Introduction
Rheumatoid arthritis (RA) is characterized by
chronic inflammatory synovitis resulting in progressive joint
destruction (1). Persistent
synovial inflammation is considered to be a characteristic feature
of RA, leading and contributing to cartilage and bone destruction,
and subsequent disability in RA (2); however, there is no effective
treatment for RA, and the precise etiology and underlying
mechanisms remain unclear. At present, various cell types have been
reported to serve distinct, complex and interconnected roles in the
synovial inflammation of RA, including monocytes/macrophages, T
cells, B cells, fibroblast-like synoviocytes (FLSs) and osteoclasts
(3,4). For example, FLSs in the intimal
lining of the synovium serve an important role in RA via the
hyperproduction of proinflammatory cytokines and matrix
metalloproteinases (MMPs), potentially resulting in cartilage
destruction (5).
Monocytes/macrophages are recognized as a prominent
joint-specific determinant; their activation may lead to
inflammation, acute phase reaction and joint destruction in acute
and chronic RA by promoting the production of key proinflammatory
cytokines, including tumor necrosis factor (TNF)-α, interleukin
(IL)-1β and MMPs (6–8). In addition, these cells are a potent
source of chemokines, which recruit additional leukocytes to the
inflamed joint to exacerbate the disease process. Furthermore,
monocytes can differentiate into osteoclasts, and have also been
reported to enhance the production of the osteoclastogenic cytokine
IL-17 by CD4+ T cells, in turn further contributing to
bone erosion, and focal and systemic osteoporosis (6,9,10).
The importance of the role of macrophages in RA is supported by
findings reporting these cells to be a reliable biomarker for
treatment response and associated with disease activity markers
(11–13). Nuclear factor-κB (NF-κB) has
received considerable attention as a key regulator of immunology,
inflammation and cancer development, and has been reported to
influence the pathogenesis and progression of RA (14,15).
Activation of NF-κB promotes the secretion of cytokines such as
TNF-α and IL-1β by the monocytes/macrophages of patients with RA
(16). Therefore, increasing
attention in the field of RA research has been placed on
monocytes/macrophages as potential therapeutic targets (7).
G-protein-coupled bile acid receptor 1 (TGR5) is a
G-protein-coupled receptor, which responds to various bile acids by
activating transcriptional networks and/or signaling cascades,
which regulate a diverse array of physiological processes,
including bile acid synthesis, lipid and carbohydrate metabolism,
carcinogenesis and inflammation (17,18).
TGR5 gene expression is diffusely distributed in the endocrine
glands, muscles, immune organs, adipocytes, spinal cord and enteric
nervous system (19). Human and
mouse TGR5 exhibit similar affinity for the different bile acid
species; the highest affinity is for lithocholic acid (LCA),
followed by deoxycholic acid, chenodeoxycholic acid and cholic acid
(20,21). Since the identification of
dedicated bile acid receptors, there has been an increased interest
in modulating these targets pharmacologically. For example, the
observation that TGR5 induces the secretion of clinically relevant
glucagon-like peptide 1 has promoted the development of a novel
class of drugs for the treatment of type 2 diabetes mellitus
(22). Regarding the potential for
manipulating TGR5 within the context of immunology and
inflammation, studies on rabbit alveolar macrophages and human
hepatic Kupffer cells have suggested that TGR5 activation
suppresses lipopolysaccharide (LPS)-induced production of the
proinflammatory cytokines TNF-α, IL-1, IL-6 and IL-8 (23,24).
The potential for targeting TGR5 in immunology has been further
validated by the finding that TGR5 activation inhibits
atherosclerosis by reducing macrophage inflammation and lipid
loading (25). Furthermore,
previous studies on mouse bone marrow-derived macrophages and human
macrophage subsets revealed that TGR5 activation negatively
regulates hepatic inflammatory responses via impaired NF-κB
signaling pathways (26,27).
RA is one of the most severe chronic diseases, which
is caused by persistent synovial inflammation and bone destruction.
TGR5 is important for inflammatory signaling pathways; however, at
present, the expression and pathophysiological role of TGR5 in RA
have not been elaborated. Therefore, in the present study, TGR5
expression in RA peripheral blood mononuclear cells (PBMCs) was
determined, and its association with clinical disease activity,
histological synovitis severity and radiological joint destruction
was investigated. Subsequently, the role of TGR5 in PBMC
inflammation and the potential mechanisms underlying its effects
were determined. Finally, the anti-arthritic and anti-inflammatory
effects of LCA on mice with collagen type II (CII)-induced
arthritis (CIA) were investigated.
Materials and methods
Patients
A total of 50 Chinese patients with RA who fulfilled
the 1987 revised criteria of the American College of Rheumatology
(ACR) (28) or the 2010
ACR/European League against Rheumatism classification criteria for
RA (29) were recruited from the
Department of Rheumatology and Orthopedics of the Hunan Cancer
Hospital (Changsha, China). All patients presented with active
disease, described as the presence of a 28-Joint Disease Activity
Score (DAS28) of ≥3.2. Age, gender or disease duration did not
differ among the patients with RA and healthy controls. In total,
11 RA patients were male and 39 were female, with a mean age of 59
(49–64) years. In the healthy control group, 14 patients were male
and 26 were female, with a mean age of 36 (29–48)
years. The date range of assessment/sample collection for both
groups was from June 2016 to October 2018. The present study was
conducted in compliance with the Helsinki Declaration principles.
All patients provided written informed consent
Clinical assessments
The clinical data of all patients with RA were
collected at baseline, including the 28-joint tender and swollen
joint counts (28TJC and 28SJC, respectively), patient and provider
global assessment of disease activity (PtGA and PrGA, respectively)
scores, visual analog scale score for pain, Chinese language
version of the Stanford Health Assessment Questionnaire (HAQ) score
(30), erythrocyte sedimentation
rate (ESR), C-reactive protein (CRP) level, rheumatoid factor (RF)
level and anti-cyclic citrullinated peptide (anti-CCP) antibody
level. Disease activity was assessed using the DAS28 with four
variables, including CRP level [DAS28 (4)-CRP] (31).
Isolation and culture of human
PBMCs
Heparinized whole blood was collected from 50
patients with RA and 40 healthy controls (HCs). PBMCs were
separated via Ficoll/Paque density gradient centrifugation at 400 ×
g for 30 min at 18–20°C. Cells were then re-suspended in α-Minimal
Essential Medium with 10% fetal bovine serum (Gibco; Thermo Fisher
Scientific, Inc.) and plated in a 37°C humidified incubator with 5%
CO2. On the following day, the suspended cells were
removed and washed thoroughly, and the adherent cells were
collected and plated. PBMCs were first treated with various
concentrations (0, 50 and 100 µM) of LCA for 60 min, and then
treated with LPS (100 ng/ml) for 12 h, mRNA was extracted, and the
culture medium was collected for ELISA analysis.
Immunofluorescence staining
The PBMCs (8×105 cells/well) were plated
on 24-well culture plates with coverslips, fixed in 4%
paraformaldehyde for 20 min at room temperature, permeabilized with
0.2% Triton X-100 for 10 min at room temperature, and blocked in
PBS containing 3% bovine serum albumin (Abcam) for 30 min at 37°C.
