Introduction
Lung cancer is the primary cause of
cancer-associated mortalities worldwide, and non-small-cell lung
cancer (NSCLC) constitutes 85% of all cases of lung cancer
(1,2). Platinum combined with paclitaxel,
gemcitabine and pemetrexed is currently the basic chemotherapy
regimen for lung cancer, but it has a low efficacy and strong side
effects (3). Patients with
epidermal growth factor (EGF) receptor, anaplastic lymphoma kinase
or other cancer-driving gene mutations initially benefit from
targeted therapy, but subsequently experience severe relapse and
drug resistance (4). Therefore,
there is an urgent need for the development of new drugs for
patients with lung cancer.
Gibberellin is a triterpene compound. It was
originally isolated from the induced Bakanae gibberella to yield an
amorphous solid. It is a broad-spectrum plant-growth regulator,
which promotes the growth and development of crops. Gibberellin is
widely distributed in the metabolites of fungi and higher plants.
It is also widely used in vegetable cultivation, agriculture and
horticulture (5).
13-Chlorine-3,15-dioxy-gibberellic acid methyl ester
(GA-13315), a small molecular compound (molecular weight 370.4)
derived from gibberellin, possesses strong anti-angiogenic and
anti-cancer effects. It inhibited recombinant human EGF-induced
chemotactic motility and capillary-like tube formation in primary
cultured human endothelial cells (6). GA-13315, at sub-toxic concentrations,
reversed the multi-drug resistance mediated by ATP binding cassette
subfamily B member 1 (7). It
inhibited proliferation and caused apoptosis in oral cancer
(8). The product of the protein
phosphatase 2 regulatory subunit Bbeta (PPP2R2B) gene belongs to
the phosphatase II regulation subunit B family, which serves
important roles in cell growth and division (9). PPP2R2B methylation is associated with
survival and prognosis in patients with gliomas (10). GA-13315 also suppressed PPP2R2B
expression in H1299 lung cancer cells (8). In vivo studies have indicated
that it had a significant inhibitory effect on the growth of human
colon cancer xenografts in HCT116 nude mice without causing any
apparent side effects (6).
Therefore, GA-13315 exhibited marked potential as a
novel therapeutic agent for lung cancer. However, the underlying
mechanism of GA-13315-mediated apoptosis remains unclear. The
present study used RNA-sequencing (RNA-Seq) to survey the
differentially expressed genes (DEGs) between cancer cells treated
with control vehicles and those treated with GA-13315. The results
indicated that GA-13315 increased tripartite motif containing 67
(TRIM67), NF-κB subunit 2 (NF-κB2) and Fas cell surface death
receptor (FAS) expression. For example, TRIM67 has been
demonstrated to be downregulated in NSCLC cancer cell lines
(11). Genetic variations of
NF-κB2 are significantly associated with NSCLC risk and overall
survival (12). FAS regulates lung
cancer cell apoptosis (13,14).
Additional investigation indicated that TRIM67 promoted the
processing of NF-κB2 into its active form, p52, to enhance the
NF-κB pathway, which serves an important role in GA-13315-induced
apoptosis.
Materials and methods
Cell culture
A549 and H460 lung carcinoma cell lines were
obtained from the Kunming Institute of Zoology, Chinese Academy of
Sciences, and cultured in high glucose Dulbecco's modified Eagle's
medium (DMEM; GE Healthcare) containing 10% fetal bovine serum
(FBS, EMD Millipore) and 1% penicillin-streptomycin (EMD Millipore)
in a 37°C incubator with 5% CO2 (Thermo Fisher
Scientific, Inc.). Cells were grown to 70% confluence prior to use
for subsequent experiments.
Cell viability assay
GA-13315 (purity >95%, measured by proton nuclear
magnetic resonance spectroscopy) was prepared by the School of
Chemical Science and Technology at Yunnan University (Kunming,
China). Cell proliferation, with or without GA-13315 treatment, was
measured using a Cell Counting Kit-8 (CCK-8) assay kit (BD
Biosciences). Briefly, cells were seeded into 96-well plates at
2,000 cells/plate, incubated overnight and treated with various
doses (4, 8, 16 and 32 ng/µl) of GA-13315 or the vehicle control
for 48 h. Then, 10 µl CCK8 solution was added to each well and
cells were incubated for an additional 2.5 h. Optical density was
measured by a microplate reader (Thermo Fisher Scientific, Inc.) at
a wavelength of 450 nm.
RNA-Seq
In total, 2 ml A549 cells (2×105
cells/ml) were plated into a 6-well plate in triplicate. A total of
3 wells were treated with 16 ng/µl GA-13315 (treated), and the
other 3 wells treated with control vehicle (dimethyl sulfoxide;
DMSO) at 37°C for 48 h. RNA was extracted with a RNeasy kit
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol. RNA libraries were prepared using an
Illumina NEB Next Ultra RNA Library Prep Kit (New England Biolabs,
Inc.; cat. no. E7530). Briefly, replicates of 5 µg total RNA were
sheared into fragments (200 nt) and reverse transcribed into cDNA,
which were then blunt-ended and phosphorylated followed by the
addition of single 3′ adenosine moiety and ligated with Illumina
adapters. Libraries were amplified by polymerase chain reaction
(PCR) using a NEB Phusion Polymerase (New England Biolabs, Inc.)
followed by purification. The thermocycling conditions were as
follows: Initial denaturation at 98°C for 30 sec; followed by 13
cycles of 98°C for 10 sec, 65°C for 30 sec, 72°C for 30 sec and by
a final extension step at 65°C for 5 min. The primers used to
amplify the libraries were the following: Forward primer,
5′-AATGATACGGCGACCACCGA-3′; and reverse primer,
5′-CAAGCAGAAGACGGCATACGA-3′. RNA-Seq was conducted using
paired-end, 100 base pair reads by HiSeq 2500 v4 100PE (Illumina,
Inc.).
DEGs
Subsequent to filtering out the low quality reads,
the remaining reads were aligned to the reference genome hg19 using
TopHat2 software (v2.0.8: http://tophat.cbcb.umd.edu/) with default parameters
(15). Fragments per kilobase of
exon model per million fragments mapped was used to quantify the
expression of each gene. Differentially expressed genes were
identified using Cuffdiff software (v2.1.0: http://cole-trapnell-lab.github.io/cufflinks/) by
comparing the GA-13315 treated group to the control vehicle treated
group. Genes that passed the |log2 fold change (FC)|>1.5 and
P<0.05 thresholds were considered to be significant DEGs
(16). Pathway analysis was
performed using Ingenuity Pathway Analysis tools (version 2018;
Qiagen Bioinformatics).
Quantitative (q)PCR
A549 or H460 cells were plated in triplicate into a
6-well plate. A total of 3 wells of cells were treated with 16
ng/µl GA-13315 (Treated) and the other three wells of cells treated
with control vehicle (DMSO) at 37°C for 48 h. RNA was extracted
with RNeasy kit (Invitrogen; Thermo Fisher Scientific, Inc.). Equal
amounts of cDNA were synthesized using the Advantage RT-for-PCR kit
(Clontech, Mountain View, CA). Briefly, the samples were mixed with
random primers and incubated at 65°C for 5 min and then on ice for
at least 1 min. Reverse transcriptase was added to each tube, mix
and incubate at 25°C for 10 min and then 42°C for 50 min, heat
inactivated at 70°C for 15 min, and then kept on ice. A total of 1
ul RNase H was added and the samples were incubated at 37°C for 20
min. The 1st strand cDNA was stored at-20°C until use for qPCR.
qPCR was performed using Power SYBR-Green PCR Master
Mix (Applied Biosystems; Thermo Fisher Scientific, Inc.). All qPCR
performed using SYBR Green was conducted at 50°C for 2 min and 95°C
for 10 min, and then 40 cycles of 95°C for 15 sec and 60°C for 1
min. The specificity of the reaction was verified by melt curve
analysis. GAPDH was used an internal control and all data were
normalized to GAPDH. Control groups for the TRIM67 siRNA
transfection and TRIM67 plasmid assays used negative control siRNA
and control empty vectors, respectively. The relative gene
expression was calculated using 2−ΔΔCq quantification
method (17). The primers used are
summarized in Table SI.
