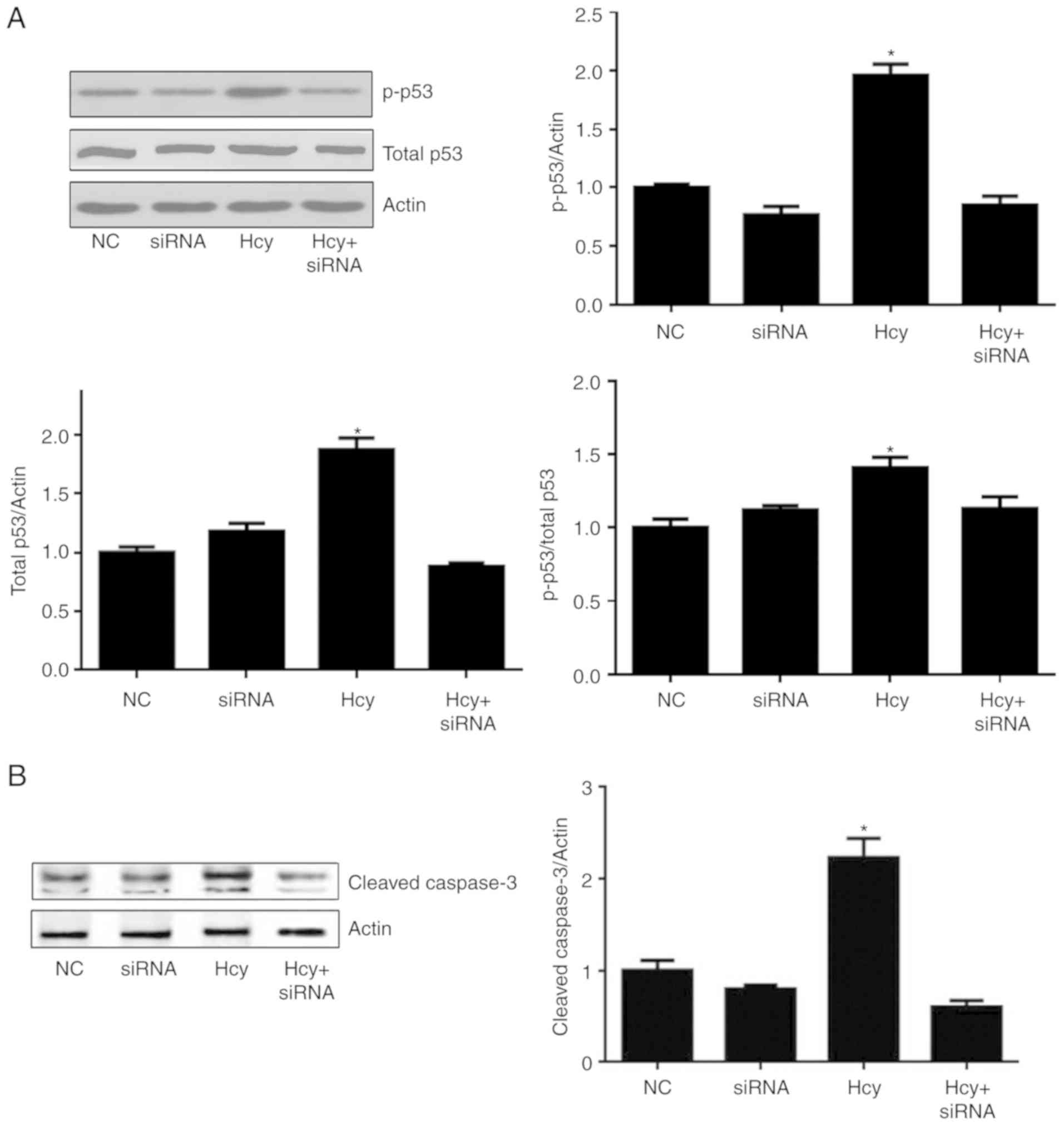
p53 is one of the most important tumor suppressor
factors in cells, and it has a key role in cell cycle regulation,
DNA repair and apoptosis. p53 can be phosphorylated by multiple
protein kinases at multiple sites, and Serine 15 is the most common
phosphorylation site of p53, leading to its transcriptional
activity during cell death. To determine whether siah-1 is critical
for Hcy-induced cell death, the expression levels of phosphorylated
and total p53 protein under different treatment conditions were
measured and normalized with actin, and p-p53 was normalized to
total p53. As shown in Fig. 5A,
Hcy increased the expression levels of p-p53 (ser15) and total p53
compared with levels in the control cells. Following siRNA
interference to knock down the expression of siah-1, the increases
in the expression of p-p53 and total p53 caused by Hcy were
significantly inhibited. The knockdown of siah-1 also inhibited the
phosphorylation of p53 at serine 15. For further confirmation, the
protein levels of cleaved caspase 3 were also measured. The results
showed that 8 mmol/l Hcy significantly upregulated the protein
expression of cleaved caspase 3, whereas siah-1 silencing with
target siRNA significantly reduced the protein expression of
cleaved caspase 3 in the Hcy-treated C6 cells (Fig. 5B).