The cells were then incubated in PBS containing rabbit anti-human
polyclonal antibody against TGR5 (10 µg/ml; ab72608; Abcam)
overnight at 4°C, and subsequently incubated with Alexa
Fluor® 633-conjugated goat anti-rabbit immunoglobulin G
secondary antibodies (1:1,000; cat. no. A-21071; Invitrogen; Thermo
Fisher Scientific, Inc.) for 1 h at 37°C. Subsequently, DAPI
(Sigma-Aldrich; Merck KGaA) was used to stain the cell nuclei at a
concentration of 1.43 µM for 3 min at room temperature, and cells
were mounted using ProLong® Gold Antifade Reagent (cat.
no. P36934; Invitrogen; Thermo Fisher Scientific, Inc.). The images
were acquired using a 160 Zeiss LSM 510 Confocal Imaging system
(Zeiss GmbH).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from PBMCs using the RNAiso
Plus reagent (Takara Bio, Inc.) according to the manufacturer's
protocols. First-strand complementary DNA (cDNA) was synthesized
from each total RNA sample using a PrimeScript 1st Strand cDNA
Synthesis kit (Takara Bio, Inc.) at 37°C for 15 min, 85°C for 5
sec. qPCR was performed using the QuantiTect™ SYBR Green PCR kit
(Takara Bio, Inc.) on the Roche LightCycler480 sequence detector
system (Roche Diagnostics) according to the manufacturer's
protocols. The qPCR conditions were as follows: Denaturation at
95°C for 30 sec, then 95°C for 5 sec and 60°C for 30 sec for 40–50
cycles. The sequences of the primers used in the study are
presented in Table I, and these
were synthesized by Invitrogen (Thermo Fisher Scientific, Inc.).
PCR amplification of the housekeeping gene β-actin was performed
for each sample as a control for sample loading. Normalization and
quantification of the PCR signals was performed by comparing the
cycle threshold value of the gene of interest, in triplicate, with
β-actin. The fold change in gene expression relative to the control
was calculated using the 2−ΔΔCq method (32). Data are presented as the mean ±
standard deviation of three independent experiments and analyzed
with the LightCycler480 software release 1.5.0 (Roche
Diagnostics).
 | Table I.Primers for reverse
transcription-quantitative polymerase chain reaction. |
Table I.
Primers for reverse
transcription-quantitative polymerase chain reaction.
| Gene | Sequence |
|---|
| β-actin | Sense:
5′-GGACTTCGAGCAAGAGATGG-3′ |
|
| Antisense:
5′-TGTGTTGGCGTACAGGTCTTTG-3′ |
| IL-6 | Sense:
5′-CTGCGCAGCTTTAAGGAGTTC-3′ |
|
| Antisense:
5′-CAATCTGAGGTGCCCATGCTA-3′ |
| IL-8 | Sense:
5′-GTGCAGAGGGTTGTGGAGAAGTTT-3′ |
|
| Antisense:
5′-TCACTGGCATCTTCACTGATTCTTG-3′ |
| IL-1β | Sense:
5′-CCAGCTACGAATCTCCGACC-3′ |
|
| Antisense:
5′-CATGGCCACAACAACTGACG-3′ |
| TNF-a | Sense:
5′-GCTAAGAGGGAGAGAAGCAACTACA-3′ |
|
| Antisense:
5′-GAAGAGGCTGAGGAACAAGCA-3′ |
| TGR5 | Sense:
5′-CCCAGGCTATCTTCCCAGC-3′ |
|
| Antisense:
5′-GCCAGGACTGAGAGGAGCA-3′ |
Western blot analysis
PBMCs were collected and lysed to obtain proteins
using a RIPA buffer (Cell Signaling Technologies, Inc.), whereas
nuclear protein was separated using a NE-PER™ Nuclear and
Cytoplasmic Extraction kit (Pierce; Thermo Fisher Scientific,
Inc.). Protein concentration was measured using bicinchoninic acid
Protein Assay kit from Bio-Rad Laboratories, Inc. Proteins (50
µg/lane) were separated by 12% SDS-PAGE under denaturing
conditions, and were electrotransferred to nitrocellulose
membranes. The membranes were then blocked with 5% non-fat milk in
0.1% TBS-Tween 20 for 1 h at room temperature, and were probed with
antibodies against TGR5 (cat. no. ab72608; Abcam), phosphorylated
(p)-IκB kinase α (IκBα; cat. no. 2859; Cell Signaling Technologies,
Inc.), total (T)-IκBα (cat. no. 9249; Cell Signaling Technologies,
Inc.) and p-NF-κB (cat. no. 3033; Cell Signaling Technologies,
Inc.; all 1:1,000); GAPDH (cat. no. 5174; Cell Signaling
Technologies, Inc.; 1:1,000) and histone H3 (cat. no. 9275; Cell
Signaling Technologies, Inc.; 1:2,000) overnight at 4°C. For the
detection of proteins, membranes were then incubated for 1 h at
room temperature with horseradish peroxidase-conjugated secondary
antibody (cat. no. E030120-01; EarthOx Life Sciences; 1:5,000).
Immunoreactive bands were visualized using an enhanced
chemiluminescence reagent (EMD Millipore). Protein bands were
semi-quantified via densitometric analyses using a G:BOX Gel &
Blot Imaging Series (Syngene Europe) and ImageQuant LAS500 (GE Life
Sciences). Phosphorylated protein expression was normalized to
total protein expression, and expressed as a fold change relative
to the untreated control. Data are presented as the means ±
standard deviation of three independent experiments.
Enzyme-linked immunosorbent assay
(ELISA)
The quantities of TNF-α (cat. no. DTA00D, R&D
Systems, Inc.), IL-1β (cat. no. DLB50, R&D Systems, Inc.), IL-6
(cat. no. D6050, R&D Systems, Inc.) and IL-8 (cat. no. D8000C,
R&D Systems, Inc.) in culture supernatants were measured via
ELISA according to the manufacturer's protocols. The quantities of
TNF-α (cat. no. MTA00B, R&D Systems, Inc.), IL-1β (cat. no.
MLB00C, R&D Systems, Inc.), IL-6 (cat. no. M6000B, R&D
Systems, Inc.) and IL-8 (cat. no. LS-F55769, LifeSpan BioSciences,
Inc.) in mouse blood samples were measured via ELISA according to
the manufacturer's protocol. Briefly, PBMCs were subjected to
different treatments. Then, mouse blood samples were centrifuged at
1,006.2 × g for 15 min to separate the blood plasma at room
temperature. The culture supernatant or the blood plasma were mixed
with the assay buffer and added to anti-TNF-α, IL-1β, IL-6 and IL-8
antibody-coated wells. Horseradish peroxidase-conjugated anti-human
TNF-α, IL-1β, IL-6, and IL-8 monoclonal antibodies were added and
incubated at 37°C for 2 h, followed by incubation with colorimetric
(tetramethylbenzidine) solution for a further 10 min. The relative
absorbance was measured at 450 nm, and three independent
experiments were performed for each condition.
Animal study
This study was conducted on male DAB/1J mice (age,
6–8 weeks; 18–22 g) obtained from Beijing HFK Bioscience Co., Ltd.
A total of 30 mice were divided into three equal groups: Negative
controls (NCs), experimental arthritis controls not treated with
LCA and experimental arthritis group treated with LCA (10
mg/kg/day; Sigma-Aldrich; Merck KGaA). The NCs did not have CIA.
The mice were housed in a room on a 12-h light/dark cycle under
specific pathogen-free conditions, at a temperature of 22–26°C and
relative humidity of 55–65%. All experiments were conducted in
compliance with the National Institutes of Health guidelines for
the care and use of laboratory animals (33). The study was approved by the
Institutional Animal and Clinical Committees of Hunan Cancer
Hospital.