Transient transfection
A set of siRNAs targeting TRIM67
(5′-GTACCATCGACGGTCTTCA-3′; 5′-CCTCGTTGCTCAGTGTGAT-3′;
5′-GCGGAGTTTGATCTGACTT-3′; Shanghai GenePharma Co., Ltd.) targeting
TRIM67 or fluorescein amidite-labeled control siRNA
(5′-TAAGGCTATGAAGAGATAC-3′; cat. no., A07001; Shanghai GenePharma
Co., Ltd.) was dissolved in ddH2O to a final
concentration of 0.05 nmol/l, aliquoted and stored at −20°C. Cells
in the logarithmic growth phase were plated into 6-well plates at
1×105 cells per well. When the cells reached 60–70%
density, transfection with 10 nM siRNA was performed using
Lipofectamine (Invitrogen; Thermo Fisher Scientific, Inc.)
according to the protocol of the manufacturer. The transfection
efficiency of control siRNA was visually inspected under a
fluorescence microscope (magnification, ×200). Culture medium (90%
DMEM + 10% FBS) was replaced 6 h after transfection. A total of 48
h after transfection, cells were harvested and RNA or protein was
extracted for analysis.
For the gene overexpression assays, whole-length
TRIM67 was synthesized and inserted into pcDNA3.1 expression vector
(GenScript). An empty vector was used as a control. When the cells
were at 60–70% confluence, gene transfection (1 µg plasmid) was
performed using Lipofectamine, as aforementioned. Transfection
efficiency was evaluated through visual inspection under a
fluorescence microscope (magnification, ×200) by transfecting 1 µg
expression vector (pEGFP-N1; Clontech Laboratories, Inc.)
containing green fluorescent protein. A total of 48 h after
transfection, cells were harvested and RNA or protein was extracted
for analysis. The transfection efficiency of knockdown or
overexpression was additionally quantified by measuring the density
of the band of western blot analysis using ImageJ software (v1.8.0;
National Institutes of Health) (Fig.
S1).
Cell apoptosis assay
Cells were transfected and GA-13315 (16 ng/µl) was
added 24 h after transfection. At 48 h after transfection, cells
were harvested for the apoptosis assay with FITC Annexin V
Apoptosis Detection Kit I (BD Biosciences). Briefly, adherent cells
were trypsinized, transferred to an Eppendorf tube and collected
via centrifugation for 10 min at 4°C at 300 × g. Cell pellets were
washed twice with pre-cooled PBS and rinsed once with 1X binding
buffer (10X; double vapor dilution). Cells were additionally
suspended in PBS to a concentration of 1×106/ml,
following which 100 µl cell suspensions (1×105) were
transferred to centrifuge tubes, mixed with 5 µl fluorescein
isothiocyanate-Annexin V and 10 µl propidium iodide, and incubated
for 15 min at room temperature in the dark. Then, 400 µl 1X binding
buffer was added to the cells 1 h prior to flow cytometry. The data
were analyzed using FlowJo software (version 10; BD Biosciences).
To investigate the role of NF-κB pathway in GA-13315 induced
apoptosis, A549 cells (2×105 cells/ml; 2 ml) were plated
into a 6-well plate in triplicate and incubated in a cell culture
incubator at 37°C and 5% CO2. After 24 h, cells were
treated with control vehicle, GA-13315 (16 ng/µl), and GA-13315
plus 500 µM NF-κB essential modulator-binding domain (NBD;
ABclonal, Inc.) for 48 h. Cells were subsequently harvested to
measure the level of apoptosis.
Western blot analysis
Total protein from the A549 cells was extracted
using radioimmunoprecipitation assay lysis buffer (Beyotime
Institute of Biotechnology) and quantified using a BCA Protein
Assay kit (Beyotime Institute of Biotechnology). Equal amounts of
proteins (15 µg) were separated via 10% SDS-PAGE and transferred
onto a 0.45 µm polyvinylidene difluoride (PVDF) membrane (EMD
Millipore) through a wet trans-blot system (Bio-Rad Laboratories,
Inc.). The membranes were then blocked for 1 h at room temperature
in 5% nonfat dried milk diluted in TBST (Bio-Rad Laboratories,
Inc.), incubated overnight at 4°C with rabbit polyclonal antibodies
against TRIM67 (1:250; cat. no., PA5-42274; Thermo Fisher
Scientific, Inc.), Fas (1:1,000; cat. no., ab82419; Abcam), or
NF-κB2 (1:2,000; cat. no., 37359; Cell Signaling Technology), and
then with a goat anti-rabbit horseradish peroxidase-conjugated
secondary antibody (1:3,000; cat. no., 12-348; Sigma-Aldrich; Merck
KGaA) for 1 h at room temperature, and analyzed using an enhanced
chemiluminescence (ECL) detection kit (Applygen Technologies,
Inc.). β-actin (1:2,000; cat. no. ab8227; Abcam) was used as the
internal control. The density of the bands was analyzed using
ImageJ software (v1.8.0; National Institutes of Health).
Dual luciferase report assay
Wild type FAS promoter (−1,025 to +25 bp) or mutant
(without NF-κB binding sites) was synthesized and inserted into a
pGL3-Basic vector (GeneScript). A FAS promoter activity assay was
performed using a dual luciferase reporter system (Promega
Corporation), according to the manufacturer's protocol. Briefly,
10,000 cells in 0.5 ml DMEM culture medium (GE Healthcare) were
seeded into 24-well plates and incubated at 37°C until the cells
had attached. Each well was co-transfected with 200 ng luciferase
plasmids (pGL3-Basic plasmids without the promotor region as the
negative control or plasmids containing FAS promoter with/without
NF-κB binding sites) and 5 ng pRL-TK plasmids (Promega Corporation)
encoding Renilla, which was used as the internal control to assess
transfection efficiency. At 24 h after transfection, whole-cell
lysates were collected and measured using a GloMax Microplate
Luminometer (Promega Corporation). Transfections were performed in
triplicate and then repeated 3 times to assure reproducibility. All
luciferase reads were normalized to the corresponding Renilla
value. Relative luciferase activity was calculated using the
following formula: Relative luciferase levels =
Luc-promoter/Luc-pGL3 basic.
Chromatin immunoprecipitation (ChIP)
assay
A ChIP assay was performed with a ChIP-IT kit (cat.
no. 53040; Active Motif). A total of 1×107 cells were
cross-linked and lysed. Chromatin was extracted and sonicated to
200–800 bp length fragments with 8 rounds of 10 sec pulses using
25% power. Normalized inputs of sheared chromatin DNA were
incubated with 3 µg negative control immunoglobulin G (1:30; cat.
no. ab171870; Abcam) or a p52 antibody (1:30; cat. no. ab7972;
Abcam) overnight at 4°C. The immunocomplex was treated with RNase A
to remove RNA and incubated at 65°C for 4 h to remove cross-links.
Proteins were removed by treating with proteinase K, and DNA was
purified and subjected to PCR detection following the
aforementioned protocol. Primers targeting the FAS promoter
(forward, CTCCATTCTCCTTCAAGACCT; reverse, GTGTGTCACTCTTGCGCGAGAT)
were used to evaluate the binding of the p52 antibody.
