Introduction
Endothelial dysfunction is a state in which the
endothelial layer of vessels undergoes structural changes and
functional impairment, which is characterized by reduced nitric
oxide (NO) synthesis, impairment of endothelial-mediated
vasodilation and dysregulated angiogenesis (1). Endothelial dysfunction is considered
as an independent predictor of perturbations in the cardiovascular
system and is the initial step in the progression of cardiovascular
diseases. This is due to its central role in the commencement of
atherosclerotic events and the progression of clinical
cardiovascular complications associated with several conditions,
such as diabetes and hypertension (2). Several factors affect endothelial
function, including insulin due to its ability to enhance the
bioavailability of endothelium-derived NO via its interaction with
vascular endothelial cell insulin receptors (3). Binding of insulin results in the
auto-phosphorylation of the receptor and subsequent downstream
activation of the PI3K/Akt signaling pathway, thereby activating
the key enzyme responsible for NO synthesis, endothelial NO
synthase (eNOS) (4,5). It has been shown that conditions
characterized by insulin resistance, such as diabetes and obesity,
are closely associated with endothelial dysfunction (3).
Protein tyrosine phosphatase (PTP) 1B is a soluble
non-transmembrane and cytosolic tyrosine-specific phosphatase that
has been implicated in several pathological conditions, including
diabetes and obesity (6,7). Due to its tyrosine phosphatase
activity, PTP1B negatively regulates the insulin receptor, thus
serving a critical role in insulin resistance (8). Accumulating evidence has suggested
that PTP1B attenuates insulin signaling, and hence, may impair the
vascular effects mediated by insulin (9). PTP1B has also been shown to contribute
to endothelial dysfunction and cardiovascular disorders (9). Several models of PTP1B genetic
deletion and pharmacological inhibition have been used to assess
this role of PTP1B. The inhibition of PTP1B has been reported to be
beneficial in restoring endothelial function and NO-mediated
vasodilation in a mouse model chronic heart failure (CHF) (10). Furthermore, deletion of PTP1B was
found to protect the mice against CHF-induced (11) and high fat diet-induced endothelial
dysfunction (12). In these models,
endothelial function was preserved, and was accompanied by an
increase in eNOS phosphorylation. In adult mice, the selective
deletion of PTP1B in endothelial cells resulted in an improvement
in mouse survival, cardiac function and cardiac hypertrophy
(13). Moreover, endothelial
PTP1B-deficient mice exhibited an improvement in vascular
angiogenic capacity, increased VEGFR phosphorylation, upregulation
of eNOS expression and a substantial decline in the development of
cardiac fibrosis (13). Although
the deletion and inhibition of PTP1B has been proven to be
beneficial for both whole-body glucose homeostasis and endothelial
function, the molecular mechanisms via which PTP1B negatively
alters endothelial functions are not yet fully understood.
PTP1B has been reported to serve a role in the
activation of the endoplasmic reticulum (ER) stress response
(14,15), which is an inflammatory pathway that
has emerged as a strong molecular link between insulin resistance
and endothelial dysfunction (1,16,17).
The ER stress response is triggered in obesity and can cause
insulin resistance and lipid accumulation (18–20),
as well as serve a crucial role in cardiovascular dysfunction
(21,22). The ER is the site for protein
synthesis and folding, and the synthesis of lipids and steroids, as
well as being a large store for intracellular calcium (17). The ER ensures that proteins are
properly folded and prevents their aggregation (23,24).
Physiological and pathological disturbances can overwhelm the ER
due to the surge in protein demand, resulting in the build-up of
misfolded proteins within the lumen of the ER. This activates an
adaptive mechanism known as the unfolded protein response (UPR),
which aims to restore ER homeostasis and increase ER folding
capacity (17,23,25–28).
The UPR mediates its actions via three major ER transmembrane
sensors: Inositol requiring enzyme-1α, activating transcription
factor (ATF)-6 and protein kinase-like ER kinase, which become
activated upon dissociating from the ER resident chaperone
immunoglobulin binding protein (BiP), also known as heat shock
protein family A (Hsp70) member 5 (HSPA5), in response to the
accumulation of unfolded proteins within the ER (17,23,25–28).
However, the sustained activation of the UPR due to continued
stress results in a switch from the adaptive UPR to the
pro-apoptotic and pro-inflammatory ER stress response, causing cell
inflammation, dysfunction and ultimately cell death (17,23,25–28).
Previous studies have shown that ER stress
activation is responsible for endothelial cell apoptosis and
decreased eNOS expression (1,17,29).
Moreover, in mice treated with angiotensin-II, endothelial function
was found to be preserved when ER stress was inhibited, as
evidenced by an enhancement in eNOS activity and phosphorylation
levels, as well as the amelioration of endothelial-mediated
vasodilation (21). The
pharmacological induction of ER stress has been observed to reduce
angiogenic capacity via a mechanism involving reactive oxygen
species and cell death in human umbilical vein endothelial cells
(HUVECs) (30). Activation of ER
stress can increase endothelial cell apoptosis via the elevated
expression level of the pro-apoptotic factor CHOP and enhanced JNK
phosphorylation (31).
PTP1B has been shown play a role in activating the
ER stress response in a complex interaction with insulin resistance
in mouse models of obesity, due to its critical location at the
surface of ER membrane. For instance, it has been demonstrated that
liver-PTP1B deletion from birth or induced in adult mice can
decrease obesity-stimulated ER stress in mouse livers (14,15).
By inducing the ER stress response, PTP1B overactivation may lead
to endothelial cell death, promotion of a prothrombotic state and
endothelial dysfunction (32).
However, to the best of our knowledge, the role of ER stress and
its related key apoptotic signals in PTP1B-mediated endothelial
dysfunction, particularly in relation to its angiogenic capacity,
have not yet been investigated. Therefore, the aim of the present
study was to examine the impact of PTP1B suppression on ER
stress-mediated endothelial cell dysfunction and impaired
angiogenic response, and to assess the contribution of ER
stress-induced apoptosis and activation of underlying apoptotic
pathways in this process.
Materials and methods
Cell culture
EA.hy926 [ATCC® CRL-2922™; American Type
Culture Collection (ATCC)] endothelial cells were cultured in DMEM
containing high glucose and 10% FBS, supplemented with 1%
penicillin/streptomycin (P/S), sodium pyruvate (1%) and L-glutamine
(1%) (all from Gibco; Thermo Fisher Scientific, Inc.). Cells were
incubated at 37°C with 5% CO2 and 95% humidity. Cells
were only cultured up to passage 20 in these experiments.
To culture primary HUVECs (ATCC®
PCS-100-010™; ATCC), cells were maintained in medium 200 containing
low serum growth supplement (2%) and supplemented with P/S (1%)
(all from Gibco; Thermo Fisher Scientific, Inc.). Cells were placed
inside an incubator at 37°C with 5% CO2 and 95%
humidity. HUVECs used in experiments were between passage 1 and 6.
Based on the regulations of the Ministry of Public Health in Qatar,
the Institutional Review Board (IRB) of Qatar University (Doha,
Qatar) has categorized the use of these commercially sourced and
de-identified HUVECs as non-human subject research and hence
exempted the study from IRB ethics review (QU-IRB 008-NR/21).
Cell treatments
For all treatments, 6-well plates were seeded with
endothelial cells (200,000 cells/well) prior to treatment and
cultured for 24 h to allow the cells to adhere. ER stress was
pharmacologically induced by incubating EA.hy926 cells and HUVECs
with 2 mM 1,4-dithiothreitol (DTT; Sigma-Aldrich; Merck KGaA) for
24 h, or 300 nM thapsigargin (TG; Thermo Fisher Scientific, Inc.)
for 5 h at 37°C, respectively. DTT is a strong ER stress inducer
that inhibits disulfide bond formation, which results in the
improper folding of proteins (33).
The unfolded proteins then accumulate within the ER lumen, thereby
activating the UPR and ER stress response (17). On the other hand, TG induces ER
stress via the inhibition of the Ca+2-ATPase pump, which
results in Ca+2 depletion from the ER stores (34). To determine the role of PTP1B in ER
stress activation, cells were treated with or without a PTP1B
inhibitor (BML-267; Abcam), which was administered at 20 µM for 1 h
at 37°C prior to treatment with DTT or TG (35).
In another set of experiments, HUVECs were
stimulated with 20 µM bradykinin (Sigma-Aldrich; Merck KGaA) for 45
min at 37°C (36), following the
treatment of cells with TG (300 nM; 5 h). Then, protein expression
and phosphorylation of eNOS was assessed via western blotting.
HUVECs were also exposed or not to TG with or without PTP1B
inhibitor, and then stimulated with insulin (Sigma-Aldrich; Merck
KGaA) (20 nM; 5 min at 37°C). Then, protein expression and
phosphorylation of Akt was assessed via western blotting.
PTP1B (cat. no. hs.Ri.PTPN1.13.3) (https://eu.idtdna.com/site/order/designtool/index/DSIRNA_PREDESIGN)
and control (cat. no. 51-01-19-08) (https://eu.idtdna.com/site/order/stock/index/trifecta)
small interfering (si) RNA duplexes were designed and synthesized
by Integrated DNA Technologies, Inc., and were used for all PTP1B
gene silencing experiments. Briefly, 50,000 HUVECs/well were seeded
into 6-well plates, followed by a 24-h incubation to allow the
cells to adhere. According to the manufacturer's protocol, cells
were transfected the following day with either control or PTP1B
siRNA duplexes (15 nM) using INTERFERin® transfection
reagent (Polyplus-transfection SA). Negative control cells were
left untreated. Cells were incubated for 24 h, after which the
medium was replaced with fresh complete medium. Cells were treated
for a total duration of 48 h, prior to treatment with 300 nM TG for
5 h. Western blot analysis showed that protein expression level of
PTP1B was not affected when cells were treated with scrambled
control siRNA duplexes (data not shown), and therefore, the
remainder of experiments were conducted using ‘untreated cells’ as
negative controls.
Western blot analysis
Following cell treatments, the medium was aspirated
and discarded, and then cells were washed with ice-cold PBS (Thermo
Fisher Scientific, Inc.). Next, EA.hy926 cells or HUVECs were lysed
in cold RIPA lysis buffer (Tris (0.5 M, pH 6.8) and SDS (20%);
Thermo Fisher Scientific, Inc.) supplemented with a cocktail of
protease and phosphatase inhibitors (Thermo Fisher Scientific,
Inc.). The protein concentration was determined using a BCA assay
(Thermo Fisher Scientific, Inc.). Equal quantities of proteins
(10–20 µg) were separated using SDS-PAGE gels (8–12%, according to
the molecular weights of targets). Proteins were then transferred
to PVDF membranes (Thermo Fisher Scientific, Inc.), which were
blocked for 1 h at room temperature in TBS containing 0.1% Tween-20
(TBST; Sigma-Aldrich; Merck KGaA) and either 5% BSA (Thermo Fisher
Scientific, Inc.) (phosphorylated targets) or dry milk. Membranes
were then washed with TBST and incubated with the following primary
antibodies overnight at 4°C: BiP (cat. no. 3183S), CHOP (cat. no.