Induction of CIA and LCA
treatment
CIA was induced according to the previously
described method by Brand et al (34) in the experimental arthritis
controls not treated with LCA and experimental arthritis group
treated with LCA mouse groups. Briefly, native chick CII (Chondrex,
Inc.) was dissolved in 10 mM acetic acid to a 1 mg/ml
concentration, and was emulsified in an equal volume of Freund's
complete adjuvant (Sigma-Aldrich; Merck KGaA). Each mouse received
two intradermal injections of emulsion (0.1 ml) containing CII (1
mg/ml) and Freund's complete adjuvant at two separated sites tail
on days 1 and 21.
For LCA treatment, LCA was dissolved and freshly
diluted in PBS. PBS and LCA (10 mg/kg/day) were orally administered
via a gastric tube once a day from day 21 to day 42 following the
first immunization. Both NCs and experimental arthritis controls
not treated with LCA were treated with PBS.
Arthritis score
The arthritis score was assessed once every 3 days
following the initiation of LCA treatment. The severity of
arthritis was measured in a double manner using the
semi-quantitative clinical scoring system described by Yoo et
al (35) (0, no arthritis; 1,
definite erythema and swelling of the ankle or one digit; 2, two or
more joints involved or mild erythema and swelling of the entire
paw; 3, erythema and swelling extending from the ankle to the
metatarsal joints of the entire paw and all digits; and 4,
ankylosing deformity with severe joint erythema and swelling). The
arthritis score for each mouse was the sum of all paw scores
(yielding a score between 0 and 16). On day 42, the mice were
sacrificed and blood was collected for serum separation.
Statistical analysis
Statistical analyses were performed using SPSS 13.0
software (SPSS, Inc.). For categorical variables, data are
presented as frequencies and percentages. For continuous variables,
data are presented as the means ± standard deviation, or the median
and interquartile range. Wilcoxon rank-sum test was used to compare
the expression of TGR5 in the PBMCs of patients with RA and HCs,
and the arthritis score in the experimental arthritis controls and
experimental arthritis controls treated with LCA. One-way or
Welch's ANOVA followed by least significant difference post-hoc
tests were performed to compare the expression of TNF-α, IL-1β,
IL-6 and IL-8 in the culture supernatant or the blood plasma, and
the expression of p-NF-κB and p-IκBα in the PBMCs treated with
various LCA or not treated with LCA. Spearman's rank order
correlation test was used to assess the correlation between TGR5
expression in RA PBMCs and clinical parameters. P<0.05 was
considered to indicate a statistically significant difference.
Results
Characteristics of the study
patients
Table II presents
the baseline demographic and clinical features of individuals in
the study. The gender ratio did not differ between the patients in
the RA and HC groups. Among the patients with RA, 48% (24/50) had
not previously taken corticosteroids or disease-modifying
anti-rheumatic drugs (DMARDs). The majority of patients had taken
only Chinese herbal medicine and/or painkillers to relieve
arthralgia. At recruitment, 18% (9/50) had taken corticosteroids
alone. The remaining 34% (17/50) received treatment with one or
more DMARD, including methotrexate, leflunomide, sulfasalazine,
hydroxychloroquine and etanercept.
| Table II.Baseline demographic and clinical
features of patients with RA and HCs. |
Table II.
Baseline demographic and clinical
features of patients with RA and HCs.
| Characteristic | Patients with RA
(n=50) | HC (n=40) |
|---|
| Demographic |
|
|
| Age
[years; median (IQR)] | 59 (49–64) | 36 (29–48) |
| Female
[n (%)] | 39 (78) | 26 (65) |
| Disease status |
|
|
| Disease
duration [months; median (IQR)] | 34 (12–106) | NA |
| ESR
[mm/h; median (IQR)] | 75 (54–105) | 11 (3–21) |
| CRP
[mg/dl; median (IQR)] | 4.02
(1.19–5.75) | 0.11
(0.02–0.23) |
|
Rheumatoid factor-positive [n
(%)] | 47 (94) | NA |
|
Anti-CCP-positive [n (%)] | 43 (86) | NA |
| DAS28
[median (IQR)] | 5.5 (4.6~6.3) | NA |
| Previous
medications [n (%)] |
|
|
|
Corticosteroids | 22 (50) | NA |
|
Methotrexate | 20 (40) | NA |
|
Leflunomide | 5 (10) | NA |
|
Sulfasalazine | 4 (8) | NA |
|
Hydroxychloroquine | 6 (12) | NA |
|
Etanercept | 2 (4) | NA |
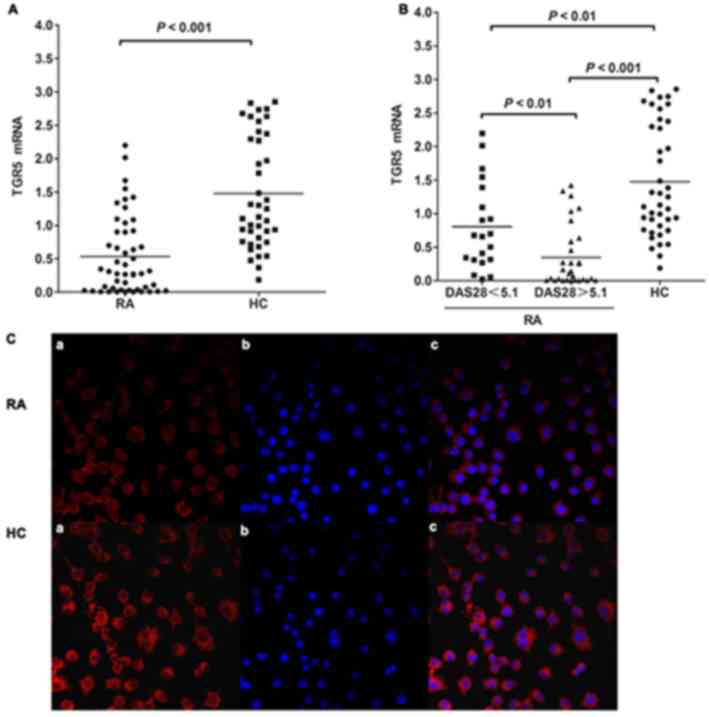
Expression of TGR5 is downregulated in
the PBMCs of patients with RA
The mRNA expression levels of TGR5 in PBMCs were
evaluated in 50 patients with RA and 40 HCs. The expression of TGR5
mRNA in the PBMCs of patients with RA was significantly decreased
compared with in the HCs (0.53±0.58 vs. 1.49±0.83; P<0.001;
Fig. 1A). Additionally, the
expression of TGR5 mRNA in the PBMCs of patients with RA with a
high DAS28 was significantly decreased compared with in patients
with a low DAS28 (0.35±0.46 vs. 0.81±0.65, respectively; P=0.002;
Fig. 1B). The expression of TGR5
mRNA in the PBMCs of the patients with RA with either a high or low
DAS28 was also significantly lower than that of the HCs (0.81±0.65
vs. 1.49±0.83, P<0.001; 0.35±0.46 vs. 1.49±0.83, P=0.002;
Fig. 1B). The number of patients
with RA with a DAS28 score of ≥5.1 was 30. Of these patients, 50%
(15/30) had never taken corticosteroids or DMARDs. At recruitment,
20% (6/30) had taken corticosteroids alone. The remaining 30%
(9/30) received treatment with one or more DMARDs, including
methotrexate, leflunomide, sulfasalazine, hydroxychloroquine or
etanercept. Patients did not exhibit any obvious side effects after
taking DMARDs. Immunofluorescence staining revealed markedly
reduced expression of TGR5 in the PBMCs of patients with RA
compared with in the PBMCs of HCs (Fig. 1C).