Statistical analysis
Data were analyzed by SPSS 19.0 software (SPSS
Inc.). and are presented as the mean ± standard deviation.
Student's t-tests were performed to evaluate the significance of
difference between two groups. Differences between multiple groups
were analyzed using analysis of variance followed by Tukey's
honestly significant difference post-hoc test was used to evaluate
the differences between groups. P<0.05 was considered to
indicate a statistically significant difference.
Results
RNA-Seq identified TRIM67, NF-κB2 and
FAS as differentially expressed genes induced by GA-13315
A cell viability assay indicated that GA-13315
caused ~50% A549 cell death at a concentration of 16 ng/µl
(Fig. 1A). Therefore, this
concentration was selected for subsequent experiments. Upon
GA-13315 treatment, a total of 250 genes were identified to be
significantly differentially expressed (FC≥1.5; P<0.05), of
which 94 genes were upregulated and 156 genes were downregulated
(Fig. 1B). A detailed list is
presented in Table SII. The
TRIM67, NFKB2 and FAS genes were selected for subsequent
investigation as they were enriched in pathway analysis. In total,
seven genes were randomly selected for qPCR verification. The
expression levels of these 10 genes were additionally confirmed
using qPCR, including TRIM67 (3.1), F-box protein 15 (2.5), NF-κB2
(2.4), Fas cell surface death receptor (3.2), baculoviral IAP
repeat containing 3 (1.9), pyruvate dehydrogenase kinase 1 (−2),
cyclin dependent kinase inhibitor 2D (−2.1), Fos proto-oncogene,
AP-1 transcription factor subunit (−3), JunB proto-oncogene, AP-1
transcription factor subunit (−2.1) and BAF chromatin remodeling
complex subunit BCL11B (−3.2) (Fig.
1C). TRIM67 has been demonstrated to be downregulated in NSCLC
cancer cell lines (11), and
pathway analysis from the present study indicated that NF-κB and
FAS pathways were involved in this process (Fig. S2). Therefore, the protein levels
of TRIM67, FAS and NF-κB2 were also examined, and it was identified
that all 3 were increased (Fig.
1D), thereby confirming the qPCR results.
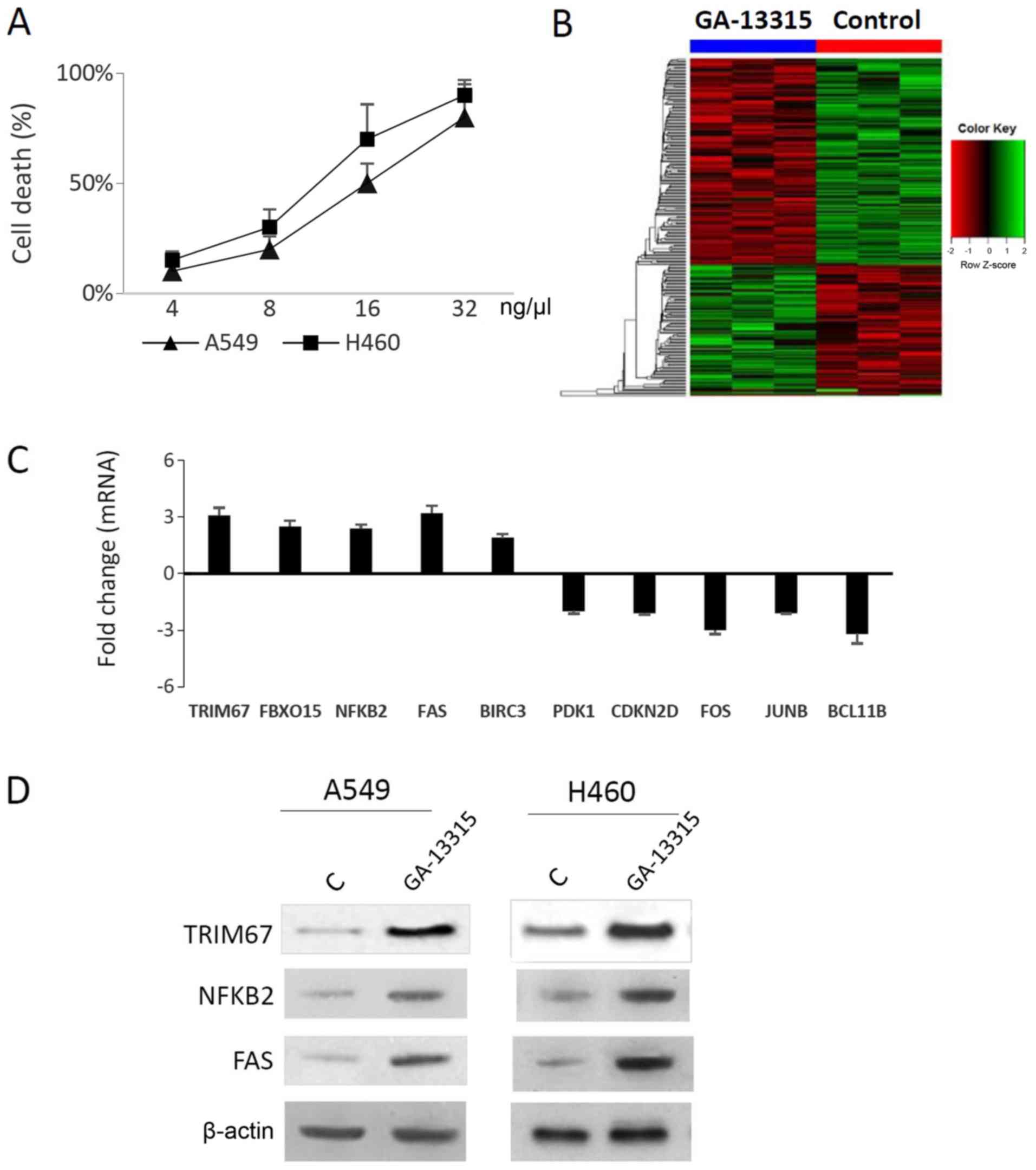 | Figure 1.RNA-seq identifies several
differentially expressed genes. (A) The lung cancer A549 and H460
cell lines were used to evaluate the effect of GA-13315. Cells were
treated with control vehicle (DMSO) or GA-13315 for 48 h, and cell
viability was measured. Cell death rate was plotted against the
GA-13315 concentration to reveal the median lethal dose for each
cell line. (B) RNA-seq identified 250 differentially expressed
genes (P<0.05; fold change >1.5). Hierarchical clustering was
performed and the differentially expressed genes were summarized as
a heatmap. (C) Quantitative polymerase chain reaction confirmed