Discussion
An important pathological feature of
neurodegenerative diseases is the apoptosis and necrosis of cells.
The abnormal apoptosis of nerve cells is the ultimate cause of
neurodegenerative diseases. In order to further investigate the
pathogenesis of neurodegenerative diseases, the present study
established a variety of cell and animal models, through which it
has been confirmed that Hcy can exert its neurotoxic effect in
various nerve cells. The detrimental effects of Hcy on neurons is
well documented in the literature, however, few studies have probed
the effects of Hcy in astrocytes. It is widely recognized that Hcy
can induce cell death, however, the specific molecular mechanism
remains to be fully elucidated. Studies have shown that Hcy-induced
cell death involves DNA damage (25), promotes the expression level of
poly-ADP-ribosome polymerase activation and mitochondrial
dysfunction following the activation of caspase-3 (26,27).
A previous study showed that the interaction between GAPDH and
siah-1 was associated with cell death in neuronal and non-neuronal
cell types (28). Therefore, it
was hypothesized that the cytotoxicity of Hcy may be associated
with its interaction with GAPDH/siah-1. In the present study, the
nuclear accumulation of GAPDH/siah-1 was detected in Hcy-treated C6
cells.
A previous study showed that Hcy concentrations
>2 mM were required to induce cell death (7), however, the mode of action remained
unknown. In the present study, C6 cells were treated with various
concentrations of Hcy to mimic the pathological changes in Hcy. The
results showed that treatment of cells with Hcy >2 mM
significantly reduced their proliferation capacity, and the higher
the concentration of Hcy, the lower the proliferation capacity,
showing a dose-dependent effect.
As a key enzyme in the glycolysis pathway, GAPDH is
also involved in DNA repair, cell membrane fusion and transport,
transcriptional regulation and apoptosis caused by oxidative
stress. In cells, GAPDH acts as an oxidant-stress sensor. It has
been reported that, under oxidative stress, GAPDH translocates to
the nucleus, leading to cell death (29). GAPDH possesses a region homologous
to a nuclear localization sequence motif (NLS), and its nuclear
transport can occur via this NLS (30). A previous study identified siah-1
as a potential carrier/shuttle protein; GAPDH binds the NLS-bearing
siah-1, forming a complex and subsequently promoting the
translocation of GAPDH from the cytosol to the nucleus (22). The present study demonstrated that
the NLS on siah-1 can promote nuclear movement of this complex. The
results showed that Hcy inhibited the survival of C6 cells, which
was at least partially dependent on the nuclear interaction between
GAPDH and siah-1.
Siah-1 is an E3-ubiquitin-ligase involved in the
ubiquitination and proteasome-mediated degradation of proteins, and
it has been shown to be involved in the regulation of a variety of
substrate proteins that are involved in different signal
transduction pathways, including cell cycle, cell differentiation,
apoptosis and neurodegenerative diseases (31). It has been suggested that siah-1 is
a potential metastatic protease of GAPDH from the cytoplasm to the
nucleus. In the present study, it was found that Hcy not only
induced a high expression of GAPDH in C6 cells, but also induced
the nuclear transposition and nuclear aggregation of GAPDH. To
verify the molecular mechanism underlying the nuclear transposition
of GAPDH, the effects of Hcy on the expression and localization of
siah-1 were also observed. The experimental results showed that
when Hcy caused the overexpression of GAPDH, it also induced the
overexpression of siah-1. Therefore, it was concluded that the
nuclear transposition of GAPDH and siah-1 is involved in the
cytotoxic processes.