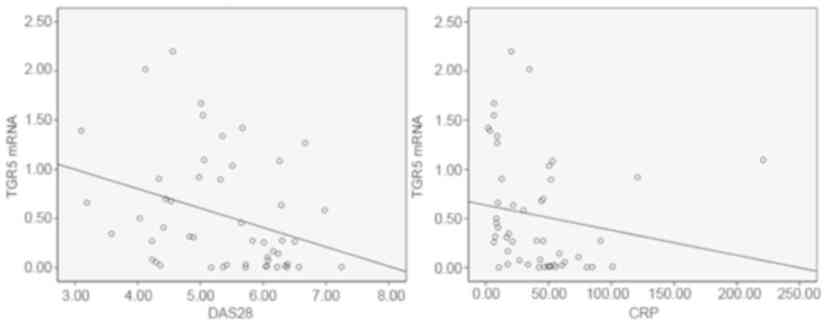
Expression of TGR5 mRNA in the PBMCs
of patients with RA demonstrates a negative correlation with
clinical parameters
Spearman's rank order correlation test revealed
significant correlations between the mRNA expression levels of TGR5
in PBMCs and the CRP level (r=−0.429, P=0.002), and the DAS28
(r=−0.383, P=0.006) of patients (Fig.
2). There was no significant correlation between TGR5 mRNA
expression and 28TJC, 28SJC, PtGA score, PrGA score, HAQ score, RF
level, anti-CCP level, ESR, the simplified disease activity index
and the clinical disease activity index (respectively: r=0.043,
P=0.763; r=−0.072 P=0.611; r=−0.011, P=0.939; r=0.185, P=0.188;
r=0.172, P=0.223; r=0.049, P=0.732; r=0.188, P=0182; r=0.067,
P=0.642; r=−0.099, P=0.544; and r=−0.106, P=0.455).
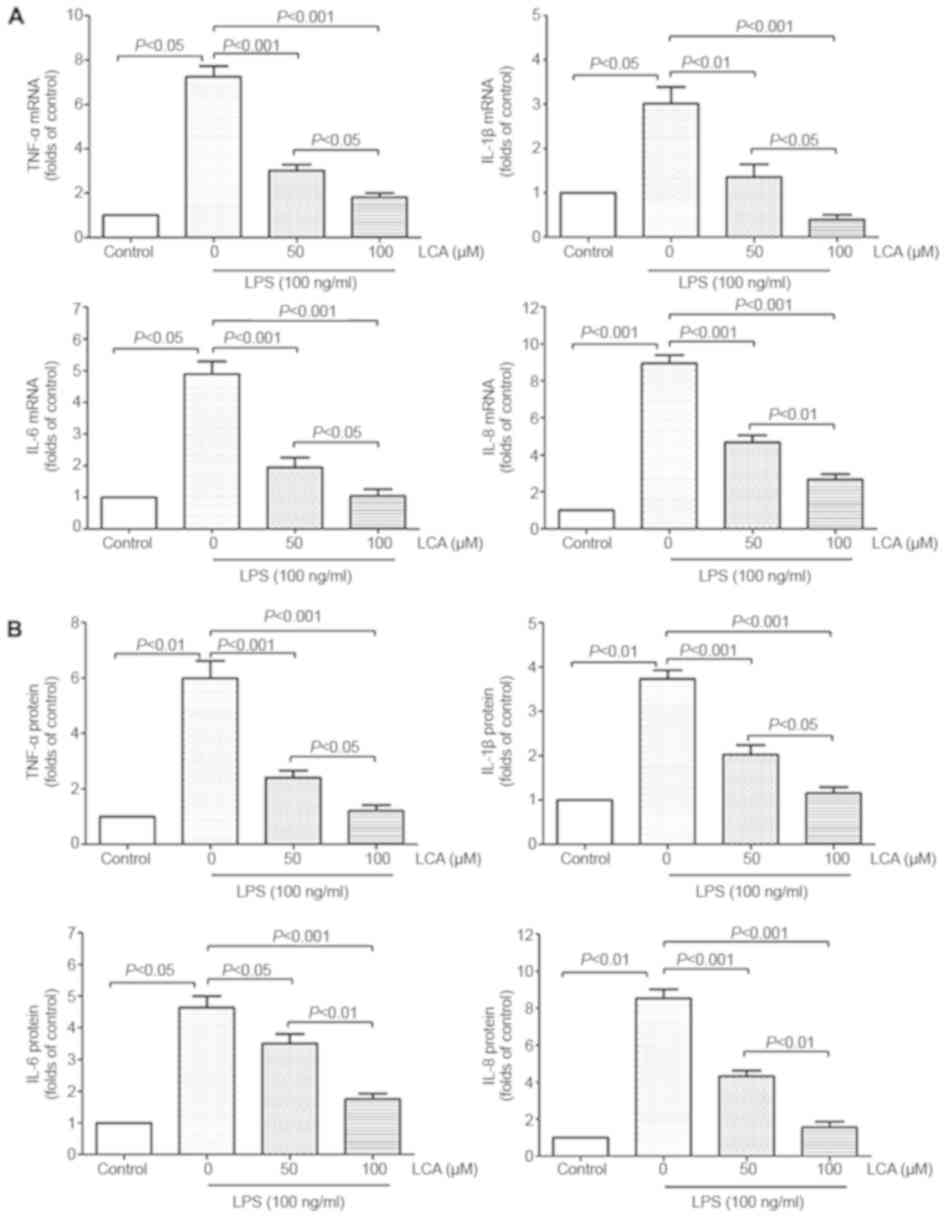
LCA attenuates LPS-induced
proinflammatory cytokine production in the PBMCs of patients with
RA
LPS is a potent endotoxin involved in the
progression of RA diseases (36).
LPS treatment (100 ng/ml) for 12 h markedly increased the mRNA and
protein expression levels of TNF-α, IL-1β, IL-6 and IL-8 in the
PBMCs of patients with RA by between 3- and 10-fold (Fig. 3). Pretreatment of PBMCs with LCA
(50 and 100 µM) for 60 min significantly inhibited LPS-induced
TNF-α, IL-1β, IL-6 and IL-8 mRNA expression and protein release
into the supernatant in a concentration-dependent manner (Fig. 3).