2895S), phosphorylated (p)-eNOS (Ser1177; cat. no. 9571S), eNOS
(cat. no. 5880S), p-Akt (Ser473; cat. no. 4060L), Akt (cat. no.
4691L), cleaved caspase-9 (cat. no. 9505S), cleaved poly
(ADP-ribose) polymerase (PARP-1; cat. no. 5625S), p-ERK 1/2 MAPK
(Thr202/204; cat. no. 9106S), ERK 1/2 MAPK (cat. no. 9102S), LC-3
I/II (cat. no. 12741S), and PTP1B (cat. no. 5311S; all 1:1,000;
Cell Signaling Technology, Inc.), caspase-12 (cat. no. ab18766;
1:1,000; Abcam) and mouse anti-β-actin (cat. no. sc-47778; 1:5,000;
Santa Cruz Biotechnology, Inc.). Membranes were then washed again
with TBST and incubated for 1 h at room temperature with the
corresponding HRP-conjugated anti-rabbit (cat. no. 7074S) or
anti-mouse (cat. no. 7076S) secondary antibodies (1:10,000; Cell
Signaling Technology, Inc.). To visualize the chemiluminescence
signal, membranes were incubated with SignalFire™ Elite ECL reagent
(Cell Signaling Technology, Inc.) and images were captured using
FluorChem™ M imaging system (ProteinSimple). Densitometry was
conducted using Scion image software 4.0 (Scion Corporation) to
assess protein band intensity.
Total RNA isolation and gene
expression analysis using reverse transcription-quantitative
(RT-q)PCR
After treatments, EA.hy926 cells or HUVECs were
washed, trypsinized and collected. The innuPREP RNA Mini kit
(Analytik Jena AG) was used to extract total RNA as per the
manufacturer's protocol. A NanoDrop 2000 system (Thermo Fisher
Scientific, Inc.) was used to determine the concentration and
quality of RNA. For reverse transcription, a RevertAid Reverse
Transcription kit (Thermo Fisher Scientific, Inc.) and an oligo
(dT) 12–18 primer were used to synthesize cDNA from a total of 500
ng RNA, following the manufacturer's instructions. A mixture of
cDNA, human primers for target genes (primer pair sequences are
summarized in Table I) and
GoTaq® qPCR Master mix (Promega Corporation) were
amplified in an Applied Biosystems 7500 Fast Real-Time PCR system
(Thermo Fisher Scientific, Inc.) (33). The thermocycling conditions were as
follows: holding at 95°C for 10 min, cycling stage (40 cycles; 95°C
for 15 sec, 60°C for 60 sec, 72°C for 40 sec), and holding stage at
95°C for 60 s, followed by melting curve analysis. Experiments were
conducted with six independent biological repeats. To assess
relative gene expression, the comparative 2−∆∆Cq method
(37) was used to analyze data.
mRNA expression was normalized against the housekeeping gene
β-actin and reported as fold-change after normalization to
control group. Sourcing of the human primers pairs was conducted
using the PrimerBank (https://pga.mgh.harvard.edu/primerbank/), followed by
their synthesis by Sigma-Aldrich (Merck KGaA).
 | Table I.List of human primer pairs used in
the present study. |
Table I.
List of human primer pairs used in
the present study.
| Target gene | Forward primer
(5′→3′) | Reverse primer
(5′→3′) |
|---|
| ATF-4 |
CCCTTCACCTTCTTACAACCTC |
TGCCCAGCTCTAAACTAAAGGA |
| β-actin |
CATGTACGTTCGTATCCAGGC |
CTCCTTAATGTCACGCACGAT |
| BiP |
CATCACGCCGTCCTATGTCG |
CGTCAAAGACCGTGTTCTCG |
| CHOP |
GAACGGCTCAAGCAGGAAATC |
TTCACCATTCGGTCAATCAGAG |
| FGF-1 |
ACACCGACGGGCTTTTATACG |
CCCATTCTTCTTGAGGCCAAC |
| FGF-2 |
AGAAGAGCGACCCTCACATCA |
CGGTTAGCACACACTCCTTTG |
| GRP94 |
GCTGACGATGAAGTTGATGTGG |
CATCCGTCCTTGATCCTTCTCTA |
| ICAM-1 |
ATGCCCAGACATCTGTGTCC |
GGGGTCTCTATGCCCAACAA |
| IL-6 |
AAATTCGGTACATCCTCGACGG |
GGAAGGTTCAGGTTGTTTTCTGC |
| IL-8 |
ACTGAGAGTGATTGAGAGTGGAC |
AACCCTCTGCACCCAGTTTTC |
| MCP-1 |
TTAAAAACCTGGATCGGAACCAA |
GCATTAGCTTCAGATTTACGGGT |
| PTP1B |
GCAGATCGACAAGTCCGGG |
GCCACTCTACATGGGAAGTCAC |
| VEGF-A |
AGGGCAGAATCATCACGAAGT |
AGGGTCTCGATTGGATGGCA |
Endothelial cell tube-like structures
formation assay
The angiogenic capacity of endothelial cells was
assessed in vitro using a Geltrex™ Matrigel matrix (Thermo
Fisher Scientific, Inc.). HUVECs were seeded into 6-well plates
(200,000 cells/well) and left for 24 h to adhere. The following
day, cells were treated with 300 nM TG for 5 h with or without 20
µM PTP1B inhibitor at 1 h prior to treatment with TG. Control cells
were left untreated. Geltrex™ was stored overnight in the
refrigerator (4°C) for thawing prior to the experiment. On the
experiment day, Geltrex™ (100 µl) was loaded onto 24-well plates
and the gel was spread uniformly. The gel was allowed to solidify
by placing the gel-coated wells in the incubator (37°C) for 30 min.
During the gel incubation time, treated cells were collected and
counted. Then, a cell suspension containing 50,000 cells (100 µl)
was added onto each well. The plates were placed back inside the
incubator. Tube formation was visualized, and images were captured
after 4 h of incubation using a phase contrast inverted microscope
at ×40 magnification (OPTIKA Srl). Web-based WimTube software from
Wimasis Image Analysis (Onimagin Technologies SCA) was used to
semi-quantitatively analyze the images. The comparison across the
experimental groups was performed by averaging the counted total
tube lengths per condition, from five random blind fields.
Tali-based assay for apoptosis and
cell cycle analysis
A Tali™ Apoptosis kit (Thermo Fisher Scientific,
Inc.) was used, according to the manufacturer's instructions, to
assess apoptosis in HUVECs challenged with TG (300 nM; 5 h) with or
without PTP1B inhibitor (20 µM) or PTP1B siRNA (15 nM, 48 h).
Untreated cells were used as controls. After treatments, cells were
collected, washed, and then pelleted via centrifugation at 1,500 ×
g (5 min; 4°C). Then, 100 µl 1X Annexin Binding Buffer (ABB) was
used to resuspend the pellet. Next, each sample was admixed with 5
µl Annexin V solution and incubated at room temperature for 20 min
in the dark. Following the incubation period, samples were
centrifuged at 1,500 × g (5 min; 4°C), and the pellets were
resuspended in fresh 1X ABB (100 µl). Finally, samples were mixed
with 1 µl PI (100 µg/ml), and the mixture was incubated for 10 min
in the dark. Stained cells (25 µl) were loaded into the Tali
Cellular Analysis Slides (Thermo Fisher Scientific, Inc.) and
imaged using Tali Image-Based Cytometer (Thermo Fisher Scientific,
Inc.), following the manufacturer's instructions. The percentages
of live, dead and apoptotic cells in each cell preparation were
included in the analysis. Representative fluorescence images are
presented to indicate apoptotic cells in green color, dead cells in
red and green (appear yellow), and live cells with little or no
fluorescence.
For cell cycle analysis, cells were harvested
following treatments, washed, and then centrifuged at 1,500 × g (5
min; 4°C). According to the manufacturer's protocol, PI solution
(Thermo Fisher Scientific, Inc.) was used to stain the cells. After
incubation in the dark for 30 min at 37°C, stained cells (25 µl)
were loaded into the Tali Cellular Analysis Slides (Thermo Fisher
Scientific, Inc.) and then imaged using Tali Image-Based Cytometer
(Thermo Fisher Scientific, Inc.), according to the manufacturer's
instructions. The percentages of cells in each phase of the cell
cycle were determined as follows: Sub-G1 (apoptotic
cells), G0/G1, S and G2/M.
Representative fluorescence images of cell cycle phases are shown:
Red (Sub-G1), orange (G0/G1), blue
(S) and green (G2/M).
Statistical analysis
The data are presented as the mean ± SEM, and ‘n’
represents the number of biological repeats. The normality of data
was tested each time using a Shapiro-Wilk normality test. GraphPad
Prism 7.01e software for Mac (GraphPad Software, Inc.) was used to
perform the statistical analyses. One-way ANOVA followed by Tukey's
multiple comparison post hoc test (for data with Gaussian
distribution) or the non-parametric Kruskal-Wallis test followed by
Dunn's multiple comparison post hoc test (data with non-Gaussian
distribution) were used to analyze the data. P<0.05 was
considered to indicate a statistically significant difference.
Results
Effect of PTP1B inhibition on ER
stress in endothelial cells
The pharmacological induction of ER stress in
EA.hy926 cells was achieved by treating cells with DTT (2 mM) for
24 h. Then, the mRNA and protein expression levels of several
markers of ER stress were assessed using qPCR and western blot
analysis, respectively (Fig. 1). To
determine the role of PTP1B in the ER stress response, a group of
cells were pre-incubated with a PTP1B inhibitor (20 µM) prior to
DTT treatment. It was found that ER stress was significantly
induced by DTT, as shown by the significant upregulation in the
mRNA expression levels of key ER stress markers, CHOP
(Fig. 1A), BiP (Fig. 1B), 94 kDa glucose-regulated protein
(GRP94) (Fig. 1C) and
ATF-4 (Fig. 1D). A
significant reduction in CHOP and GRP94 gene
expression was observed after PTP1B inhibition (Fig. 1A and C). Similarly, EA.hy926 cells
treated with DTT exhibited an increase in protein expression levels
of CHOP and BiP (Fig. 1E).
Moreover, PTP1B inhibition caused a significant decrease in protein
expression level of CHOP, while the protein expression of BiP was
not affected (Fig. 1E left and
right panels).