that TRIM67, FBXO15, NF-κB2, FAS and BIRC3 were overexpressed;
while PDK1, CDKN2D, FOS, JUNB and BCL11B were downregulated in
GA-13315-treated A549 cells. (D) Western blot analysis. Total
proteins from the vehicle control (DMSO) and GA-13315-treated cell
lines were examined using TRIM67, NF-κB2 or FAS antibody. β-actin
was used as the loading control. The results indicated that TRIM67,
NF-κB2 and FAS protein levels were increased in both A549 and H460
cells. RNA-seq, RNA-sequencing; GA-13315,
13-chlorine-3,15-dioxy-gibberellic acid methyl ester; DMSO,
dimethyl sulfoxide; TRIM67, tripartite motif containing 67; FBXO15,
F-box protein 15; NF-κB2, NF-κB subunit 2; FAS, Fas cell surface
death receptor; BIRC3; baculoviral IAP repeat containing 3; PDK1,
pyruvate dehydrogenase kinase 1; CDKN2D, cyclin dependent kinase
inhibitor 2D; FOS, Fos proto-oncogene, AP-1 transcription factor
subunit; JUNB, JunB proto-oncogene, AP-1 transcription factor
subunit; BCL11B, BAF chromatin remodeling complex subunit
BCL11B. |
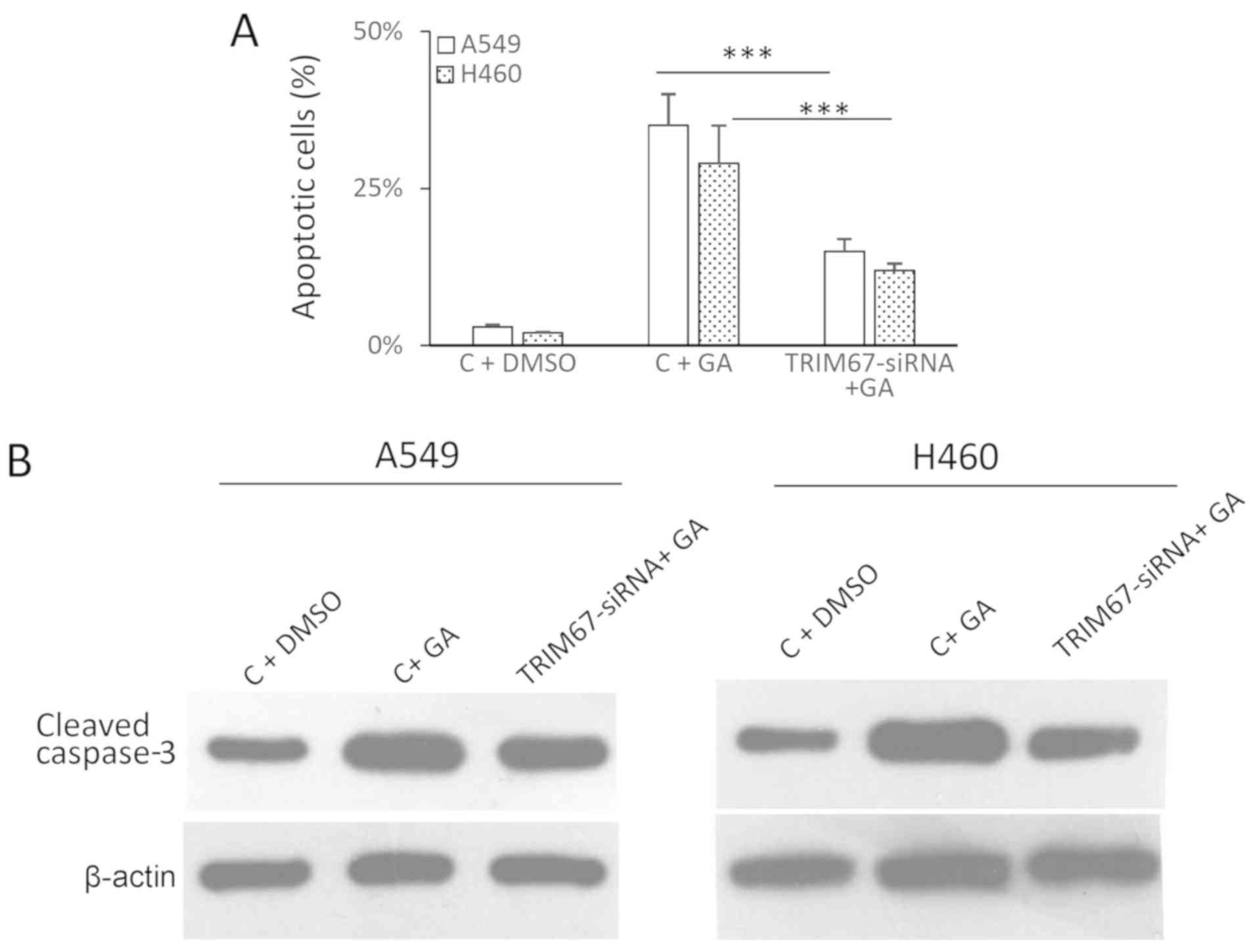
TRIM67 mediates GA-13315-induced cell
apoptosis
To investigate the association between TRIM67 and
GA-13315-induced apoptosis, TRIM67 expression was silenced using
siRNA and the levels of apoptosis were assessed using Annexin V
staining and cleaved caspase-3. Annexin V staining indicated that
silencing TRIM67 decreased the GA-13315-induced apoptosis rate from
50 to 15% in A549 cells (P<0.001) and from 35 to 12% in H460
cells (P<0.001; Fig. 2A).
Exemplary images of flow cytometry are presented in Fig. S3. This observation was
additionally confirmed by western blot analysis, which indicated
that the silencing of TRIM67 decreased the levels of
GA-13315-induced cleaved caspase-3 in both cell lines examined
(Fig. 2B). Therefore, these
results suggested a role for TRIM67 in mediating GA-13315-induced
apoptosis.
TRIM67 promotes NF-κB2 processing and
enhances the NF-κB pathway
Pathway analysis indicated that the NF-κB complex
was enhanced in GA-13315-treated cells. As NF-κB2 is a precursor
protein that requires additional processing to convert into its
active forms (18), and TRIM67
belongs to the TRIM family and possesses ubiquitin E3 ligase
activity (19), whether TRIM67
affected NF-κB2 processing was examined. Forced overexpression of
TRIM67 increased p52 but decreased the levels of NF-κB2 precursor
p100 (Fig. 3A), while knockdown of
TRIM67 increased p100 levels but decreased p52 levels (Fig. 3B), suggesting that TRIM67 promoted
NF-κB2 processing. The results from the qPCR analysis indicated
that the alteration of TRIM67 had no effect on the NF-κB2 mRNA
level (Fig. 3C and D), suggesting
that TRIM67 regulates NF-κB2 at the protein level.
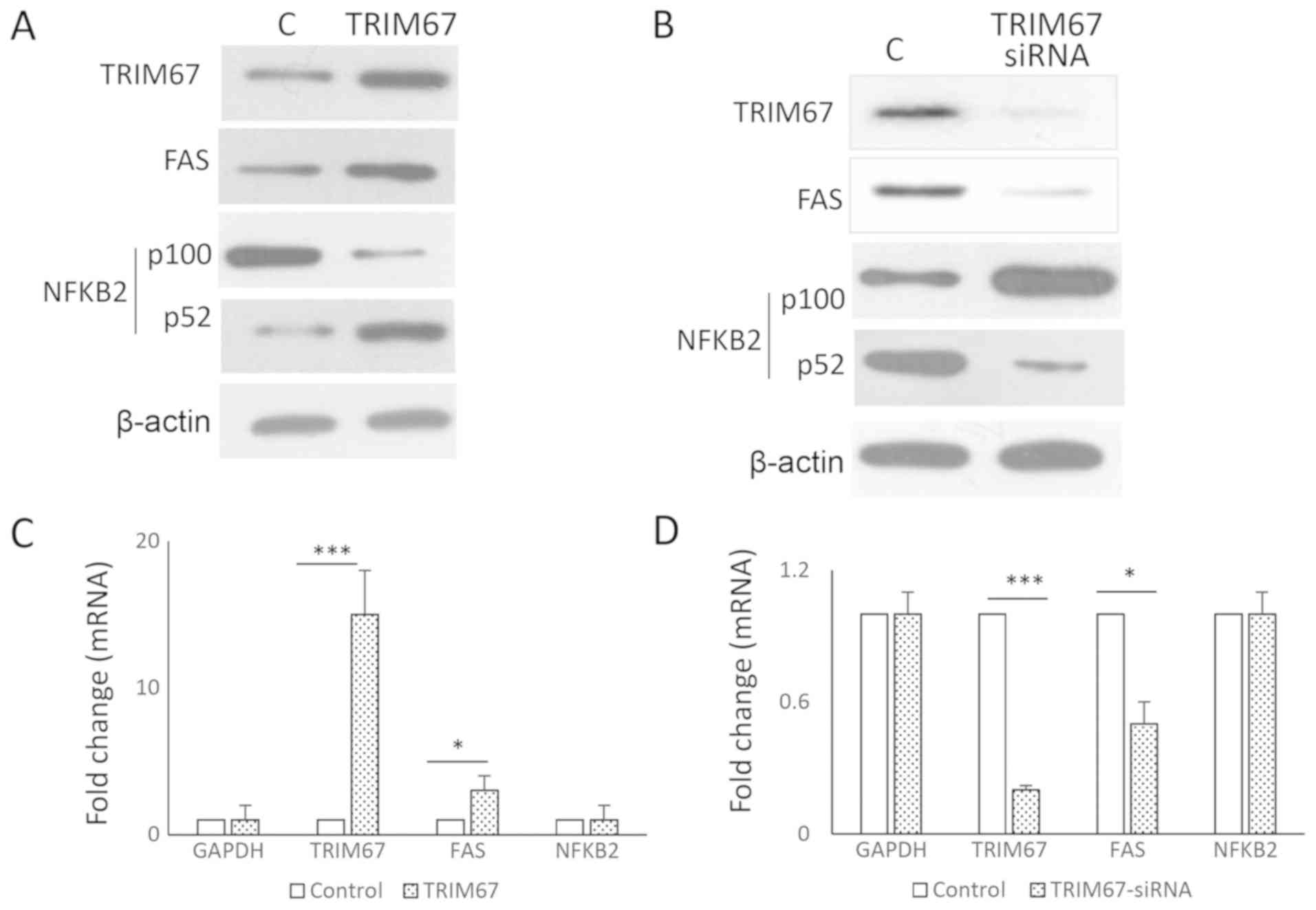 | Figure 3.TRIM67 promotes NF-κB2 processing.