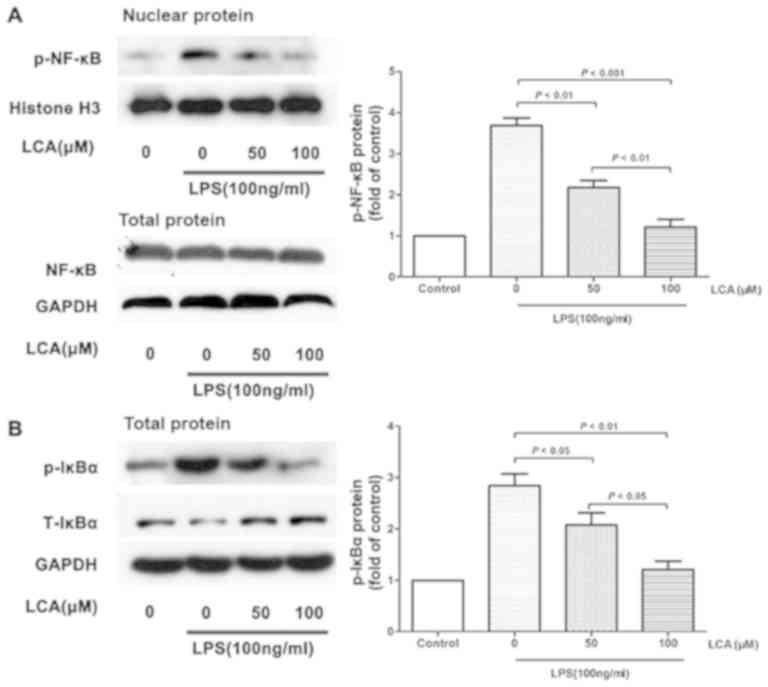
LCA inhibits LPS-induced NF-κB and
IκBα activation in the PBMCs of patients with RA
To further investigate the mechanism underlying the
effects of LCA on the inhibition of LPS-induced proinflammatory
cytokine production, NF-κB and IκBα signaling was measured via
western blot analysis. LCA treatment significantly decreased the
phosphorylation of NF-κB and IκBα, but not the total protein levels
of IκBα (Fig. 4).
Effects of LCA on DAB/1J mice with
CIA
Mice with CIA have been frequently utilized to
investigate arthritic diseases, as they possess various
pathological features similar to those of human RA (37). There was no macroscopic evidence of
paw erythema or edema in the normal blank control mice. As
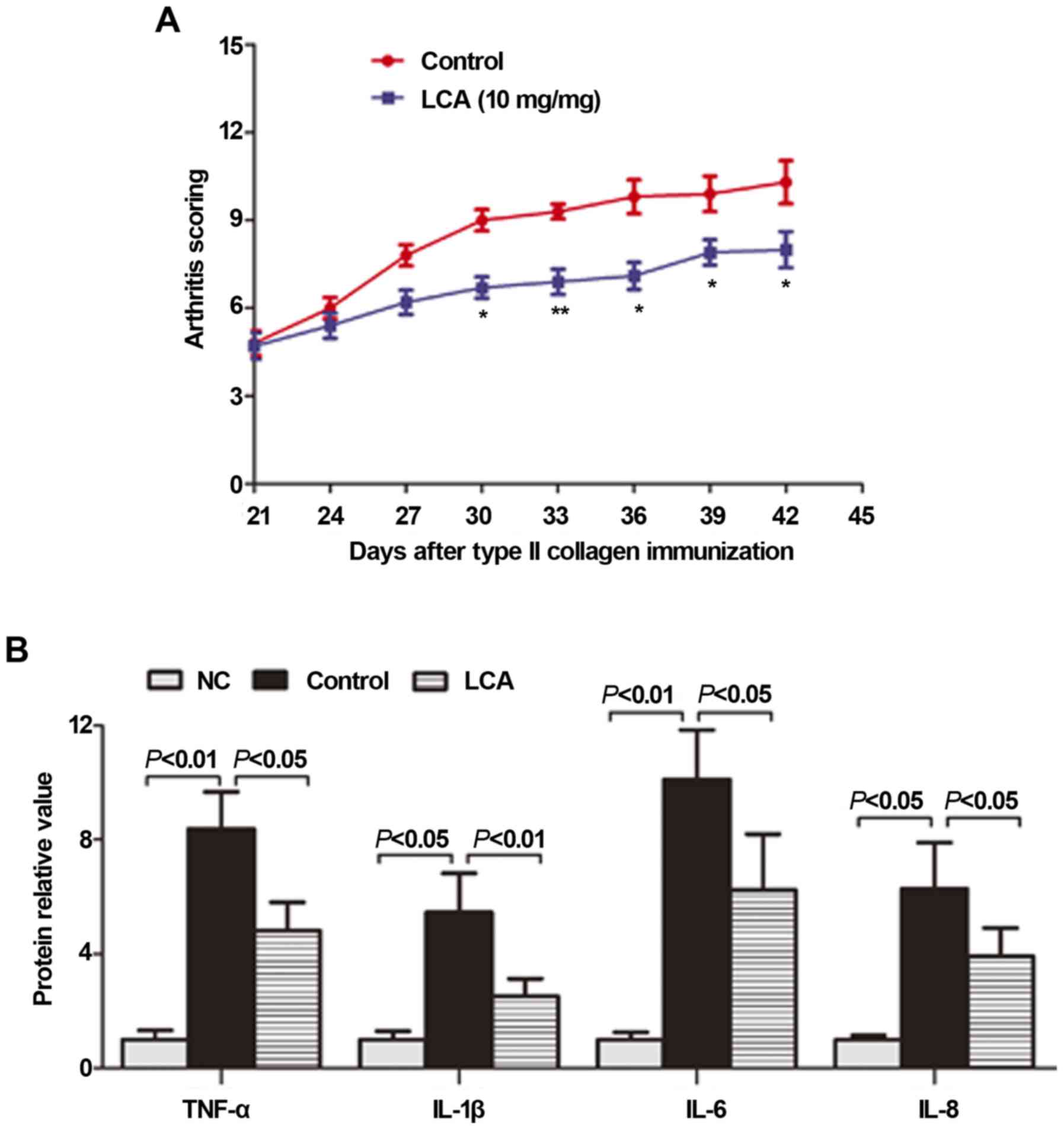
presented in Fig. 5A, arthritis
symptoms in the mice with CIA progressed and maintained a high
intensity from day 21 to day 42 following immunization; this was
notably relieved following treatment with LCA (10 mg/kg/day). From
day 30, the arthritis score was significantly decreased in the LCA
treatment group compared with in the CIA model group (P<0.05 or
0.01).
To demonstrate its anti-inflammatory effects in
vivo, LCA was evaluated for its inhibition of the production of
proinflammatory cytokines in mice with CIA. Cytokines, such as
TNF-α, IL-1β, IL-6 and IL-8, are associated with the progression
and severity of arthritis (16).
On day 42, the mice were sacrificed, and blood was collected for
serum separation and subsequent ELISA detection. As presented in
Fig. 5B, the serum levels of the
proinflammatory cytokines TNF-α, IL-1β, IL-6 and IL-8 were
significantly increased in the non-treated CIA mice compared with
in the non-arthritic group. The increased TNF-α, IL-1β, IL-6, and
IL-8 production was significantly suppressed by LCA treatment
compared with in the non-treated mice with CIA.
Discussion
RA is an inflammatory disease characterized by
chronic inflammatory synovitis resulting in progressive joint
destruction. The pathogenesis of RA involves a diverse range of
cells, including monocytes/macrophages, T cells, B cells and FLSs,
which produce a large number of proinflammatory cytokines, MMPs and
other proteolytic enzymes. In addition, previous studies have
indicated that the presence of inflammatory cells is positively
correlated with synovitis score, further demonstrating the
involvement of inflammatory cells in the pathophysiology of RA
(38,39).