As a tumor suppressor, siah-1 encodes nuclear
protein and promotes apoptosis (32). It has been demonstrated that siah-1
can bind to GAPDH and then translocate from the cytoplasm to the
nucleus, inducing the accumulation of GAPDH and siah-1 in the
nucleus. The nuclear GAPDH/siah-1 complex may stabilize siah-1 and
enhance its E3 ligase activity, thereby causing cell death
(21). To investigate whether
siah-1 is involved in the nuclear translocation of GAPDH following
Hcy treatment, the expression of siah-1 was silenced with a
specific siRNA. The results showed that silencing siah-1 inhibited
the nuclear accumulation of GAPDH, and inhibited the Hcy-induced
impairment of C6 cells. This suggests that siah-1 is a key factor
in Hcy-induced cell damage.
Suarez et al (28) reported that the association between
GAPDH and siah-1, in turn, results in nuclear translocation and
accumulation of the complex in the nucleus, leading to cell death.
The findings also demonstrated that siah-1 is a novel regulator of
GAPDH. The present study investigated the role of siah-1 in the
Hcy-induced impairment of C6 cells. In the absence of siah-1, the
cytotoxic effect of Hcy against C6 cells was significantly reduced.
p53 is a tumor suppressor protein that regulates the expression of
a variety of genes, including apoptosis, growth inhibition,
differentiation and accelerated DNA repair, genotoxicity and aging
following cellular stress. As the transcriptional activity of p53
is regulated by phosphorylation, the present study examined the
effect of siah-1 knockdown on the phosphorylation of p53 at serine
15. The experiments confirmed that Hcy induced an increase in the
protein expression of p-p53 and total p53 in the cells. Siah-1
knockdown inhibited the phosphorylation of p53, indicating that
siah-1 may regulate the transcriptional activity of p53. The
results suggested that siah-1 may regulate the phosphorylation of
p53 and thereby stabilize p53. The role of siah-1 in this process
has not been determined and may be associated with the transport of
GAPDH to the nucleus. Caspase-3 enzyme activity is a common marker
of apoptosis. In the present study, Hcy significantly upregulated
the expression of cleaved caspase 3, which was inhibited following
siah-1 silencing. This suggested that Hcy caused cell death by an
increase in GAPDH/siah-1 association. The present study is the
first, to the best of our knowledge, to examine the possible
association between Hcy and the nuclear accumulation of siah-1.
Taken together, the results confirmed that Hcy
reduced the activity of C6 cells and induced cell damage. The
overexpression of siah-1 and GAPDH proteins and their nuclear
aggregation were also demonstrated during cell injury. Interference
of the expression of siah-1 can reduce the Hcy-induced nuclear
aggregation of GAPDH and inhibit the Hcy-induced overexpression of
p-p53 (ser15) and caspase 3. The results suggest that siah-1 may be
the key factor in cell damage induced by Hcy, and interfering with
the expression of siah-1 can inhibit the resulting cell injury.
Therefore, interference of the siah-1 gene may be a key molecular
approach to neuroprotection, preventing or delaying the occurrence
and progress of neurodegenerative diseases. Further investigations
are required to clearly identify the role of nuclear siah-1 in the
process of cell damage.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the
National Natural Science Foundation of China (grant nos. 81771131
and 81571033) and by the Science and Technology Commission of
Shanghai Municipality (grant nos. 17411950100 and 17411967500).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YT made substantial contributions to the conception
and design of the study. XZ conducted and supervised the
experiments. XT and LG conducted the experiments and wrote the
manuscript. AJ and YW prepared the figures, analyzed and
interpreted the data, and drafted the manuscript. All authors
critically revised the manuscript, and read and approved the final
version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interest.