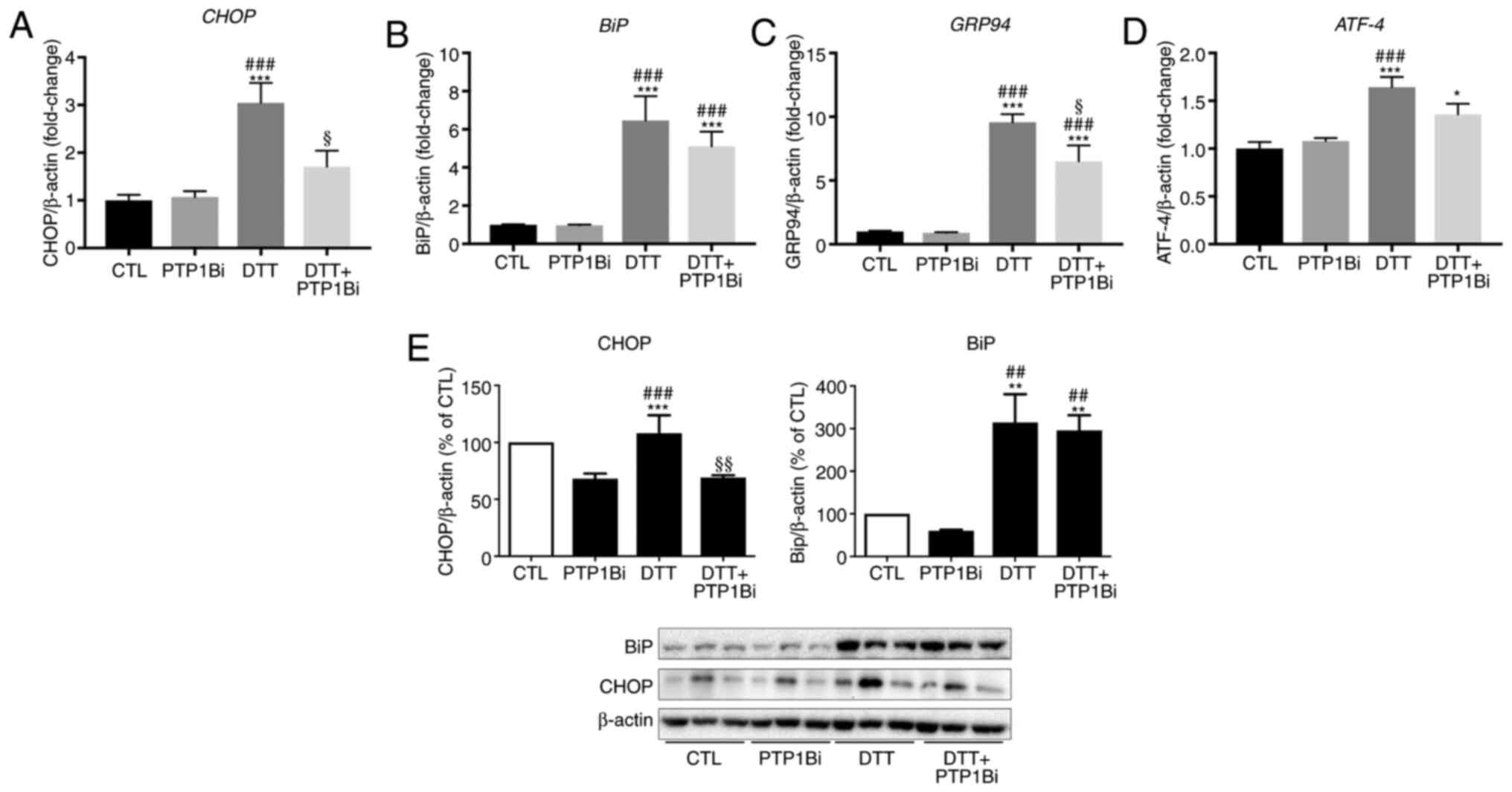 | Figure 1.Impact of PTP1B inhibition on
pharmacologically-induced endoplasmic reticulum stress in EA.hy926
cells. Reverse transcription-quantitative PCR analysis to assess
mRNA expression of (A) CHOP, (B) BiP, (C)
GRP94 and (D) ATF-4 and western blot analysis to
assess protein expression levels of (E) CHOP and BiP in EA.hy926
endothelial cells exposed to DTT (2 mM, 24 h) with or without
PTP1Bi (BML, 20 µM, added 1 h prior to treatment. (A-D) mRNA
expression was normalized against the housekeeping gene
β-actin (n=6 in each group). (E) Bars represent the pooled
densitometry data of protein expression of CHOP (left panel) and
BiP (right panel) normalized to loading control β-actin and
expressed as percentage (%) of untreated group (CTL) (n=4 in each
group). All data are presented as the mean ± SEM. *P<0.05,
**P<0.01, ***P<0.001 vs. CTL group; ##P<0.01,
###P<0.001 vs. PTP1Bi group; §P<0.05,
§§P<0.01 vs. DTT group. PTP1B, protein tyrosine
phosphatase 1B; BiP, endoplasmic reticulum resident chaperone
immunoglobulin binding protein; GRP94, 94 kDa glucose-regulated
protein; ATF-4, activating transcription factor 4; DTT,
1,4-dithiothreitol; CTL, control; PTP1Bi, PTP1B inhibitor. |
To further validate these results, HUVECs (primary
endothelial cells model), were treated with TG (300 nM) for 24 h
with or without PTP1B inhibitor. TG treatment resulted in a
significant increase in mRNA expression levels of CHOP
(Fig. 2A), BiP (Fig. 2B), GRP94 (Fig. 2C) and ATF-4 (Fig. 2D). Furthermore, inhibition of PTP1B
significantly prevented the TG-induced effects on all the markers
(Fig. 2A-D). Similar to the effects
observed with gene expression, after treatment with TG, HUVECs
showed a significant enhancement in protein expression levels of
CHOP (Fig. 2E, left panel) and BiP
(Fig. 2E, right panel). Similarly
to EA.hy926 cells, PTP1B inhibition significantly reduced CHOP
protein expression, while it had no impact on the protein
expression of BiP (Fig. 2E).
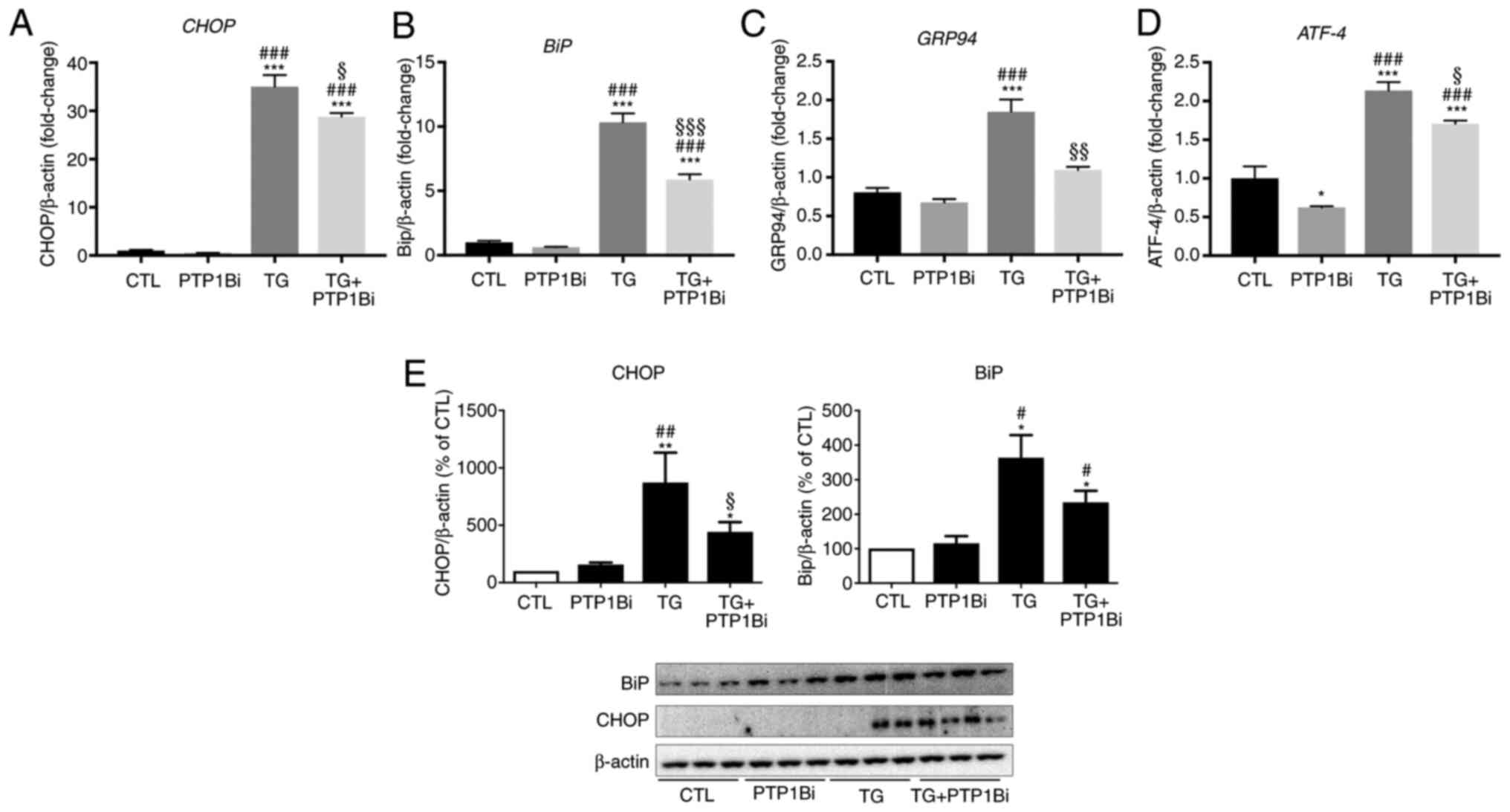 | Figure 2.Impact of PTP1B inhibition on
pharmacologically-induced endoplasmic reticulum stress in HUVECs.
Reverse transcription-quantitative PCR analysis to assess mRNA
expression of (A) CHOP, (B) BiP, (C) GRP94 and
(D) ATF-4 and western blot analysis to assess protein
expression levels of (E) CHOP and BiP in HUVECs exposed to TG (TG;
300 nM, 5 h) with or without PTP1Bi (BML, 20 µM, added 1 h prior to
treatment). (A-D) mRNA expression was normalized against the
housekeeping gene β-actin (n=6 in each group). (E) Bars
represent the pooled densitometry data of protein expression of
CHOP (left panel) and BiP (right panel) normalized to loading
control β-actin and expressed as percentage (%) of untreated group
(CTL) (n=4 in each group). All data are presented as the mean ±
SEM. *P<0.05, **P<0.01, ***P<0.001 vs. CTL group;
#P<0.05, ##P<0.01,
###P<0.001 vs. PTP1Bi group; §P<0.05,
§§P<0.01 and §§§P<0.001 vs. TG group.
PTP1B, protein tyrosine phosphatase 1B; HUVECs, human umbilical
vein endothelial cells; BiP, endoplasmic reticulum resident
chaperone immunoglobulin binding protein; GRP94, 94 kDa
glucose-regulated protein; ATF-4, activating transcription factor
4; TG, thapsigargin; CTL, control; PTP1Bi, PTP1B inhibitor. |
Effect of PTP1B inhibition on eNOS and
Akt activation in HUVECs subjected to ER stress
The activity of eNOS is a hallmark of endothelial
cell function (9,17). To investigate the impact of PTP1B on
endothelial function, it is important to assess the effects of
PTP1B on the NO signaling pathway. As PTP1B inhibition exerted no
changes in the expression level of phosphorylated eNOS (Ser1177) at
the basal levels (data not shown), HUVECs were stimulated with
bradykinin following the treatment of cells with TG.