(A) A549 cells were transfected with either control (empty vector)
or TRIM67 plasmid. Proteins were harvested 24 h after transfection
for western blot analysis. The results indicated that TRIM67
overexpression decreased the levels of p100, the precursor of
NF-κB2, but increased p52, the active form of NF-κB2. FAS protein
levels were also increased. (B) By contrast, TRIM67 siRNA decreased
p52 and FAS levels, but increased p100 protein levels. (C) A549
cells were transfected with either control (empty vector) or TRIM67
plasmid. Total RNA was extracted and qPCR was performed using GAPDH
as the internal control. Student's t-test was used to evaluate the
gene expression difference between two groups (control vs. TRIM67
plasmid). Results are expressed as the mean ± standard deviation.
*P<0.05 and ***P<0.001. (D) A549 cells were transfected with
either control (non-targeting siRNA) or TRIM67 siRNA. Total RNA was
extracted and qPCR performed to evaluate gene expression. GAPDH was
used as internal control. Student's t-test was used to evaluate the
gene expression difference between two groups (control vs. TRIM67
siRNA). Results are expressed as the mean ± SD. *P<0.05 and
***P<0.001. TRIM67, tripartite motif containing 67; NF-κB2,
NF-κB subunit 2; FAS, Fas cell surface death receptor; siRNA, small
interfering RNA; qPCR, quantitative polymerase chain reaction. |
GA-13315 induces FAS upregulation and
cell apoptosis via the NF-κB pathway
Whether the NF-κB pathway is involved in
GA-13315-induced FAS upregulation and apoptosis was then
investigated. An NF-κB essential NBD peptide was used to inhibit
the NF-κB pathway, and it was identified that it suppressed the
GA-13315-induced cell apoptosis rate, decreasing it from 35 to 12%
in A549 cells (P<0.001) and from 32 to 11% in H460 cells
(Fig. 4A). The anti-apoptotic
effects of NBD peptide on GA-13315-induced cell apoptosis were
additionally confirmed by western blot analysis on cleaved
caspase-3 (Fig. 4B). Both RNA and
protein levels of FAS were decreased, suggesting that
GA-13315-induced apoptosis involved NF-κB-induced FAS (Fig. 4B and C). The effect of NBD peptide
on TRIM67 and NFBK2 were also examined; the results indicated that
neither mRNA nor protein levels were changed (Fig. 4B and C).
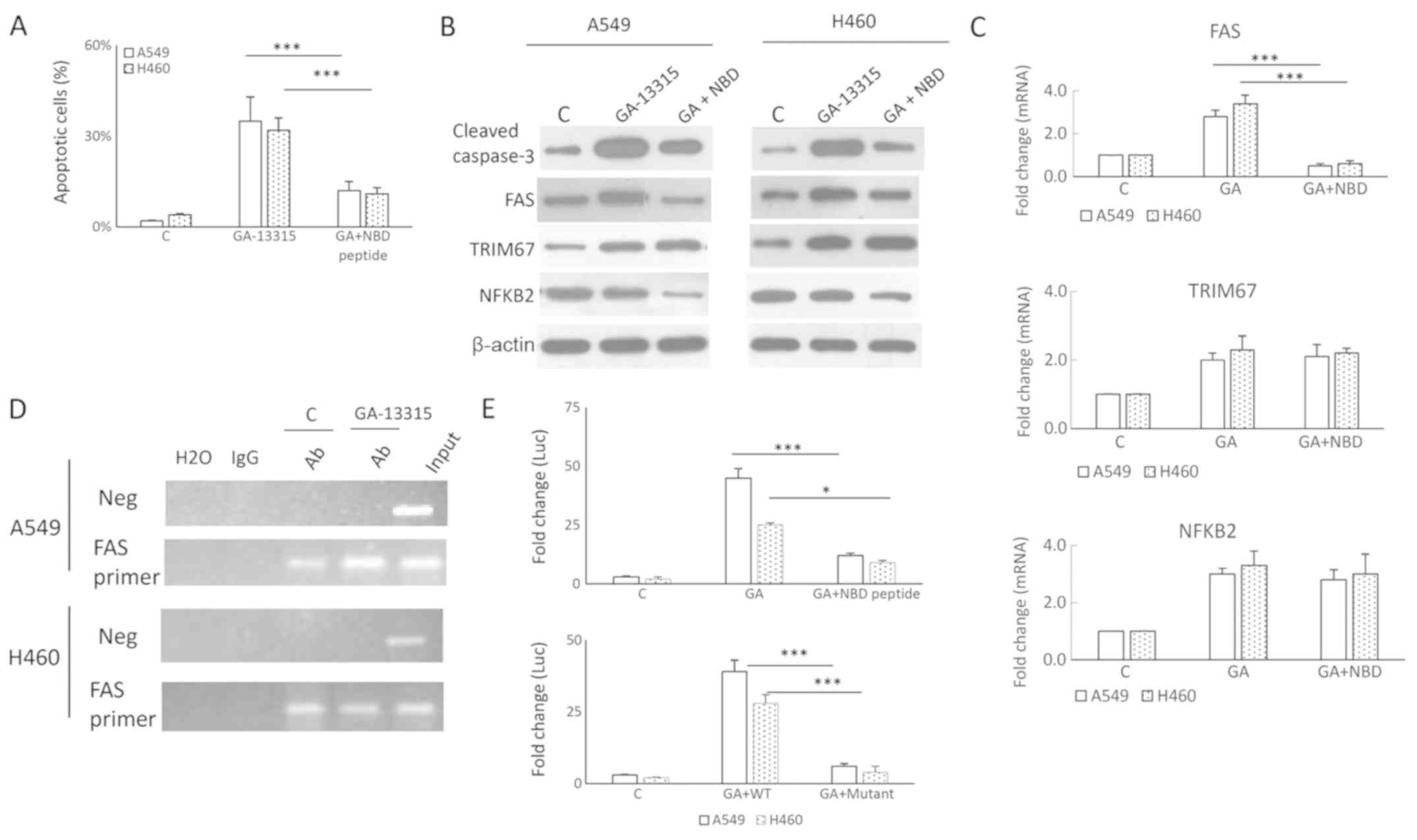 | Figure 4.NF-κB mediates GA-13315-induced FAS
and apoptosis. (A) NF-κB pathway inhibitor NBD peptide suppressed
GA-13315-induced apoptosis, as measured by an Annexin V assay. The
apoptosis rate was calculated by dividing Annexin V-positive cells
by total cells. A one-way ANOVA followed by Tukey's honestly
significant difference post-hoc test was used to evaluate the
difference between the groups in each cell line. Results are
expressed as the mean ± SD. ***P<0.001. (B) NBD peptide
treatment decreased the levels of GA-13315-induced cleaved
caspase-3. (C) A quantitative polymerase chain reaction assay
indicated that NBD suppressed GA-13315-induced FAS mRNA expression,
but exhibited no effects on the mRNA levels of TRIM67 and NF-κB2.