Monocytic/macrophagic inflammation is central to
almost all aspects that contribute to the development of RA, as it
is regarded as the primary source of proinflammatory cytokines,
including TNF-α and IL-1β, which serve important roles in the
communication between the various inflammatory cells involved in RA
(40,41). TGR5 has been identified as an
important component of the bile acid signaling network, and its
activation has been reported to control numerous physiological
pathways associated with metabolic homeostasis, bile acid synthesis
and inflammation. Furthermore, emerging evidence has demonstrated
that TGR5 is expressed in various immune cells, including
monocytes, alveolar macrophages and Kupffer cells, and it has been
reported that its activation modulates inflammatory responses in
these cells (23,24,26);
however, whether TGR5 serves a role in the regulation of PBMC
inflammation in RA has yet to be fully investigated. In the present
study, it was revealed that TGR5 was expressed in primary PBMCs,
and that TGR5 expression was significantly downregulated in the
PBMCs of patients with RA compared with in PBMCs from HCs.
Additionally, TGR5 expression in PBMCs exhibited significantly
negative correlations with CRP level and DAS28. Therefore, it was
hypothesized that decreased TGR5 expression may potentially be
involved in the pathogenesis of RA.
To investigate the effects of TGR5 on PBMC-mediated
inflammation, LPS was used to induce inflammation in PBMCs, and LCA
was administered to activate TGR5. It was observed that
pretreatment of PBMCs with LCA potently downregulated LPS-induced
TNF-α, IL-1β, IL-6 and IL-8 expression, and inhibited NF-κB and
IκBα phosphorylation in a concentration-dependent manner; this
finding is consistent with previous observations demonstrating that
TGR5 activation attenuates proinflammatory cytokine production in
monocytes/macrophages via suppression of the nuclear translocation
of NF-κB (23,27,42).
Högenauer et al (42)
indicated that TGR5 activation inhibits LPS-induced TNF-α and IL-12
release from PBMCs and in mice. Additionally, it has been reported
that TGR5 activation does not affect production of the
anti-inflammatory cytokine IL-10, it only suppresses the secretion
of proinflammatory cytokines TNF-α, IL-6 and IL-12p40 by primary
human macrophages (27).
Therefore, inhibition of the release of different proinflammatory
cytokines may be associated with activation of TGR5 by various
agonists in human monocytes/macrophages, with or without RA. The
present findings indicated that TGR5 is a potential suppressor of
NF-κB-dependent inflammatory responses; however, the nature of the
interaction between TGR5 and NF-κB/IκBα in the PBMCs of patients
with RA remains unknown. It has been reported that TGR5 serves an
important role in immunological regulation via cyclic adenosine
monophosphate, a second messenger with immunoregulatory functions
(25,43). Additionally, a previous study
indicated that TGR5 promotes the activation of cytosolic protein
β-arrestin 2, which interacts with IκBα following bacterial antigen
stimulation in mouse livers, suppressing the production of TNF-α
(26). Therefore, further studies
into the mechanisms underlying the modulation of the activity of
NF-κB and IκBα in the PBMCs of patients with RA by TGR5 are
required.
NF-κB is a family of ubiquitously expressed
transcription factors that serve crucial roles in the majority of
inflammatory and immune responses (15). Constitutive NF-κB activation has
been associated with the pathogenesis of numerous diseases,
including inflammatory and rheumatic diseases (RA and
osteoarthritis), atherosclerosis, asthma, multiple sclerosis,
chronic inflammatory demyelinating inflammatory bowel disease and
diabetes (14). Additionally,
studies on emerging animal models of inflammatory arthritis support
the hypothesis that NF-κB is a predominant adaptor in the
development and progression of arthritis in vivo (44,45).
In the present study, it was revealed that TGR5 activation
significantly inhibited NF-κB activity in vitro, implying
that TGR5 activation may potentially alleviate arthritis symptoms
and inflammation of RA in vivo. CIA mouse models with
pathological features similar to those of human RA are used to
study arthritic diseases. It was observed that TGR5 activation by
LCA significantly suppressed inflammation and the progression of
arthritis in mice with CIA, as determined by their arthritis score.
RA is characterized by chronic inflammatory synovitis and joint
destruction; thus, future experiments will aim to identify the
mechanisms underlying the effects of TGR5 on synovitis and joint
destruction in patients with RA or mice with CIA.
In RA, activated immune cells secrete large
quantities of proinflammatory cytokines. TNF-α is a potent inducer
of inflammation, and IL-6 and IL-1β are downstream mediators of
TNF-α, together serving significant roles in the progression of
inflammatory synovitis and joint destruction in RA (46,47).
IL-8 is involved in leukocyte recruitment to a diseased synovium
(48). Agents that inhibit TNF-α,
IL-6 and IL-1 are frequently used in clinical settings
therapeutically (49). In the
present study, TGR5 activation demonstrated therapeutic effects
against RA by downregulating the levels of TNF-α, IL-1β, IL-6 and
IL-8 in the plasma of mice with CIA in vivo. TGR5 activation
also significantly suppressed LPS-induced TNF-α, IL-1β, IL-6 and
IL-8 expression in PBMCs in vitro. These results indicated
that TGR5 activation may inhibit proinflammatory cytokine
expression in monocytes/macrophages and further reduce inflammation
in RA; however, the influence of TGR5 on other immune cells and
tissues of patients with RA or mice with CIA remains unclear and
requires further study. Monocytes can differentiate into
proinflammatory microbicidal M1 macrophages or anti-inflammatory M2
macrophage subtypes (50).
Macrophages are stimulated to differentiate into M1 macrophages by
infectious microorganism-associated molecules such as LPS and
inflammatory cytokines such as interferon-γ (51). M1 macrophages can produce
inflammatory cytokines, including interleukin IL-1β, TNF-a and IL-6
(52). Högenauer et al
(42) demonstrated that TGR5
activation stabilizes the phenotype of noninflammatory M2-like type
cells during the differentiation of monocytes into macrophages. The
effect of TGR5 on macrophage differentiation in mice with CIA
requires further investigation.
Collectively, TGR5 activation attenuated the
expression of TNF-α, IL-1β, IL-6 and IL-8 via inhibition of NF-κB
activity in LPS-stimulated PBMCs in vitro, decreased the
levels of TNF-α, IL-1β, IL-6 and IL-8 in the plasma of mice with
CIA in vivo, and hindered the progression of arthritis in
mouse models of CIA, as determined by the arthritis score. RA has
also been reported to have improved during jaundice, suggesting
that TGR5 activation may provide a novel target for the treatment
of RA (53–55). Future studies should investigate
cases of RA with obstructive jaundice, to further determine the
potential role and effects of TGR5 in RA.
Acknowledgements
Not applicable.
Funding
The present study was supported by a grant from the
National Natural Science Foundation of China (grant no. 81401631)
and Education of Zhejiang Province (grant no. Y201737942).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JJZ and ZYL participated in study development and
collection of patient samples, performed experiments, and actively
wrote the first draft of the manuscript. CLL was involved in
collecting and analyzing part of the clinical data. LMZ
participated in analyzing the data from in vitro cell
experiments and revising the manuscript. All authors approved the
final manuscript.
Ethics approval and consent to
participate
The study protocol was approved by the the
Institutional Animal and Clinical Committees of Hunan Cancer
Hospital and written informed consent was obtained from all
participants in the study.