Bradykinin-treated cells showed a significant increase in eNOS
phosphorylation at its activator site (Ser1177) compared to
controls (Fig. 3A). However, in
cells treated with TG, bradykinin failed to enhance eNOS
phosphorylation, and eNOS phosphorylation levels were reduced
compared with the untreated control group (Fig. 3A). Of note, the pretreatment of
cells with the PTP1B inhibitor significantly prevented the
TG-induced effects.
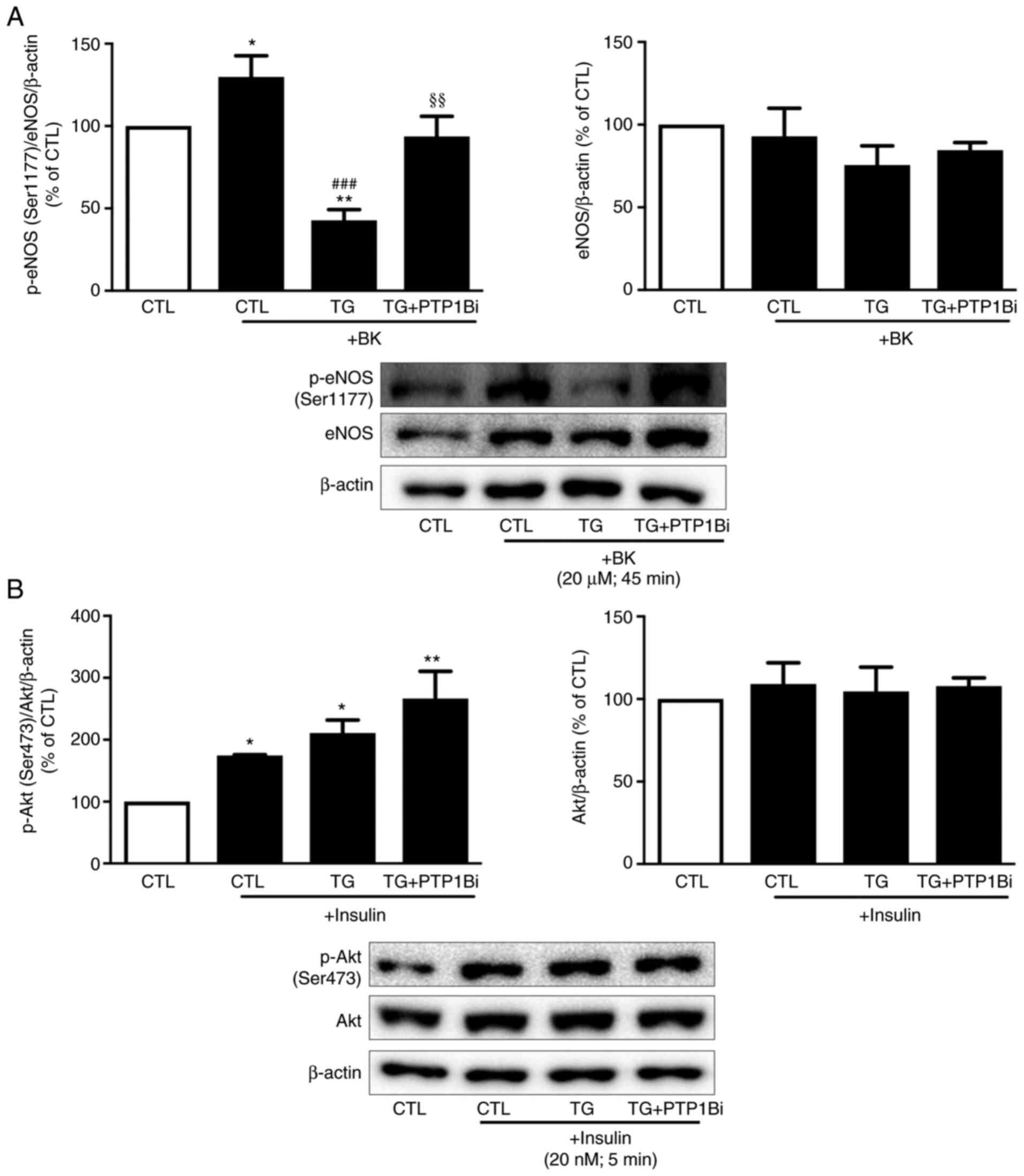 | Figure 3.Impact of PTP1B inhibition on eNOS
and Akt phosphorylation in HUVECs subjected to endoplasmic
reticulum stress. Western blot analysis to assess protein
expression levels of (A) p-eNOS (Ser1177) and total eNOS, and (B)
p-Akt (Ser473) and total Akt in HUVECs treated with TG (300 nM for
5 h) in the presence or absence of PTP1Bi (BML, 20 µM, added 1 h
prior to treatment), followed by the incubation of cells with
either (A) BK (20 µM, 45 min) or (B) insulin (20 nM, 5 min). Left
bars represent the pooled densitometry data of p-proteins
normalized to respective total protein (eNOS or Akt) and to loading
control β-actin. Right bars show pooled densitometry data of total
proteins (eNOS or Akt) normalized to loading control β-actin. Data
are expressed as percentage (%) of untreated group (CTL) (n=4 in
each group). All data are presented as the mean ± SEM. *P<0.05,
**P<0.01 vs. CTL group; ###P<0.001 vs. CTL + BK
group; §§P<0.01 vs. TG + BK group. PTP1B, protein
tyrosine phosphatase 1B; HUVECs, human umbilical vein endothelial
cells; eNOS, endothelial nitric oxide synthase; p-, phosphorylated;
TG, thapsigargin; BK, bradykinin; CTL, control; PTP1Bi, PTP1B
inhibitor. |
As insulin signaling serves a critical role in the
maintenance of endothelial cell function (9), the impact of PTP1B inhibition on the
insulin response was assessed. As shown in Fig. 3B, induction of ER stress by TG did
not alter insulin-mediated Akt phosphorylation. However, the
inhibition of PTP1B enhanced the insulin response, as shown by
increased p-Akt expression following insulin treatment (Fig. 3B).
Effect of PTP1B inhibition on the mRNA
expression levels of pro-inflammatory genes in endothelial cells
subjected to ER stress
ER stress and endothelial dysfunction are closely
associated with cellular inflammation (17). Endothelial dysfunction precedes and
imitates inflammatory response, where, at their surface,
dysfunctional endothelial cells expose various adhesion molecules,
such as intracellular molecular adhesion (ICAM)-1, together with
the release of multiple pro-inflammatory cytokines and chemokines,
including IL-6, IL-8, and monocyte chemoattractant protein (MCP)-1.
In this context, PTP1B is involved in the regulation of
inflammatory responses (9,38). Therefore, the impact of PTP1B
inhibition on the mRNA expression levels of key inflammatory
molecules was assessed in endothelial cells exposed to ER
stressors. It was identified that the exposure of HUVECs to TG (300
nM; 5 h) caused a significant increase in the mRNA expression
levels of IL-6 (Fig. 4A),
IL-8 (Fig. 4B),
ICAM-1 (Fig. 4C) and
MCP-1 (Fig. 4D). In
addition, PTP1B inhibition partially prevented this increase for
all these molecules except MCP-1 (Fig. 4A-D), showing a significant effect.
Consistent with these observations, the treatment of EA.hy926
endothelial cells with DTT (2 mM; 24 h) also increased the mRNA
expression levels of IL-6 (Fig.
4E) and IL-8 (Fig. 4F),
which were both partially prevented in the presence of PTP1B
inhibitor. These data demonstrated the protective effect of PTP1B
inhibition against the ER stress-mediated increased in the mRNA
expression levels of pro-inflammatory factors in endothelial
cells.
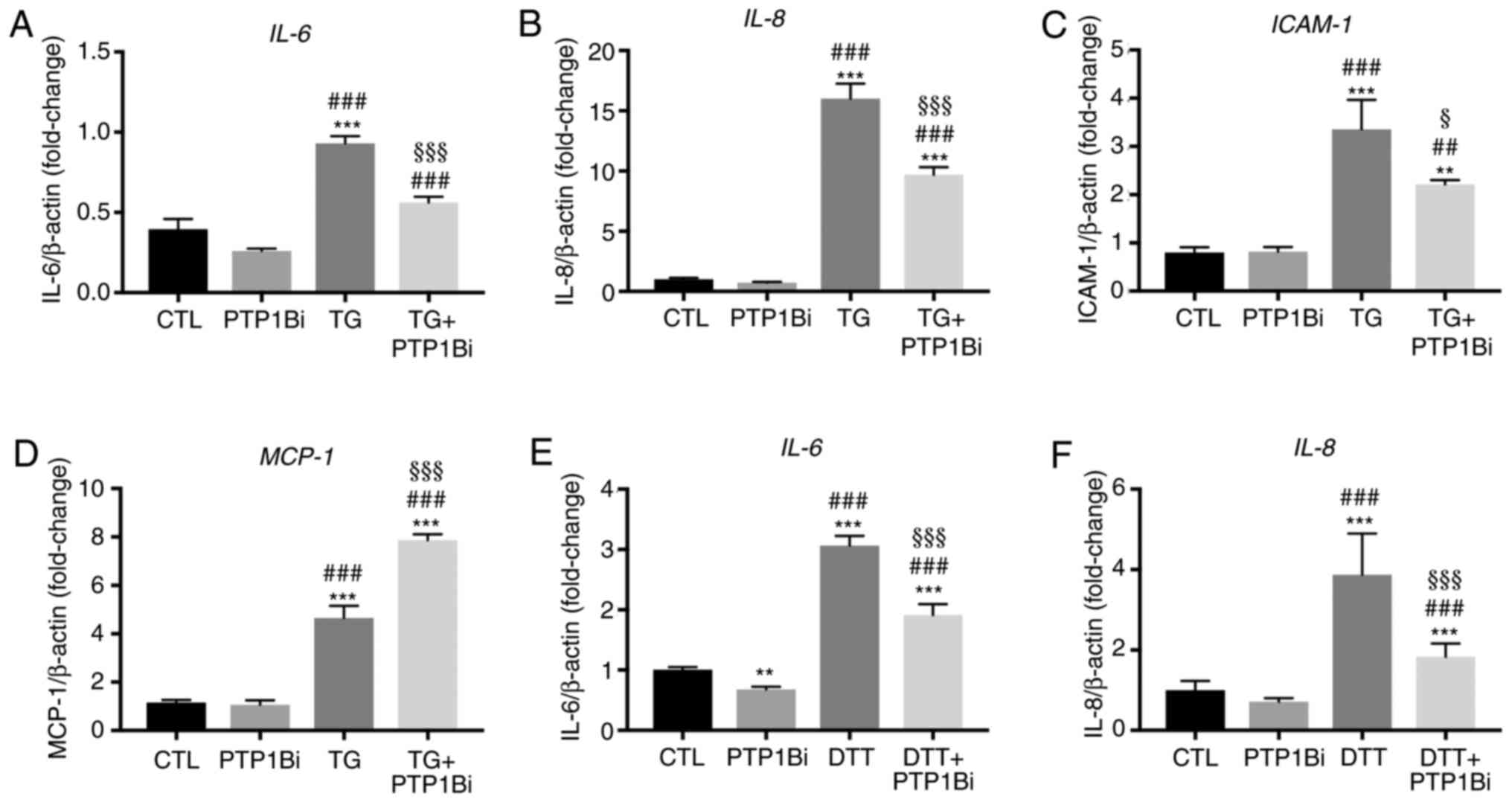 | Figure 4.Impact of PTP1B inhibition on the
mRNA expression levels of pro-inflammatory genes in endothelial
cells subjected to endoplasmic reticulum stress. (A-D) Human
umbilical vein endothelial cells were exposed to TG (300 nM, 5 h)
with or without PTP1Bi (BML, 20 µM, added 1 h prior to treatment).