(D) Chromatin immunoprecipitation demonstrated that GA-13315
treatment increased p52 binding to the FAS promoter. (E) A
luciferase reporter vector containing the FAS promoter (FAS-luc)
was used to evaluate activity changes in the FAS promoter. Cells
treated with the control vehicle or GA-13315 were co-transfected
with an empty vector or FAS-luc. Raw data were first normalized to
the Renilla signal, and fold change was calculated by dividing the
FAS-luc values by empty vector values. The results indicated that
deletion of the NF-κB binding site on the FAS promoter decreased
promoter activity induced by GA-13315. A one-way ANOVA followed by
Tukey's honestly significant difference post-hoc test was used to
evaluate the difference between groups in each cell line. Results
are expressed as the mean ± SD. *P<0.05 and ***P<0.001. NBD,
NEMO-binding domain; ANOVA, analysis of variance; SD, standard
deviation; FAS, Fas cell surface death receptor; GA/GA-13315,
13-chlorine-3,15-dioxy-gibberellic acid methyl ester; TRIM67,
tripartite motif containing 67; NF-κB2, NF-κB subunit 2; Ab,
antibody; Neg, negative control. |
There is an NF-κB binding motif on the FAS promoter.
A ChIP assay was used to investigate whether GA-13315 promoted
NF-κB binding to the FAS promoter. As indicated in Fig. 4D, the binding of NF-κB to the FAS
promoter was increased. A luciferase reporter assay was also used
to determine whether GA-13315 activated FAS through NF-κB.
Luciferase reporter plasmids containing the FAS promoter with or
without NF-κB binding site were used. The results indicated that
the FAS promoter activity was increased by 45-fold in A549 cells
and 25-fold in H460 cells upon GA-13315 treatment. Treatment with
the NF-κB inhibitor NDB peptide decreased the induction to 15-fold.
Deletion of NF-κB binding site also markedly decreased the
luciferase activity (Fig. 4E),
confirming the critical role of NF-κB in mediating GA-13315-induced
FAS. Similar results were observed in the lung cancer H460 cells.
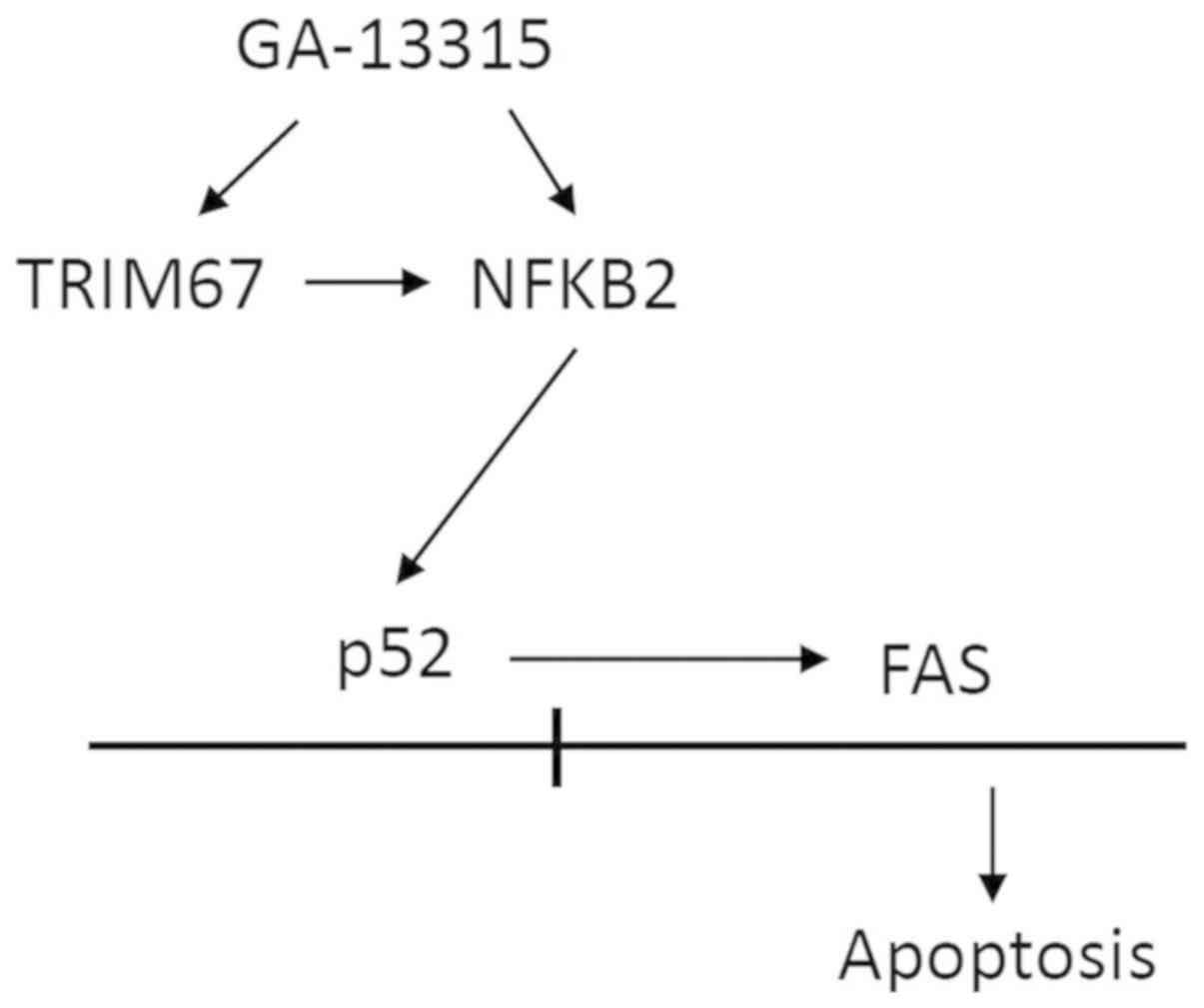
Colelctively, the present study suggested that GA-13315 induced
apoptosis via TRIM67, NF-κB2 and FAS. In addition, TRIM67 promoted
the NF-κB pathway and enhanced FAS expression (Fig. 5).
Discussion
The present study identified 250 differentially
expressed genes between GA-13315-treated A549 lung cancer cells and
controls. TRIM67, NF-κB2 and FAS expression were confirmed to be
increased at both the mRNA and protein levels upon GA-13315
treatment. Pathway analysis indicated a central role for the NF-κB
complex in GA-13315-induced apoptosis. Additional investigation
indicated that TRIM67 promoted NF-κB2 gene processing, which in
turn promoted the NF-κB pathway and subsequently increased FAS
expression.