References
|
1
|
Kim H and Lee KJ: Serum homocysteine
levels are correlated with behavioral and psychological symptoms of
Alzheimer's disease. Neuropsychiatr Dis Treat. 10:1887–1896.
2014.PubMed/NCBI
|
|
2
|
Kamat PK, Vacek JC, Kalani A and Tyagi N:
Homocysteine induced cerebrovascular dysfunction: A link to
Alzheimer's disease etiology. Open Neurol J. 9:9–14. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sharma M, Tiwari M and Tiwari RK:
Hyperhomocysteinemia: Impact on neurodegenerative diseases. Basic
Clin Pharmacol Toxicol. 117:287–296. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jin Y and Brennan L: Effects of
homocysteine on metabolic pathways in cultured astrocytes.
Neurochem Int. 52:1410–1415. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kruman II, Kumaravel TS, Lohani A,
Pedersen WA, Cutler RG, Kruman Y, Haughey N, Lee J, Evans M and
Mattson MP: Folic acid deficiency and homocysteine impair DNA
repair in hippocampal neurons and sensitize them to amyloid
toxicity in experimental models of Alzheimer's disease. J Neurosci.
22:1752–1762. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Oldreive CE and Doherty GH: Neurotoxic
effects of homocysteine on cerebellar Purkinje neurons in vitro.
Neurosci Lett. 413:52–57. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Maler JM, Seifert W, Hüther G, Wiltfang J,
Rüther E, Kornhuber J and Bleich S: Homocysteine induces cell death
of rat astrocytes in vitro. Neurosci Lett. 347:85–88. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Škovierová H, Mahmood S, Blahovcová E,
Hatok J, Lehotský J and Murín R: Effect of homocysteine on survival
of human glial cells. Physiol Res. 64:747–754. 2015.PubMed/NCBI
|
|
9
|
Turnquist C, Horikawa I, Foran E, Major
EO, Vojtesek B, Lane DP, Lu X, Harris BT and Harris CC: p53
isoforms regulate astrocyte-mediated neuroprotection and
neurodegeneration. Cell Death Differ. 23:1515–1528. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dallérac G and Rouach N: Astrocytes as new
targets to improve cognitive functions. Prog Neurobiol. 144:48–67.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hsu CC, Cheng CH, Hsu CL, Lee WJ, Huang SC
and Huang YC: Role of vitamin B6 status on antioxidant defenses,
glutathione, and related enzyme activities in mice with
homocysteine-induced oxidative stress. Food Nutr Res. 59:257022015.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Itakura M, Nakajima H, Semi Y, Higashida
S, Azuma YT and Takeuchi T: Glyceraldehyde-3-phosphate
dehydrogenase aggregation inhibitor peptide: A potential
therapeutic strategy against oxidative stress-induced cell death.
Biochem Biophys Res Commun. 467:373–376. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fang M, Jin A, Zhao Y and Liu X:
Homocysteine induces glyceraldehyde-3-phosphate dehydrogenase
acetylation and apoptosis in the neuroblastoma cell line Neuro2a.
Braz J Med Biol Res. 49:e45432016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ban R, Matsuzaki H, Akashi T, Sakashita G,
Taniguchi H, Park SY, Tanaka H, Furukawa K and Urano T: Mitotic
regulation of the stability of microtubule plus-end tracking
protein EB3 by ubiquitin ligase SIAH-1 and Aurora mitotic kinases.