Then, relative mRNA expression of (A) IL-6, (B) IL-8,
(C) ICAM-1 and (D) MCP-1, was determined and
normalized against housekeeping gene β-actin (n=6 per group). (E
and F) EA.hy926 cells were exposed to DTT (2 mM, 24 h) with or
without PTP1Bi (BML, 20 µM, added 1 h prior to treatment). Then,
relative mRNA expression of (E) IL-6 and (F) IL-8 was
determined and normalized against housekeeping gene β-actin (n=6
per group). All data are presented as the mean ± SEM. **P<0.01,
***P<0.001 vs. CTL group; ##P<0.01,
###P<0.001 vs. PTP1Bi group; §P<0.05,
§§§P<0.001 vs. TG group. PTP1B, protein tyrosine
phosphatase 1B; ICAM-1, intracellular molecular adhesion-1; MCP-1,
monocyte chemoattractant protein-1; TG, thapsigargin; DTT,
1,4-dithiothreitol; CTL, control; PTP1Bi, PTP1B inhibitor. |
Effect of PTP1B inhibition on the
angiogenic capacity of endothelial cells subjected to ER
stress
Another hallmark of endothelial function is the
angiogenic capacity of endothelial cells, which is the capacity of
endothelial cells to re-arrange themselves to create a tube-like
structure on a 3-diemnsional matrix. This capacity is mainly
regulated under the effects of VEGF-A activity (30). To study the impact of PTP1B
inhibition on the angiogenic capacity of endothelial cells, HUVECs
treated with TG with or without a PTP1B inhibitor were collected
and seeded on top of a Matrigel matrix layer, then incubated for a
further 4–6 h, allowing them time to form tube-like structures. It
was demonstrated that TG significantly impaired the ability of the
HUVECs to form tube-like structures (Fig. 5A), as evidenced by the reduction in
the average length of tubes formed (Fig. 5B), indicating impairment in
angiogenic capacity. The pre-treatment of cells with the PTP1B
inhibitor prevented the deleterious effects of TG on tube-like
structure formation (Fig. 5A and
B).
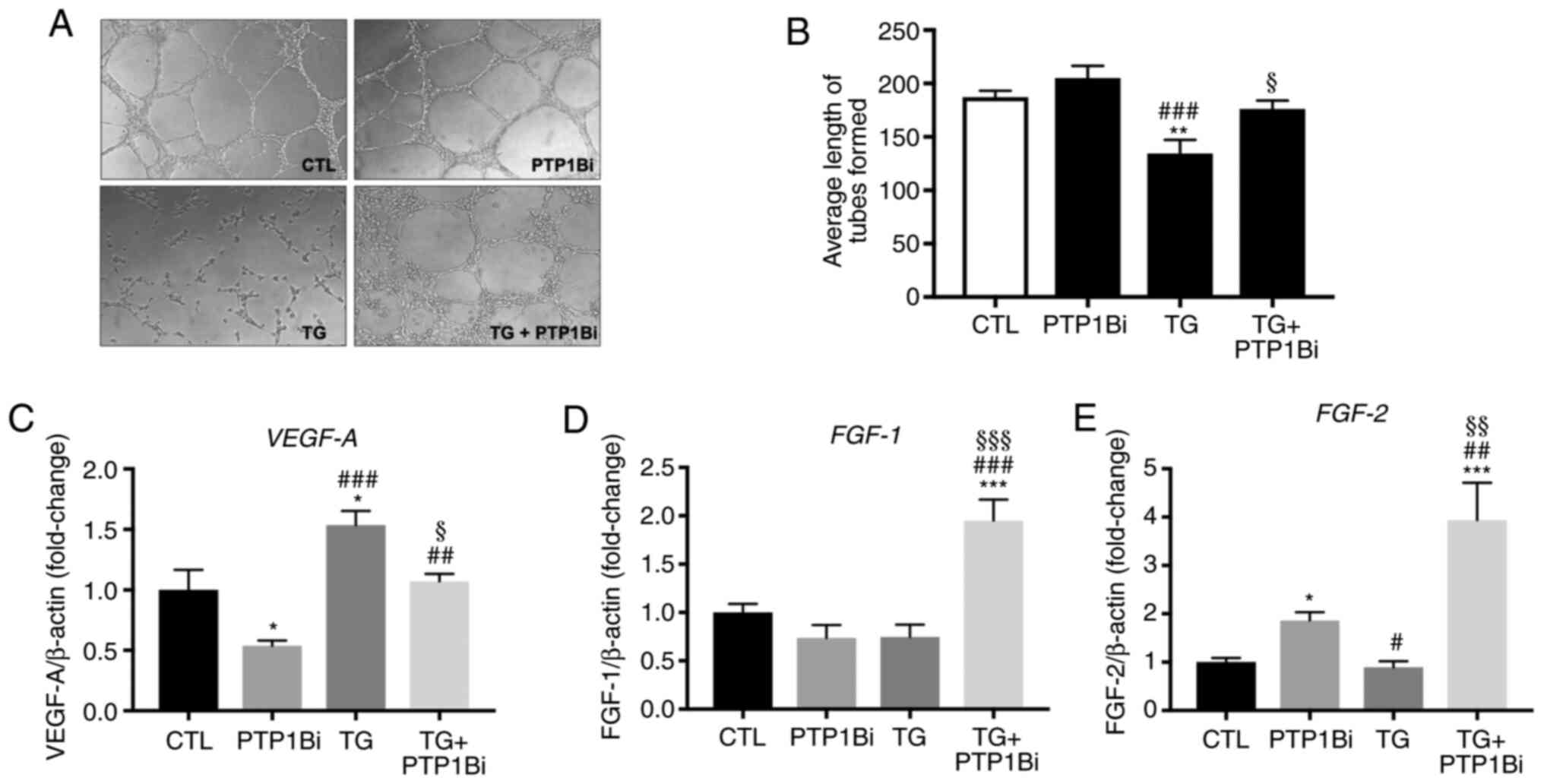 | Figure 5.Impact of PTP1B inhibition on the
angiogenic capacity of HUVECs subjected to pharmacological
endoplasmic reticulum stress. (A) Matrigel-based tube formation
assay of HUVECs treated with TG (TG; 300 nM, 5 h) with or without
PTP1Bi (BML, 20 µM, added 1 h prior to treatment). Images are
representative of three independent experiments. (B) Bars represent
pooled data of the quantification of angiogenic capacity, expressed
as the average length of tubes formed that were counted in five
random fields for each well using WimTube software (n=3 per group).
Relative mRNA expression of (C) VEGF-A, (D) FGF-1 and
(E) FGF-2, normalized against housekeeping gene β-actin (n=6
per group). All data are presented as the mean ± SEM. *P<0.05,
**P<0.01, ***P<0.001 vs. CTL group; #P<0.05,
##P<0.01, ###P<0.001 vs. PTP1Bi group;
§P<0.05, §§P<0.01,
§§§P<0.001 vs. TG group. PTP1B, protein tyrosine
phosphatase 1B; HUVECs, human umbilical vein endothelial cells; TG,
thapsigargin; PTP1Bi, PTP1B inhibitor; FGF, fibroblast growth
factor; CTL, control. |
Subsequently, the mRNA expression levels of various
pro-angiogenic molecules were assessed. As presented in Fig. 5C, the treatment of cells with TG
induced an upregulation in VEGF-A mRNA expression, while the
pre-treatment of cells with a PTP1B inhibitor significantly reduced
this effect. On the other hand, PTP1B inhibition did not affect the
mRNA expression levels of fibroblast growth factor (FGF)-1
(Fig. 5D) and moderately increased
those of FGF-2 (Fig. 5E).
Although the treatment of HUVECs with TG alone did not affect mRNA
expression of FGF-1 (Fig.
5D) and FGF-2 (Fig. 5E)
compared with the control group, the pre-treatment of cells with a
PTP1B inhibitor enhanced the mRNA expression levels of both
FGF-1 and FGF-2 in the presence of TG (Fig. 5D and E) compared with all other
groups.
Effect of PTP1B inhibition and
silencing on ER stress-mediated apoptosis
ER stress-induced apoptosis is a major contributor
to the loss of endothelial cells, which predisposes individuals to
strokes and cardiovascular complications (39). Therefore, the current study assessed
the impact of PTP1B suppression on the survival of HUVECs exposed
to pharmacological ER stress. As shown in Fig. 6A, cell cycle analysis revealed that
TG-mediated ER stress induction resulted in cell cycle progression
disruption, as evidenced by the increase in the percentage of cells
in sub-G1 phase (apoptotic cells) and a reduction in the
proportion of cells in G0/G1 phase, in
addition to the increase in the percentage of cells in
G2/M phase of the cycle, compared with both untreated
cells and those pre-treated with PTP1B inhibitor alone. Of note,
PTP1B inhibition prevented all the effects of ER stress activation
on cell cycle progression (Fig.
6A).