In order to gain a general understanding of the
observed effects of GA-13315 in lung cancer, 2 distinct lung cancer
cell lines (A549 and H460) were used. It was demonstrated that
knockdown of TRIM67 resulted in a significant decrease of FAS and
GA-13315-induced apoptosis, suggesting a critical role for TRIM67
in regulating apoptosis. In addition, the NF-κB inhibitor (NBD
peptide) also decreased GA-13315-induced apoptosis, suggesting a
key role for NF-κB in GA-13315-induced apoptosis. Suppressing
TRIM67 expression also inhibited the NF-κB pathway. TRIM67
knockdown decreased the active form of NF-κB2, p52, suggesting that
TRIM67 promoted the processing of NF-κB2 into its active form, p52,
which may then enhance NF-κB signaling. As GA-13315 already
enhanced TRIM67 overexpression, the additional overexpression
exhibited limited effects on p100 and p52 protein levels following
exposure to GA13315 (data not shown). The role of NF-κB2 (p52) in
mediating GA-13,315-induced FAS was demonstrated at multiple
levels. Firstly, GA-13315 increased NF-κB2 (p52) binding to the FAS
promoter. Secondly, deletion of the KB binding motif from the FAS
promoter decreased GA-13315-induced FAS promoter activity in a
luciferase reporter assay. Finally, inhibition of the NF-κB
activator IκB kinase using NBD peptide decreased GA-13315-induced
FAS expression. A number of chemopreventive compounds, in
particular dietary compounds, including phenolic compounds,
carotenoids, iridoids, nitrogen compounds, organosulfur compounds,
phytosterols, essential oil compounds, polyunsaturated fatty acids
and dietary fiber induce apoptosis of cancer cells. The
proapoptotic properties of GA-13315 suggest potential
chemopreventive effects.
TRIM67, located at chromosome 11q22-23, encodes a
783 amino acid protein with ListBox zinc-finger motifs and an
adjacent leucine-zipper motif. TRIM67 was identified to be
significantly downregulated in NSCLC cell lines (11), which was additionally confirmed by
Hao et al (20), who
suggested that TRIM67 may be a tumor suppressor gene. The functions
of TRIM67 are largely unclear. Studies investigating TRIM protein
members have indicated that TRIM proteins belong to a family of
structurally conserved, rapidly evolving proteins (21,22).
TRIM proteins, as a type of E3 ubiquitin-linked enzyme, participate
in cell cycle regulation, cell apoptosis, signal transduction,
protein processing and transportation, proteasome-mediated protein
degradation and responses to viral infection, all of which are
important biological processes (21,22).
The results of the present study indicating that TRIM67 promoted
the NF-κB pathway are consistent with previous data that TRIM
family members regulate the NF-κB pathway (19,23).
The results of the present study are also consistent
with the previous data that NF-κB regulates FAS transcription and
FAS-mediated apoptosis (24).
There are two types of NF-κB pathways, the canonical and
non-canonical pathways, distinguished by the core components of
NF-κB transcription factors (25).
In the canonical pathway, following stimulation from Toll-like
receptor ligands and inflammatory cytokines such as tumor necrosis
factor and interleukin 1β, NF-κB1 (p50), which is processed from
its precursor form NF-κB1 (p105), forms a heterodimer with
transcription factor p65 and proto-oncogene c-Rel (26,27).
The non-canonical pathway is activated by ligands for lymphotoxin β
receptor, receptor activator of NF-κB and cluster of
differentiation 40, which induce NF-κB2 (p52) processing from the
(p100) precursor and heterodimerization with transcription factor
RelB (28,29). NF-κB dimers recognize κB-binding
motifs on the promoters of target genes, and then activate or
repress their transcription in a context-dependent manner (26,27).
As NF-κB2 belongs to the non-canonical NF-κB pathway, the results
of the present study additionally suggested that the non-canonical
NF-κB pathway may similarly regulate apoptosis, and that TRIM67 is
likely a new regulator of the non-canonical NF-κB pathway, which
warrants further investigation. NF-κB is involved in drug-induced
apoptosis and the promotion of cancer aggressiveness, depending on
the signaling context (30,31),
which may be the reason that the upregulation of NF-κB targeted
genes, including tumor protein P53, was not observed in the RNA-Seq
analysis of GA-13315-treated cells. Other TRIM family members may
also interact with NF-κB to regulate apoptosis, which requires
additional investigation.
The results of the present study also indicated that
the apoptosis rate of A549 cells increased significantly along with
the increase in FAS expression levels following treatment with
GA-13315. However, various death receptors have been identified to
serve a role in apoptosis signaling. Tumor necrosis factor receptor
1 (32), cytopathic avian receptor
1 (33), nerve growth factor
receptor (34), death receptor
(DR)3 (35), DR4 and DR5 (36) are present, in addition to the most
common FAS/FASL. Whether other cell death pathways are also
involved requires further investigation. Similarly, TRIM67 may
regulate other signaling pathways to modulate GA-13315-induced
apoptosis. TRIM67 has been demonstrated to modulate the RAS
signaling pathway in neural cells (37). Whether the TRIM67-RAS signaling
pathway is involved in regulating GA-13315-induced apoptosis also
requires further investigation.
The present study revealed novel mechanisms
underlying GA-13315-induced apoptosis. Due to limited resources,
the experimental design may not be ideally rigorous, and therefore,
the results should be interpreted with caution. For example, there
might be other mechanisms for GA-13315-induced apoptosis that
require verification. The evaluation of GA-13315-induced apoptosis
in additional cancer cell lines may provide an improved
understanding of the mechanisms involved. How GA-13315 increases
TRIM67 and NF-κB2 gene expression, and how TRIM67 regulates NF-κB2
processing, require additional detailed investigation.
The present study indicated that GA-13315-induced
apoptosis involved TRIM67, NF-κB2 and FAS. TRIM67 promoted the
NF-κB pathway and enhanced FAS expression, which may be an
underlying mechanism for GA-13315-induced apoptosis in lung cancer
cells.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81460559) and the
Key Scientific and Technological Achievements Cultivation Project
of Kunming Medical University (grant no. CGPY201502).
Availability of data and materials
The RNA-Seq data, and qPCR primer sequence and
pathway analysis results are included as supplemental data
(Tables SI and SII, Figs.
S1, S2 and S3). All other data and materials are
available upon request.
Authors' contributions
JC synthesized and provided the compound GA-13315.
CQ, JC and RL conceived and supervised the study and RL and CQ
designed the experiments. CQ, RL and YC performed the experiments,
and YC provided reagents. TS and JH analyzed the data. RL, YC and
JH wrote the manuscript, and CQ, JC and YC edited and revised the
manuscript. All authors discussed and reviewed the results, and
approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
NSCLC
|
non-small-cell lung cancer
|
|
EGF
|
epithelial growth factor
|
|
FC
|
fold change
|
|
PVDF
|
polyvinylidene difluoride
|
References
|
1
|
Negoita S, Feuer EJ, Mariotto A, Cronin
KA, Petkov VI, Hussey SK, Benard V, Henley SJ, Anderson RN, Fedewa
S, et al: Annual report to the nation on the status of cancer, part
II: Recent changes in prostate cancer trends and disease
characteristics. Cancer. 124:2801–2814. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cronin KA, Lake AJ, Scott S, Sherman RL,
Noone AM, Howlader N, Henley SJ, Anderson RN, Firth AU, Ma J, et
al: Annual report to the nation on the status of cancer, part I:
National cancer statistics. Cancer. 124:2785–2800. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lam VK and Papadimitrakopoulou V: Master
protocols in lung cancer: Experience from lung master protocol.