Patient consent for publication
Written informed consent was obtained from all
participants.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
RA
|
rheumatoid arthritis
|
|
PBMC
|
peripheral blood mononuclear cell
|
|
HC
|
healthy control
|
|
TGR5
|
G-protein-coupled bile acid receptor
1
|
|
RF
|
rheumatoid factor
|
|
CRP
|
C-reactive protein
|
|
ESR
|
erythrocyte sedimentation rate
|
|
MMP
|
matrix metalloproteinase
|
|
anti-CCP
|
anti-cyclic citrullinated peptide
|
|
DAS28
|
28-Joint Disease Activity Score
|
|
ELISA
|
enzyme-linked immunosorbent assay
|
|
HAQ
|
Health Assessment Questionnaire
|
|
RT-qPCR
|
reverse transcription-quantitative
polymerase chain reaction
|
|
NF-κB
|
nuclear factor-κB
|
|
TNF-α
|
tumor necrosis factor-α
|
|
IL
|
interleukin
|
|
IκBα
|
IκB kinase α
|
|
CIA
|
collagen II-induced arthritis
|
|
ACR
|
American College of Rheumatology
|
|
28TJC
|
28-joint tender joint count
|
|
28SJC
|
28-joint swollen joint count
|
|
PtGA
|
patient global assessment of disease
activity
|
|
PrGA
|
provider global assessment of disease
activity
|
|
cDNA
|
complementary DNA
|
|
CII
|
collagen type II
|
|
DMARD
|
disease-modifying anti-rheumatic
drug
|
|
FLS
|
fibroblast-like synoviocyte
|
|
LCA
|
lithocholic acid
|
|
LPS
|
lipopolysaccharide
|
References
|
1
|
Siebert S, Tsoukas A, Robertson J and
McInnes I: Cytokines as therapeutic targets in rheumatoid arthritis
and other inflammatory diseases. Pharmacol Rev. 67:280–309. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Choy EH and Panayi GS: Cytokine pathways
and joint inflammation in rheumatoid arthritis. N Engl J Med.
344:907–916. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Karsdal MA, Woodworth T, Henriksen K,
Maksymowych WP, Genant H, Vergnaud P, Christiansen C, Schubert T,
Qvist P, Schett G, et al: Biochemical markers of ongoing joint
damage in rheumatoid arthritis-current and future applications,
limitations and opportunities. Arthritis Res Ther. 13:2152011.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wang Q, Ma Y, Liu D, Zhang L and Wei W:
The roles of B cells and their interactions with fibroblast-like
synoviocytes in the pathogenesis of rheumatoid arthritis. Int Arch
Allergy Immunol. 155:205–211. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lefevre S, Meier FM, Neumann E and
Muller-Ladner U: Role of synovial fibroblasts in rheumatoid
arthritis. Curr Pharm Des. 21:130–141. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Roberts CA, Dickinson AK and Taams LS: The
interplay between monocytes/macrophages and CD4(+) T cell subsets
in rheumatoid arthritis. Front Immunol. 6:5712015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Davignon JL, Hayder M, Baron M, Boyer JF,
Constantin A, Apparailly F, Poupot R and Cantagrel A: Targeting
monocytes/macrophages in the treatment of rheumatoid arthritis.
Rheumatology (Oxford). 52:590–598. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kinne RW, Stuhlmuller B and Burmester GR:
Cells of the synovium in rheumatoid arthritis. Macrophages.
Arthritis Res Ther. 9:2242007. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Schett G: Cells of the synovium in
rheumatoid arthritis. Osteoclasts. Arthritis Res Ther. 9:2032007.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yokota K: Inflammation and osteoclasts.
Nihon Rinsho Men'eki Gakkai kaishi. 40:367–376. 2017.(In Japanese).
View Article : Google Scholar
|
|
11
|
Alam J, Jantan I and Bukhari SNA:
Rheumatoid arthritis: Recent advances on its etiology, role of
cytokines and pharmacotherapy. Biomed Pharmacother. 92:615–633.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mulherin D, Fitzgerald O and Bresnihan B:
Synovial tissue macrophage populations and articular damage in
rheumatoid arthritis. Arthritis Rheum. 39:115–124. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yanni G, Whelan A, Feighery C and
Bresnihan B: Synovial tissue macrophages and joint erosion in
rheumatoid arthritis. Ann Rheum Dis. 53:39–44. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Aravilli RK, Vikram SL and Kohila V:
Phytochemicals as potential antidotes for targeting NF-κB in
rheumatoid arthritis. 3 Biotech. 7:2532017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Roman-Blas JA and Jimenez SA: NF-κB as a
potential therapeutic target in osteoarthritis and rheumatoid
arthritis. Osteoarthritis and Cartilage. 14:839–848. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
McInnes IB and Schett G: Cytokines in the
pathogenesis of rheumatoid arthritis. Nat Rev Immunol. 7:429–442.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Schaap FG, Trauner M and Jansen PL: Bile
acid receptors as targets for drug development. Nat Rev
Gastroenterol Hepatol. 11:55–67. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lin TH, Tang CH, Wu K, Fong YC, Yang RS
and Fu WM: 15-deoxy-Δ(12,14)-prostaglandin-J2 and ciglitazone
inhibit TNF-α-induced matrix metalloproteinase 13 production via
the antagonism of NF-κB activation in human synovial fibroblasts. J
Cell Physiol. 226:3242–3250. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yu DD, Sousa KM, Mattern DL, Wagner J, Fu
X, Vaidehi N, Forman BM and Huang W: Stereoselective synthesis,
biological evaluation, and modeling of novel bile acid-derived
G-protein coupled bile acid receptor 1 (GP-BAR1, TGR5) agonists.
Bioorg Med Chem. 23:1613–1628. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pols TW: TGR5 in inflammation and
cardiovascular disease. Biochem Soc Trans. 42:244–249. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Russell DW and Setchell KD: Bile acid
biosynthesis. Biochemistry. 31:4737–4749. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Pols TW, Noriega LG, Nomura M, Auwerx J
and Schoonjans K: The bile acid membrane receptor TGR5 as an
emerging target in metabolism and inflammation. J Hepatol.
54:1263–1272. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kawamata Y, Fujii R, Hosoya M, Harada M,
Yoshida H, Miwa M, Fukusumi S, Habata Y, Itoh T, Shintani Y, et al:
A G protein-coupled receptor responsive to bile acids. J Biol Chem.
278:9435–9440. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Keitel V, Donner M, Winandy S, Kubitz R
and Haussinger D: Expression and function of the bile acid receptor
TGR5 in Kupffer cells. Biochem Biophys Res Commun. 372:78–84. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Pols TW, Nomura M, Harach T, Lo Sasso G,
Oosterveer MH, Thomas C, Rizzo G, Gioiello A, Adorini L,
Pellicciari R, et al: TGR5 activation inhibits atherosclerosis by
reducing macrophage inflammation and lipid loading. Cell Metab.
14:747–757. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang YD, Chen WD, Yu D, Forman BM and
Huang W: The G-protein-coupled bile acid receptor, Gpbar1 (TGR5),
negatively regulates hepatic inflammatory response through
antagonizing nuclear factor κ light-chain enhancer of activated B
cells (NF-κB) in mice. Hepatology. 54:1421–1432. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Haselow K, Bode JG, Wammers M, Ehlting C,
Keitel V, Kleinebrecht L, Schupp AK, Häussinger D and Graf D: Bile
acids PKA-dependently induce a switch of the IL-10/IL-12 ratio and
reduce proinflammatory capability of human macrophages. J Leukoc
Boil. 94:1253–1264. 2013. View Article : Google Scholar
|
|
28
|
Arnett FC, Edworthy SM, Bloch DA, McShane
DJ, Fries JF, Cooper NS, Healey LA, Kaplan SR, Liang MH and Luthra
HS: The American Rheumatism Association 1987 revised criteria for
the classification of rheumatoid arthritis. Arthritis Rheum.