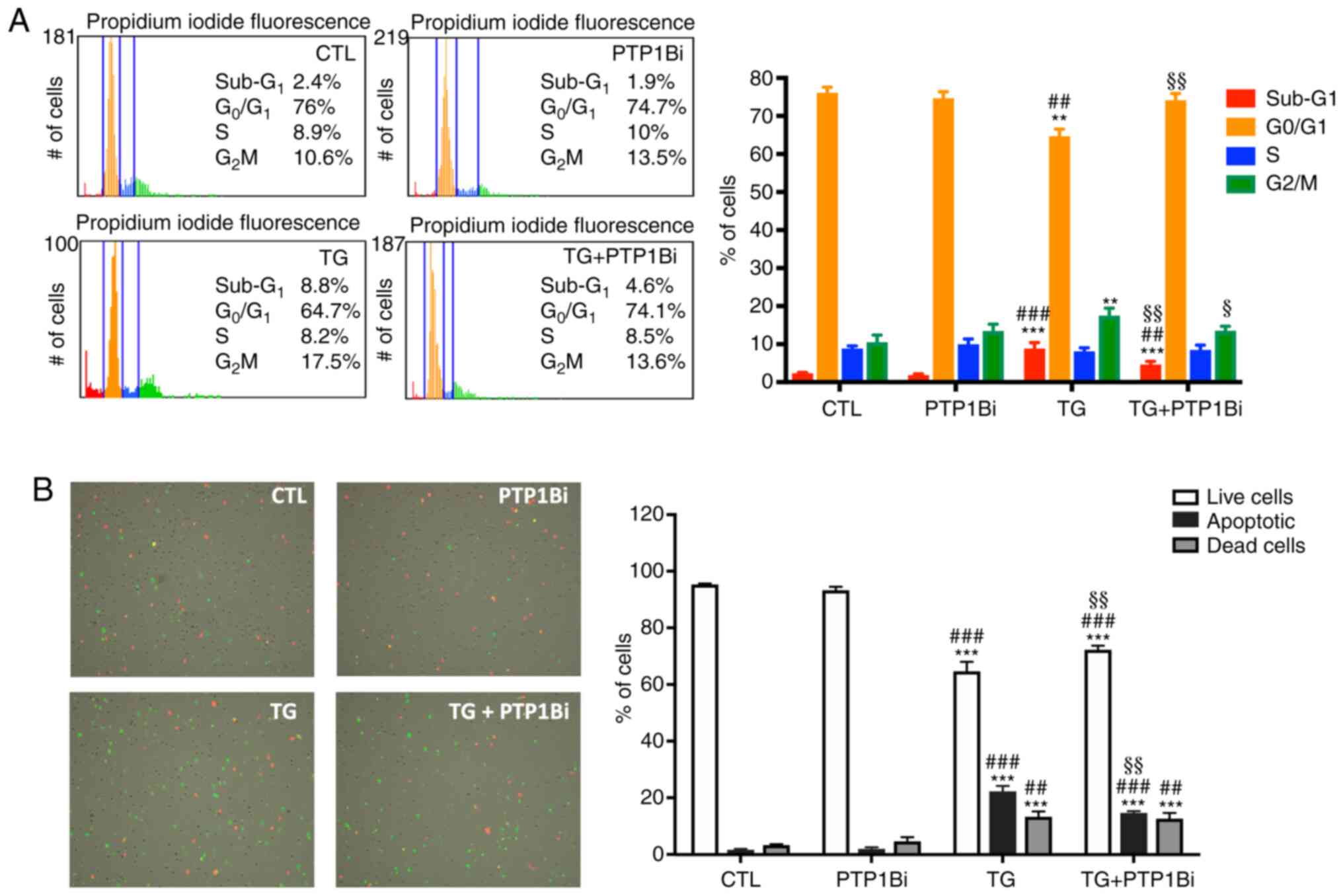 | Figure 6.Impact of PTP1B inhibition on cell
survival of HUVECs subjected to pharmacological endoplasmic
reticulum stress. (A) Tali image-based cytometer analysis of cell
cycle in HUVECs treated with TG (300 nM, 5 h) treated with or
without PTP1Bi (BML, 20 µM, added 1 h prior to treatment) (n=4 in
each group). Bars represent the percentages of cells in each phase
of cell cycle: Sub-G1 (apoptotic cells),
G0/G1, S and G2/M. Representative
fluorescence images of cell cycle phases are shown: Red
(Sub-G1 or apoptotic), orange (G0/G1), blue
(S) and green (G2/M). (B) Tali image-based cytometer
analysis of apoptosis in HUVECs treated with TG (300 nM, 5 h)
treated with or without PTP1Bi (BML, 20 µM, added 1 h prior to
treatment) (n=4 in each group). Bars represent the percentages of
live, apoptotic and dead cells. Representative fluorescence images
are shown to indicate apoptotic cells in green color, dead cells in
red and green (appear yellow), and live cells with little or no
fluorescence. Average percentage of cells in each phase of the cell
cycle is shown on each Tali representative fluorescence image of
each experimental group. All data are presented as the mean ± SEM.
**P<0.01, ***P<0.001 vs. CTL group; ##P<0.01,
###P<0.001 vs. PTP1Bi group; §P<0.05,
§§P<0.01 vs. TG group. PTP1B, protein tyrosine
phosphatase 1B; HUVECs, human umbilical vein endothelial cells; TG,
thapsigargin; PTP1Bi, PTP1B inhibitor; CTL, control. |
Subsequently, the impact of PTP1B inhibition on the
number of apoptotic cells following exposure to TG was
investigated. The results demonstrated that TG caused a significant
reduction in the number of live HUVECs, while at the same time
increasing the number of dead and apoptotic cells (Fig. 6B). Moreover, the inhibition of PTP1B
resulted in a marked decrease in the number of apoptotic cells,
thus reducing the pro-apoptotic actions of TG (Fig. 6B). This finding indicated the
beneficial role of PTP1B inhibition in reducing ER stress-mediated
apoptosis. To further examine the role of PTP1B in this mechanism,
instead of pharmacologically inhibiting PTP1B, its protein
expression was knocked down using specific siRNA duplexes. As shown
in Fig. 7A, the treatment of HUVECs
with PTP1B siRNA duplexes significantly reduced PTP1B mRNA
expression compared with untreated cells and those incubated with
scrambled siRNA. HUVECs were incubated with or without PTP1B siRNA
for 48 h to knockdown PTP1B protein expression, followed by
incubation with or without TG. As presented in Fig. 7B, TG-induced ER stress caused a
slight, but statistically significant, increase in PTP1B protein
expression compared with the control group, which was not observed
in the PTP1B knockdown groups. The exposure of HUVECs to TG caused
the number of live cells to decrease significantly, while
increasing the proportion of apoptotic cells (Fig. 7C), which was similar to what was
observed with PTP1B inhibitor as depicted in Fig. 6B. This pro-apoptotic effect caused
by TG was partially, but significantly prevented in endothelial
cells subjected to PTP1B knockdown (Fig. 7C).
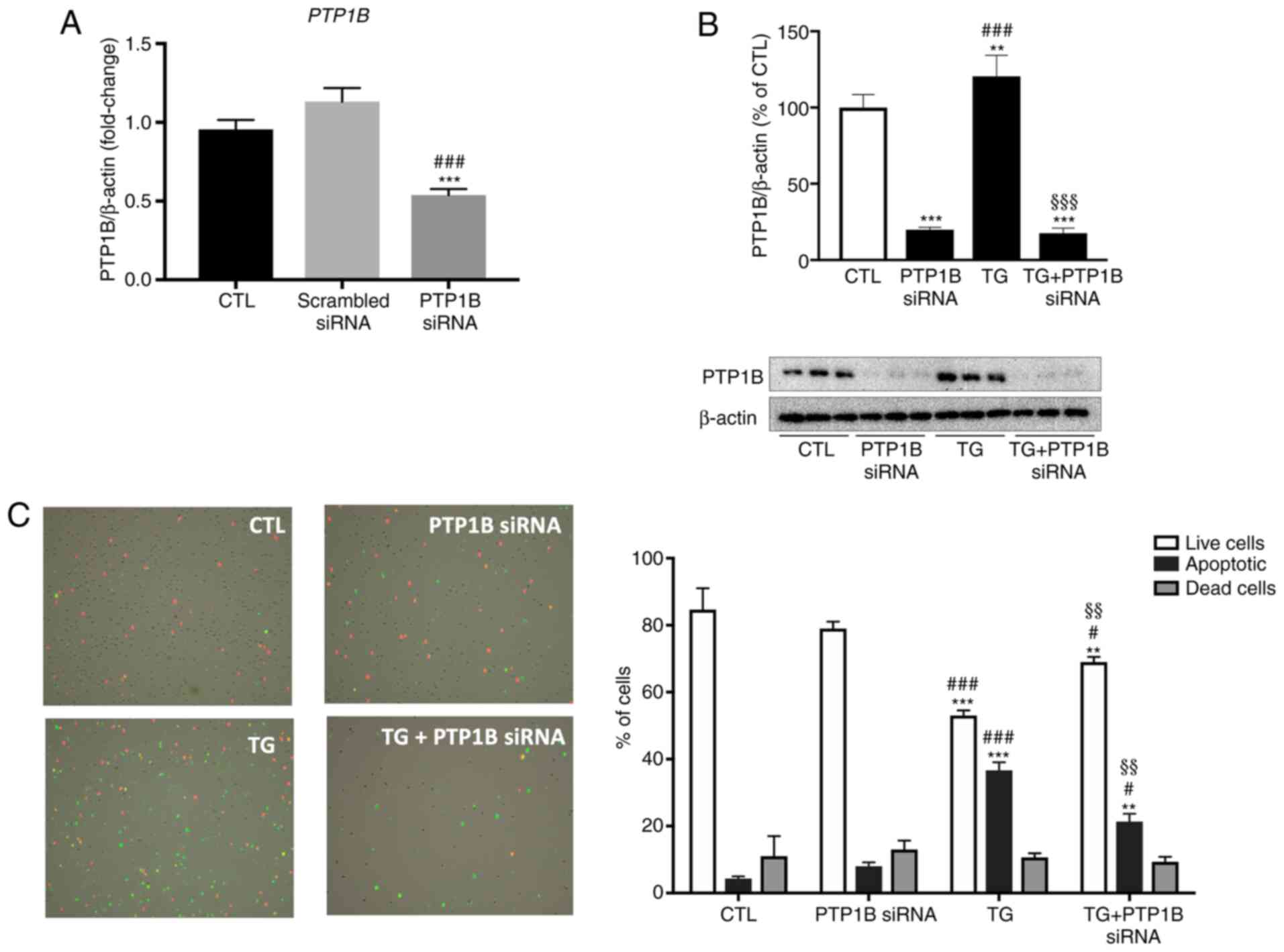 | Figure 7.Impact of PTP1B silencing on
apoptosis of HUVECs subjected to pharmacological endoplasmic
reticulum stress. (A) Relative mRNA expression of PTP1B in
untreated HUVECs and cells treated with either scrambled or
PTP1B siRNA duplexes (15 nM for 72 h) normalized against
housekeeping gene β-actin (n=6 per group). (B) Western blot
analysis to assess protein expression levels of PTP1B in HUVECs
incubated with or without PTP1B siRNA duplexes (15 nM for 72
h) in the presence or absence of TG (300 nM, 5 h). Bars represent
the pooled densitometry data normalized to housekeeping protein
β-actin and expressed as percentage (%) of untreated group (CTL)
(n=4 in each group). (C) Tali image-based cytometer analysis for
apoptosis of HUVECs treated with TG (300 nM, 5 h) with or without
PTP1B siRNA duplexes (n=4 in each group). Bars represent the
percentages of live, apoptotic and dead cells. Representative
fluorescence images are shown to indicate apoptotic cells in green
color, dead cells in red and green (appear yellow), and live cells
with little or no fluorescence. All data are presented as the mean
± SEM. **P<0.01, ***P<0.001 vs. CTL group;
#P<0.05, ###P<0.001 vs. scrambled siRNA
or PTP1B siRNA group; §§P<0.01,
§§§P<0.001 vs. TG group. PTP1B, protein tyrosine
phosphatase 1B; HUVECs, human umbilical vein endothelial cells;
siRNA, small interfering RNA; TG, thapsigargin; CTL, control. |
To investigate the role PTP1B serves in ER
stress-mediated cell death, the protein expression levels of
several apoptotic effectors were determined in HUVECs subjected to
pharmacological ER stress with or without PTP1B inhibitor. While no
changes in the protein expression levels of cleaved caspase-9
(Fig. 8A) and caspase-12 (Fig. 8B) were caused by TG, the
pre-treatment of cells with a PTP1B inhibitor significantly reduced
the expression level of caspase-12 (Fig. 8B) compared with the control or TG
alone groups. Moreover, TG increased the protein expression level
of cleaved PARP-1, while PTP1B inhibition prevented this effect
(Fig. 8C).