Curr Opin Oncol. 30:92–97. 2018.PubMed/NCBI
|
|
4
|
Lemjabbar-Alaoui H, Hassan OU, Yang YW and
Buchanan P: Lung cancer: Biology and treatment options. Biochim
Biophys Acta. 1856:189–210. 2015.PubMed/NCBI
|
|
5
|
Chen J, Sun Z, Zhang Y, Zeng X, Qing C,
Liu J, Li L and Zhang H: Synthesis of gibberellin derivatives with
anti-tumor bioactivities. Bioorg Med Chem Lett. 19:5496–5499. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhang Y, Zhang H, Chen J, Zhao H, Zeng X,
Zhang H and Qing C: Antitumor and antiangiogenic effects of
GA-13315, a gibberellin derivative. Invest New Drugs. 30:8–16.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mo J, Kang M, Ye JX, Chen JB, Zhang HB and
Qing C: Gibberellin derivative GA-13315 sensitizes
multidrug-resistant cancer cells by antagonizing ABCB1 while
agonizes ABCC1. Cancer Chemother Pharmacol. 78:51–61. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shen S and Tang J: Effects and mechanism
of GA-13315 on the proliferation and apoptosis of KB cells in oral
cancer. Oncol Lett. 14:1460–1463. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mayer RE, Hendrix P, Cron P, Matthies R,
Stone SR, Goris J, Merlevede W, Hofsteenge J and Hemmings BA:
Structure of the 55-kDa regulatory subunit of protein phosphatase
2A: Evidence for a neuronal-specific isoform. Biochemistry.
30:3589–3597. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Majchrzak-Celińska A, Słocińska M,
Barciszewska AM, Nowak S and Baer-Dubowska W: Wnt pathway
antagonists, SFRP1, SFRP2, SOX17, and PPP2R2B, are methylated in
gliomas and SFRP1 methylation predicts shorter survival. J Appl
Genet. 57:189–197. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhan W, Han T, Zhang C, Xie C, Gan M, Deng
K, Fu M and Wang JB: TRIM59 promotes the proliferation and
migration of non-small cell lung cancer cells by upregulating cell
cycle related proteins. PLoS One. 10:e01425962015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dimitrakopoulos FD, Antonacopoulou AG,
Kottorou AE, Maroussi S, Panagopoulos N, Koukourikou I, Scopa C,
Kalofonou M, Koutras A, Makatsoris T, et al: NF-κB2 genetic
variations are significantly associated with non-small cell lung
cancer risk and overall survival. Sci Rep. 8:52592018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Siena L, Pace E, Ferraro M, Di Sano C,
Melis M, Profita M, Spatafora M and Gjomarkaj M: Gemcitabine
sensitizes lung cancer cells to Fas/FasL system-mediated killing.
Immunology. 141:242–255. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mou H, Moore J, Malonia SK, Li Y, Ozata
DM, Hough S, Song CQ, Smith JL, Fischer A, Weng Z, et al: Genetic
disruption of oncogenic Kras sensitizes lung cancer cells to fas
receptor-mediated apoptosis. Proc Natl Acad Sci USA. 114:3648–3653.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kim D, Pertea G, Trapnell C, Pimentel H,
Kelley R and Salzberg SL: TopHat2: Accurate alignment of
transcriptomes in the presence of insertions, deletions and gene
fusions. Genome Biol. 14:R362013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Seyednasrollah F, Laiho A and Elo LL:
Comparison of software packages for detecting differential
expression in RNA-seq studies. Brief Bioinform. 16:59–70. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Savinova OV, Hoffmann A and Ghosh G: The
Nfkb1 and Nfkb2 proteins p105 and p100 function as the core of
high-molecular-weight heterogeneous complexes. Mol Cell.
34:591–602. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Rajsbaum R, García-Sastre A and Versteeg
GA: TRIMmunity: The roles of the TRIM E3-ubiquitin ligase family in
innate antiviral immunity. J Mol Biol. 426:1265–1284. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hao L, Du B and Xi X: TRIM59 is a novel
potential prognostic biomarker in patients with non-small cell lung
cancer: A research based on bioinformatics analysis. Oncol Lett.
14:2153–2164. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Meroni G and Diez-Roux G: TRIM/RBCC, a
novel class of ‘single protein RING finger’ E3 ubiquitin ligases.
Bioessays. 27:1147–1157. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Esposito D, Koliopoulos MG and Rittinger
K: Structural determinants of TRIM protein function. Biochem Soc
Trans. 45:183–191. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Uchil PD, Hinz A, Siegel S, Coenen-Stass
A, Pertel T, Luban J and Mothes W: TRIM protein-mediated regulation
of inflammatory and innate immune signaling and its association
with antiretroviral activity. J Virol. 87:257–272. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu F, Bardhan K, Yang D, Thangaraju M,
Ganapathy V, Waller JL, Liles GB, Lee JR and Liu K: NF-κB directly
regulates fas transcription to modulate fas-mediated apoptosis and
tumor suppression. J Biol Chem. 287:25530–25540. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Pomerantz JL and Baltimore D: Two pathways
to NF-kappaB. Mol Cell. 10:693–695. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gilmore TD: Introduction to NF-kappaB:
Players, pathways, perspectives. Oncogene. 25:6680–6684. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hayden MS and Ghosh S: Shared principles
in NF-kappaB signaling. Cell. 132:344–362. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cildir G, Low KC and Tergaonkar V:
Noncanonical NF-κB signaling in health and disease. Trends Mol Med.
22:414–429. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sun SC: The noncanonical NF-κB pathway.
Immunol Rev. 246:125–140. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Müller I, Beissert S and Kulms D:
Anti-apoptotic NF-κB and ‘gain of function’ mutp53 in concert act
pro-apoptotic in response to UVB+IL-1 via enhanced TNF production.
J Invest Dermatol. 135:851–860. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kaltschmidt B, Kaltschmidt C, Hofmann TG,
Hehner SP, Dröge W and Schmitz ML: The pro-or anti-apoptotic
function of NF-kappaB is determined by the nature of the apoptotic
stimulus. Eur J Biochem. 267:3828–3835. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
You BR, Han BR and Park WH:
Suberoylanilide hydroxamic acid increases anti-cancer effect of
tumor necrosis factor-α through up-regulation of TNF receptor 1 in
lung cancer cells. Oncotarget. 8:17726–17737. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Brojatsch J, Naughton J, Rolls MM, Zingler
K and Young JA: CAR1, a TNFR-related protein, is a cellular
receptor for cytopathic avian leukosis-sarcoma viruses and mediates
apoptosis. Cell. 87:845–855. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhou X, Hao Q, Liao P, Luo S, Zhang M, Hu
G, Liu H, Zhang Y, Cao B, Baddoo M, et al: Nerve growth factor
receptor negates the tumor suppressor p53 as a feedback regulator.
Elife. 5(pii): e150992016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Choi KE, Hwang CJ, Gu SM, Park MH, Kim JH,
Park JH, Ahn YJ, Kim JY, Song MJ, Song HS, et al: Cancer cell
growth inhibitory effect of bee venom via increase of death
receptor 3 expression and inactivation of NF-kappa B in NSCLC
cells. Toxins (Basel). 6:2210–2228. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chen M, Wang X, Zha D, Cai F, Zhang W, He
Y, Huang Q, Zhuang H and Hua ZC: Apigenin potentiates TRAIL therapy
of non-small cell lung cancer via upregulating DR4/DR5 expression
in a p53-dependent manner. Sci Rep. 6:354682016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yaguchi H, Okumura F, Takahashi H, Kano T,
Kameda H, Uchigashima M, Tanaka S, Watanabe M, Sasaki H and
Hatakeyama S: TRIM67 protein negatively regulates ras activity
through degradation of 80K-H and induces neuritogenesis. J Biol
Chem. 287:12050–12059. 2012. View Article : Google Scholar : PubMed/NCBI
|