31:315–324. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Aletaha D, Neogi T, Silman AJ, Funovits J,
Felson DT, Bingham CO III, Birnbaum NS, Burmester GR, Bykerk VP,
Cohen MD, et al: 2010 Rheumatoid arthritis classification criteria:
An American college of rheumatology/european league against
rheumatism collaborative initiative. Arthritis Rheum. 62:2569–2581.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Koh ET, Seow A, Pong LY, Koh WH, Chan L,
Howe HS, Lim TH and Low CK: Cross cultural adaptation and
validation of the Chinese health assessment questionnaire for use
in rheumatoid arthritis. J Rheumatol. 25:1705–1708. 1998.PubMed/NCBI
|
|
31
|
Anderson J, Caplan L, Yazdany J, Robbins
ML, Neogi T, Michaud K, Saag KG, O'Dell JR and Kazi S: Rheumatoid
arthritis disease activity measures: American college of
rheumatology recommendations for use in clinical practice.
Arthritis Care Res (Hoboken). 64:640–647. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Jones-Bolin S: Guidelines for the care and
use of laboratory animals in biomedical research. Curr Protoc
Pharmacol Appendix 4: Appendix 4B. 2012. View Article : Google Scholar
|
|
34
|
Brand DD, Latham KA and Rosloniec EF:
Collagen-induced arthritis. Nat Protoc. 2:1269–1275. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yoo SA, Park BH, Park GS, Koh HS, Lee MS,
Ryu SH, Miyazawa K, Park SH, Cho CS and Kim WU: Calcineurin is
expressed and plays a critical role in inflammatory arthritis. J
Immunol. 177:2681–2690. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lee H, Nah SS, Chang SH, Kim HK, Kwon JT,
Lee S, Cho IH, Lee SW, Kim YO, Hong SJ and Kim HJ: PER2 is
downregulated by the LPS-induced inflammatory response in
synoviocytes in rheumatoid arthritis and is implicated in disease
susceptibility. Mol Med Rep. 16:422–428. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Cho YG, Cho ML, Min SY and Kim HY: Type II
collagen autoimmunity in a mouse model of human rheumatoid
arthritis. Autoimmun Rev. 7:65–70. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Mo YQ, Dai L, Zheng DH, Zhu LJ, Wei XN,
Pessler F, Shen J and Zhang BY: Synovial infiltration with
CD79a-positive B cells, but not other B cell lineage markers,
correlates with joint destruction in rheumatoid arthritis. J
Rheumatol. 38:2301–2308. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhu LJ, Dai L, Zheng DH, Mo YQ, Ou-Yang X,
Wei XN, Shen J and Zhang BY: Upregulation of tumor necrosis factor
receptor-associated factor 6 correlated with synovitis severity in
rheumatoid arthritis. Arthritis Res Ther. 14:R1332012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Smeets TJ, Barg EC, Kraan MC, Smith MD,
Breedveld FC and Tak PP: Analysis of the cell infiltrate and
expression of proinflammatory cytokines and matrix
metalloproteinases in arthroscopic synovial biopsies: Comparison
with synovial samples from patients with end stage, destructive
rheumatoid arthritis. Ann Rheum Dis. 62:635–638. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Proudman SM, Cleland LG and Mayrhofer G:
Effects of tumor necrosis factor-alpha, interleukin 1beta, and
activated peripheral blood mononuclear cells on the expression of
adhesion molecules and recruitment of leukocytes in rheumatoid
synovial xenografts in SCID mice. J Rheumatol. 26:1877–1889.
1999.PubMed/NCBI
|
|
42
|
Högenauer K, Arista L, Schmiedeberg N,
Werner G, Jaksche H, Bouhelal R, Nguyen DG, Bhat BG, Raad L, Rauld
C and Carballido JM: G-protein-coupled bile acid receptor 1
(GPBAR1, TGR5) agonists reduce the production of proinflammatory
cytokines and stabilize the alternative macrophage phenotype. J Med
Chem. 57:10343–10354. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ichikawa R, Takayama T, Yoneno K, Kamada
N, Kitazume MT, Higuchi H, Matsuoka K, Watanabe M, Itoh H, Kanai T,
et al: Bile acids induce monocyte differentiation toward
interleukin-12 hypo-producing dendritic cells via a TGR5-dependent
pathway. Immunology. 136:153–162. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Makarov SS: NF-kappa B in rheumatoid
arthritis: A pivotal regulator of inflammation, hyperplasia, and
tissue destruction. Arthritis Res. 3:200–206. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Mor A, Abramson SB and Pillinger MH: The
fibroblast-like synovial cell in rheumatoid arthritis: A key player
in inflammation and joint destruction. Clin Immunol. 115:118–128.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Zheng Y, Sun L, Jiang T, Zhang D, He D and
Nie H: TNFalpha promotes Th17 cell differentiation through IL-6 and
IL-1beta produced by monocytes in rheumatoid arthritis. J Immunol
Res. 2014:3853522014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Xin W, Huang C, Zhang X, Xin S, Zhou Y, Ma
X, Zhang D, Li Y, Zhou S, Zhang D, et al: Methyl salicylate
lactoside inhibits inflammatory response of fibroblast-like
synoviocytes and joint destruction in collagen-induced arthritis in
mice. Br J Pharmacol. 171:3526–3538. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Szekanecz Z, Kim J and Koch AE: Chemokines
and chemokine receptors in rheumatoid arthritis. Semin Immunol.
15:15–21. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Smolen JS, Landewe R, Bijlsma J, Burmester
G, Chatzidionysiou K, Dougados M, Nam J, Ramiro S, Voshaar M, van
Vollenhoven R, et al: EULAR recommendations for the management of
rheumatoid arthritis with synthetic and biological
disease-modifying antirheumatic drugs: 2016 update. Ann Rheum Dis.
76:960–977. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Italiani P and Boraschi D: From monocytes
to M1/M2 macrophages: Phenotypical vs. Functional differentiation.
Front Immunol. 5:5142014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Dey A, Allen J and Hankey-Giblin PA:
Ontogeny and polarization of macrophages in inflammation: Blood
monocytes versus tissue macrophages. Front Immunol. 5:6832015.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Gordon S and Taylor PR: Monocyte and
macrophage heterogeneity. Nat Rev Immunol. 5:953–964. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Crocker I, Lawson N and Fletcher J: Effect
of pregnancy and obstructive jaundice on inflammatory diseases: The
work of P S hench revisited. Ann Rheum Dis. 61:307–310. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Rutkauskaite E, Zacharias W, Schedel J,
Müller-Ladner U, Mawrin C, Seemayer CA, Alexander D, Gay RE, Aicher
WK, Michel BA, et al: Ribozymes that inhibit the production of
matrix metalloproteinase 1 reduce the invasiveness of rheumatoid
arthritis synovial fibroblasts. Arthritis Rheum. 50:1448–1456.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Ma JD, Zhou JJ, Zheng DH, Chen LF, Mo YQ,
Wei XN, Yang LJ and Dai L: Serum matrix metalloproteinase-3 as a
noninvasive biomarker of histological synovitis for diagnosis of
rheumatoid arthritis. Mediators Inflammation. 2014:1792842014.
View Article : Google Scholar
|