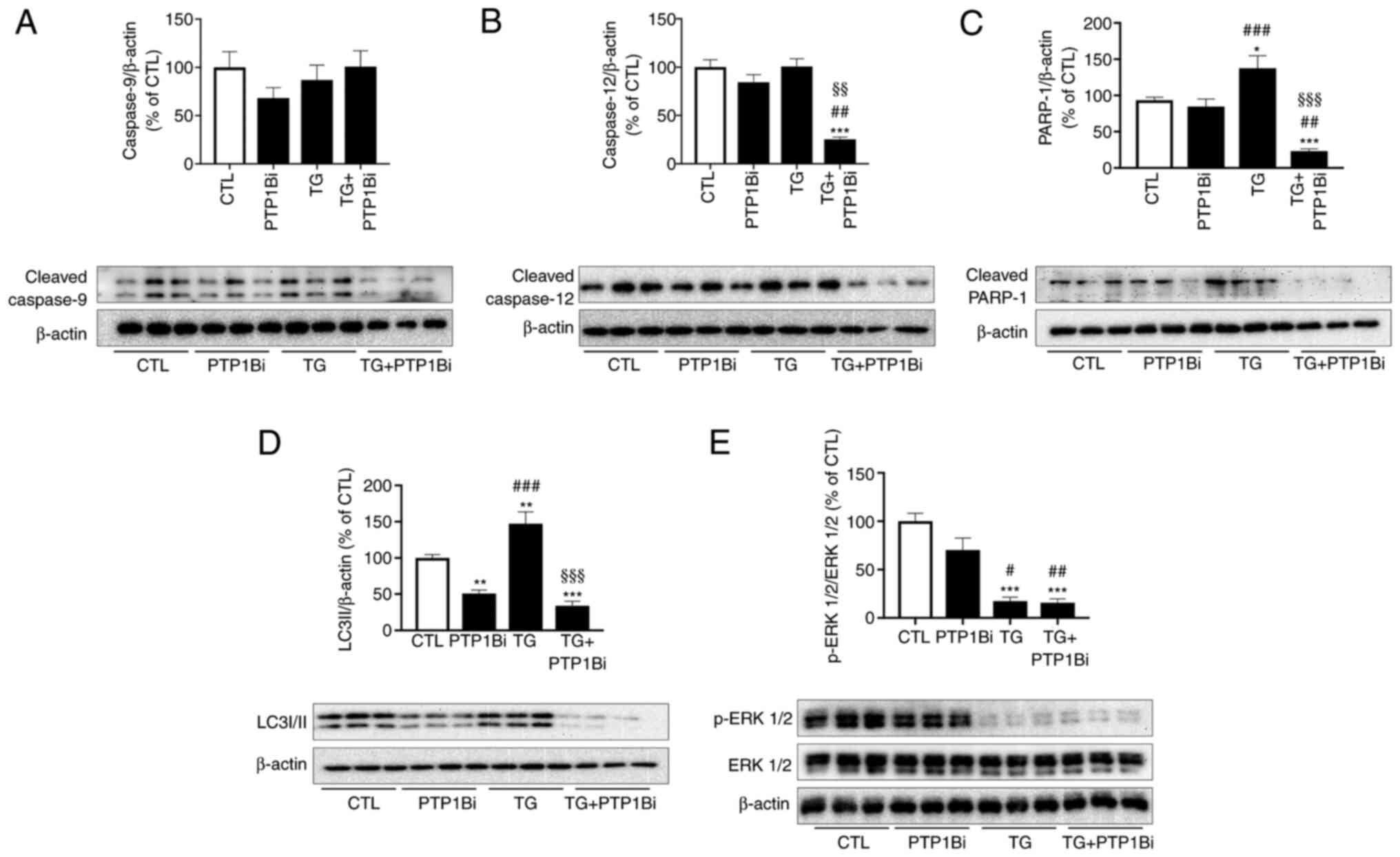 | Figure 8.Impact of PTP1B inhibition on
apoptotic signals in HUVECs subjected to pharmacological
endoplasmic reticulum stress. Western blot analysis to assess
protein expression levels of (A) cleaved caspase-9, (B) cleaved
caspase-12, (C) cleaved PARP-1, (D) LC3I/II and (E) p-ERK 1/2 in
HUVECs treated with TG (300 nM, 5 h) with or without PTP1Bi (BML,
20 µM, added 1 h prior to treatment). Bars represent the pooled
densitometry data of protein expression normalized to respective
total protein (ERK 1/2) or to loading control protein β-actin and
expressed as percentage (%) of untreated group (CTL) (n=5 in each
group). All data are presented as the mean ± SEM. *P<0.05,
**P<0.01, ***P<0.001 vs. CTL group; #P<0.05,
##P<0.01, ###P<0.001 vs. PTP1Bi group;
§§P<0.01, §§§P<0.001 vs. TG group.
PTP1B, protein tyrosine phosphatase 1B; HUVECs, human umbilical
vein endothelial cells; PARP-1, poly (ADP-ribose) polymerase-1; p-,
phosphorylated; TG, thapsigargin; PTP1Bi, PTP1B inhibitor; CTL,
control. |
As PTP1B inhibition was beneficial in reducing ER
stress-mediated apoptosis, and hence, in promoting endothelial cell
survival, the effect of PTP1B inhibition on ER stress-mediated
autophagy, which serves a central role in cell survival (40), was also assessed. HUVECs were
treated with TG with or without a PTP1B inhibitor, and then the
protein expression levels of the autophagy marker LC3-II were
determined. It was identified that TG significantly increased the
expression level of LC3-II (Fig.
8D), indicating enhanced autophagy as a result of ER stress
induction. Furthermore, PTP1B inhibition prevented this increased
expression of the autophagy marker caused by TG (Fig. 8D), which may serve a key role in the
improved survival of endothelial cells.
To further evaluate the implication of PTP1B in ER
stress-mediated cell death, the impact of PTP1B inhibition on a
major pro-proliferative signal, namely ERK1/2, was assessed. The
results demonstrated that TG-induced ER stress impaired the
phosphorylation of ERK1/2 (Fig.
8E), which may contribute to cell cycle arrest and apoptosis
observed with TG administration. Surprisingly, the inhibition of
PTP1B failed to correct this molecular alteration caused by TG
(Fig. 8E).
Discussion
The present study aimed to investigate the molecular
mechanisms underlying PTP1B-mediated endothelial dysfunction, by
assessing the role of the crosstalk between PTP1B and ER stress in
the development of endothelial dysfunction. The current study
examined the benefits and impact of PTP1B silencing or inhibition
on providing protection to endothelial cells against ER
stress-mediated impairment of eNOS activation, insulin response,
angiogenic capacity and, most importantly, ER stress-induced
apoptosis, as well as determined the underlying pathways.
To the best of our knowledge, the present study
reported for the first time that PTP1B inhibition protected HUVECs
from ER stress-mediated impaired angiogenic capacity of endothelial
cells. It was found that TG-induced ER stress abolished the ability
of HUVECs to form capillary-like structures when incubated on a
3-dimentional extracellular Matrigel matrix. Moreover, PTP1B
inhibition prevented the impairment caused by TG-induced ER stress
and restored the angiogenic capacity of HUVECs. To obtain an
improved understanding of the crosstalk between PTP1B and ER stress
in the impairment of endothelial function, the current study
investigated the impact of PTP1B blockade on the NO signaling
pathway by determining at the levels of p-eNOS and p-Akt in HUVECs
challenged with TG and stimulated with bradykinin and insulin,
respectively. While ER stress induction by TG reduced
bradykinin-stimulated phosphorylation of eNOS at the activator site
(Ser1177), it did not affect the response of endothelial cells to
insulin, as evident by the phosphorylation of Akt (Ser473).
However, PTP1B inhibition partially restored the
bradykinin-stimulated phosphorylation of eNOS at Ser1177 and
enhanced insulin-stimulated Akt phosphorylation. In addition, PTP1B
inhibition and deletion protected HUVECs against the pro-apoptotic
effects of TG-induced ER stress, as demonstrated by the reduced
number of apoptotic cells and blunted increase in the expression
levels of key apoptotic effectors. Furthermore, one of the most
important findings was that PTP1B inhibition protected endothelial
cells against ER stress-induced autophagy without affecting other
pro-apoptotic or proliferative signaling responses.
ER stress is implicated in a wide range of
pathologies, such as diabetes, neurodegenerative disorders and, of
particular interest, endothelial dysfunction. ER stress induction
was confirmed by determining the expression levels of several ER
stress markers. The present study observed a significant increase
in the expression levels of CHOP, BiP, ATF-4 and
GRP94. These results were in line with those of Thiebaut
et al (41) who reported an
increase in the expression levels of p-eukaryotic initiation
factor-2α, HSPA5 and ATF-6. However, in contrast to the current
study, these authors reported no change in the expression of
pro-apoptotic molecule CHOP. This discrepancy may be due to the use
of different ER stress inducers (tunicamycin vs. DTT and TG in the
present study). With regards to cell models used in this study,
EA.hy926 are an immortalized cell line derived from HUVECs, while
HUVECs, are primary endothelial cells and thus are more
physiologically relevant. Initially, the present study determined
the impact of PTP1B inhibition on ER stress response caused by both
DTT and TG in immortalized (EA.hy926) and primary HUVECs cells. It
was observed that PTP1B blockade reduced mRNA overexpression of
pro-inflammatory molecules and ER stress markers caused by both DTT
and TG. Therefore, for the rest of the study, TG was used as the ER
stress inducer due to its strong response compared with DTT, and
primary HUVECs were used as the cell model for functional
experiments as they had higher physiological relevance compared
with EA.hy926 cells.