J Biol Chem. 284:28367–28381. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Winter M, Sombroek D, Dauth I,
Moehlenbrink J, Scheuermann K, Crone J and Hofmann TG: Control of
HIPK2 stability by ubiquitin ligase Siah-1 and checkpoint kinases
ATM and ATR. Nat Cell Biol. 10:812–824. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Xu Z, Sproul A, Wang W, Kukekov N and
Greene LA: Siah1 interacts with the scaffold protein POSH to
promote JNK activation and apoptosis. J Biol Chem. 281:303–312.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bruzzoni-Giovanelli H, Fernandez P, Veiga
L, Podgorniak MP, Powell DJ, Candeias MM, Mourah S, Calvo F and
Marín M: Distinct expression patterns of the E3 ligase SIAH-1 and
its partner Kid/KIF22 in normal tissues and in the breast tumoral
processes. J Exp Clin Cancer Res. 29:102010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
House CM, Frew IJ, Huang HL, Wiche G,
Traficante N, Nice E, Catimel B and Bowtell DD: A binding motif for
Siah ubiquitin ligase. Proc Natl Acad Sci USA. 100:3101–3106. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
House CM, Hancock NC, Möller A, Cromer BA,
Fedorov V, Bowtell DD, Parker MW and Polekhina G: Elucidation of
the substrate binding site of Siah ubiquitin ligase. Structure.
14:695–701. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
He HT, Fokas E, You A, Engenhart-Cabillic
R and An HX: Siah1 proteins enhance radiosensitivity of human
breast cancer cells. BMC Cancer. 10:4032010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hara MR, Agrawal N, Kim SF, Cascio MB,
Fujimuro M, Ozeki Y, Takahashi M, Cheah JH, Tankou SK, Hester LD,
et al: S-nitrosylated GAPDH initiates apoptotic cell death by
nuclear translocation following Siah1 binding. Nat Cell Biol.
7:665–674. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yego EC and Mohr S: siah-1 protein is
necessary for high glucose-induced glyceraldehyde-3-phosphate
dehydrogenase nuclear accumulation and cell death in muller cells.
J Biol Chem. 285:3181–3190. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lee JT, Wheeler TC, Li L and Chin LS:
Ubiquitination of alpha-synuclein by Siah-1 promotes
alpha-synuclein aggregation and apoptotic cell death. Hum Mol
Genet. 17:906–917. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kruman II, Culmsee C, Chan SL, Kruman Y,
Guo Z, Penix L and Mattson MP: Homocysteine elicits a DNA damage
response in neurons that promotes apoptosis and hypersensitivity to
excitotoxicity. J Neurosci. 20:6920–6926. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tawfik A and Smith SB: Increased ER stress
as a mechanism of retinal neurovasculopathy in mice with severe
hyperhomocysteinemia. Austin J Clin Ophthalmol.
1:10232014.PubMed/NCBI
|
|
27
|
Ho PI, Ortiz D, Rogers E and Shea TB:
Multiple aspects of homocysteine neurotoxicity: Glutamate
excitotoxicity, kinase hyperactivation and DNA damage. J Neurosci
Res. 70:694–702. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Suarez S, McCollum GW, Jayagopal A and
Penn JS: High glucose-induced retinal pericyte apoptosis depends on
association of GAPDH and Siah1. J Biol Chem. 290:28311–28320. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sen N, Hara MR, Kornberg MD, Cascio MB,
Bae BI, Shahani N, Thomas B, Dawson TM, Dawson VL, Snyder SH and
Sawa A: Nitric oxide-induced nuclear GAPDH activates p300/CBP and
mediates apoptosis. Nat Cell Biol. 10:866–873. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kwon HJ, Rhim JH, Jang IS, Kim GE, Park SC
and Yeo EJ: Activation of AMP-activated protein kinase stimulates
the nuclear localization of glyceraldehyde 3-phosphate
dehydrogenase in human diploid fibroblasts. Exp Mol Med.
42:254–269. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhao Y, Li Q, Jin A, Cui M and Liu X: E3
ubiquitin ligase Siah-1 downregulates synaptophysin expression
under high glucose and hypoxia. Am J Transl Res. 7:15–27.
2015.PubMed/NCBI
|
|
32
|
Roperch JP, Lethrone F, Prieur S, Piouffre
L, Israeli D, Tuynder M, Nemani M, Pasturaud P, Gendron MC, Dausset
J, et al: SIAH-1 promotes apoptosis and tumor suppression through a
network involving the regulation of protein folding, unfolding, and
trafficking: Identification of common effectors with p53 and
p21(Waf1). Proc Natl Acad Sci USA. 96:8070–8073. 1999. View Article : Google Scholar : PubMed/NCBI
|