ER stress activation is closely associated with
cellular inflammation. Endothelial dysfunction precedes and
imitates inflammatory response, where, at their surface,
dysfunctional endothelial cells expose various adhesion molecules,
such as ICAM-1, together with the release of multiple
pro-inflammatory cytokines and chemokines, including IL-6, IL-8 and
MCP-1 (42). The present study
demonstrated that exposure of endothelial cells to TG enhanced the
mRNA expression levels of IL-6, IL-8, MCP-1, and
ICAM-1, while pre-treatment of cells with a PTP1B inhibitor
significantly reduced the effects of TG on the mRNA expression
levels of IL-6, IL-8 and ICAM-1. The expression
levels of adhesion molecules and the secretion of pro-inflammatory
cytokines by activated endothelial cells can attract and facilitate
the passage of monocytes through the vascular wall and promote a
pro-inflammatory environment (43).
In this context, PTP1B has been shown to be involved in the
regulation of inflammatory responses (9,38).
Moreover, specific deletion of PTP1B in mouse microphages has been
revealed to protect animals from both obesity and
lipopolysaccharide-mediated inflammatory response, where
microphages collected from myeloid-PTP1B deficient mice had lower
levels of TNF-α and a higher expression of IL-10, an
anti-inflammatory cytokine (44).
It has also been reported that mice deficient for PTP1B
specifically in microphages and deficient for apolipoprotein E
(ApoE) in the whole body exhibited smaller atherosclerotic plaque
size and higher plasma levels of IL-10 and prostaglandin E2,
compared with ApoE-deficient mice only (45). In the current study, no animal
studies were performed, which might be perceived as a limitation;
however, the present findings are in line with our previous
observations using PTP1B tissue-specific knockout animals
(14,44).
A recent study investigated the ER stress-mediated
endothelial dysfunction (46);
however, this previous study only examined endothelium-dependent
relaxation in response to acetylcholine, without assessing the
impact on NO signaling or endothelial angiogenic capacity. NO is an
important mediator and reflector of endothelial function (17). To study the impact of PTP1B
inhibition on ER stress-mediated endothelial dysfunction, the
present study assessed the impact of PTP1B deletion on NO
signaling. Moreover, the phosphorylation levels eNOS and Akt in
response to bradykinin and insulin, respectively, were evaluated in
TG-challenged HUVECs. It was found that TG-induced ER stress
resulted in reduced bradykinin-stimulated eNOS phosphorylation at
Ser1177, while it did not affect the phosphorylation of Akt.
Furthermore, PTP1B inhibition partially restored
bradykinin-stimulated eNOS phosphorylation and enhanced the
phosphorylation of Akt, showing a significant effect. The present
results are in line with the effects observed in human aortic
endothelial cells challenged with palmitate, a saturated free fatty
acid known to induce ER stress, which resulted in a reduction in
eNOS and Akt phosphorylation, which were restored by
tauroursodeoxycholic acid, a chemical chaperone known to alleviate
the ER stress response (47).
However, in disagreement with the current study, in this previous
study ER stress induction reduced Akt phosphorylation in response
to insulin (47). One possible
reason for this difference is the different cell line used, human
aortic endothelial cells compared with HUVECs in the current
report. Another reason for discrepancy could be the use of
different ER stress inducers, palmitate-induced Toll-like receptor
4 in the study by Kim et al (47) compared with the use of TG in the
present study. While the current data suggested that eNOS may be
targeted by PTP1B, with our current experimental setting, it is
difficult to assess the exact relationship between them. On the one
hand, eNOS has been shown to be phosphorylated at tyrosine residues
(48), which makes it a plausible
target for dephosphorylation by PTP1B; however, answering this may
require additional investigation, such as a phospho-proteomic
approach, since the targeting of tyrosine residues is yet to be
fully elucidated. Furthermore, eNOS has a complex role in
endothelial dysfunction as its expression and phosphorylation are
not the only regulators of its function since eNOS can also be
uncoupled and instead of producing NO, it can start producing
superoxide radicals under certain stress conditions (49).
The current study also assessed the angiogenic
capacity of endothelial cells, where it was found that TG-induced
ER stress markedly impaired the angiogenic ability of HUVECs. This
was in accordance with other studies that revealed that ER stress
induction impairs angiogenesis (1,30,50). A
previous study revealed that the isolated aorta from rats exposed
to TG displayed a significant inhibition in micro-vessel formation
compared with controls. In the same study, the proliferation and
migration of HUVECs were also impaired by TG-induced ER stress
(50). Our previous study reported
that exposure of HUVECs to TG impaired angiogenic capacity via a
mechanism involving oxidative stress, and that this effect was
prevented by 4-Phenylbutyric (PBA), a chemical chaperone that
improves ER homeostasis (24).
Moreover, the present study identified that PTP1B inhibition in
HUVECs prevented the impairment of angiogenic capacity caused by TG
and restored the tube-like formation ability of endothelial cells.
This finding was in accordance with a study by Zhang et al
(51), who showed that the
treatment of human microvascular endothelial cells with high
glucose (30 mM) for 12 h, followed by overlaying the cells on a
Matrigel-coated dish, resulted in an impairment in tube formation
capacity. Furthermore, this effect was largely prevented by PTP1B
inhibition (51). The present study
examined the mRNA expression levels of the pro-angiogenic factors
VEGF-A, FGF-1 and FGF-2, and it was observed that TG
treatment caused a decrease in the expression level of
VEGF-A, while PTP1B blockade prevented this. In addition, an
increase in the mRNA expression levels of FGF-1 and
FGF-2 was observed in cells challenged with TG in the
presence of a PTP1B inhibitor, compared with control cells. The
increase in VEGF-A mRNA expression in the TG group may be a
compensatory mechanism to the impaired angiogenic response. PTP1B
has been shown to negatively regulate VEGFR rather than affecting
the expression level of VEGF (52).
Overexpression of PTP1B was reported to decrease the
phosphorylation of both VEGFR2 and Akt in bovine aortic endothelial
cells following exposure to VEGF; however, the silencing of PTP1B
enhanced the phosphorylation of these targets (52). Furthermore, the genetic deficiency
of PTP1B was found to improve the angiogenic response in hearts
from a mouse model of myocardial infarction via a mechanism
involving improved VEGFR2 signaling (53).
The UPR response initially aims at restoring ER
homeostasis and alleviating cellular stress; however, the sustained
activation of UPR leads to ER stress and shifts the response from
survival to apoptosis and cell death (1,17).
Thus, the present study examined the impact of PTP1B inhibition and
silencing on preventing ER stress-mediated endothelial cell death.
As expected, HUVECs treated with TG displayed a significantly high
percentage of apoptotic cells. This was in line with our previous
observations in HUVECs exposed to TG (30) and findings by Huang et al
(31), which revealed that vascular
endothelial cells exposed to TG had an increased rate of cell
apoptosis (31). The present study
also demonstrated that both PTP1B deletion and inhibition reduced
the proportion of apoptotic cells, thereby protecting the cells
from the pro-apoptotic effects of TG. Similarly to the current
observations, PTP1B inhibition was reported to protect cells
against apoptosis in a human neuroblastoma cell line exposed to
tunicamycin (54). In addition,
mouse embryonic fibroblasts isolated from whole-body PTP1B
deficient mice were observed to be resistant to tunicamycin-induced
ER stress-mediated apoptosis (55).
To further determine the apoptotic sub-pathways involved in this
process, the current study assessed protein expression levels of
several apoptotic effectors, including cleaved caspase-9,
caspase-12 (specific to ER stress) and PARP-1. An increasing trend
in the expression levels of these effectors was observed in
response to TG-induced ER stress. Similar results have been
obtained in vascular endothelial cells treated with TG, where an
increase in the expression levels of cleaved caspase-9 and −12 and
PARP-1 was observed (31).
Furthermore, we have previously observed that treatment of HUVECs
with TG caused an increase in the expression of cleaved PARP-1, and
executioner caspase-3 and caspase-7, which was prevented in cells
pretreated with ER stress inhibitor, the chemical chaperone PBA
(30). A similar pattern was
observed in cells treated with high glucose, which also enhanced
the enzymatic activity of caspase-3 and −7 (30).
There is a complex, tightly controlled and two-way
relationship between MAPK and ER stress response (56,57).
The present study demonstrated that TG reduced the activation of
ERK 1/2. In general, the activation of the ERK 1/2 pathway is
considered to encourage cell survival (56,57).
Therefore, the reduction in the activation of ERK 1/2 caused by TG
may contribute to cell growth arrest and reduced survival of
endothelial cells. In the present study, the molecular alterations
observed in the phosphorylation of ERK 1/2 were not prevented in
the presence of the PTP1B inhibitor. This may suggest that the
protective effects of PTP1B inhibition against ER stress-mediated
apoptosis may be mediated via other signaling pathways, such as
enhanced eNOS activity and the PI3K/Akt pro-survival pathway, a
reduced autophagic response and a reduced expression of
pro-inflammatory molecules, in addition to a possible enhancement
in the insulin response in endothelial cells.
Autophagy is a crucial physiological process aiming
to maintain a healthy cellular environment and homeostasis.
However, excessive activation of autophagy results in cell death
and has been associated with the pathogenesis of several
cardiovascular diseases (40). The
present study demonstrated that PTP1B inhibition protected
endothelial cells from ER stress-mediated autophagy, as evidenced
by reduced expression of its marker (LC3-II). The current study
identified the excessive activation of autophagy in response to ER
stress induction by TG and found that PTP1B inhibition completely
prevented the activation of autophagy. These results are in line
with those of Wang et al (58), who revealed that induction of ER
stress in vivo via an injection of tunicamycin into mice
resulted in the upregulation of LC3-II expression, which was
significantly attenuated by genetic PTP1B deletion (58).
In conclusion, the present study provided evidence
on the critical role of PTP1B in ER stress-mediated endothelial
dysfunction, which was characterized by reduced angiogenic capacity
via a mechanism involving reduced eNOS activation and cell
survival. These findings demonstrated the therapeutic potential of
targeting PTP1B in cardiovascular ischemic conditions.
Acknowledgements
Not applicable.
Funding
This work was supported by Qatar University grant
(grant. no. QUCG-CPH-20/21-3) and awards from Qatar National
Research Fund (a member of Qatar Foundation; grant. nos.
NPRP-8-1750-3-360 and UREP24-016-3-004). SSA is supported by a
graduate assistantship from the office of graduate studies (Qatar
University).
Availability of data and materials
The datasets used and/or analyzed during the
current study are available from the corresponding author on
reasonable request.
Authors' contributions
AA conceptualized the present study, designed the
research plan, and acquired resources and funding. AA supervised
the experiments and collected the data. SSA, MP, HEG and MH
performed the experiments and collected data. AA, SSA, MP, MAE, AZ
and HMK performed formal data analysis. AA and SSA wrote the
manuscript and prepared the figures with editing and input from all
authors. MAE, AZ and HMK revised the manuscript critically for
important intellectual content. AA and SSA confirm the authenticity
of all the raw data. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Maamoun H, Benameur T, Pintus G, Munusamy
S and Agouni A: Crosstalk between oxidative stress and endoplasmic
reticulum (ER) stress in endothelial dysfunction and aberrant
angiogenesis associated with diabetes: A focus on the protective
roles of heme oxygenase (HO)-1. Front Physiol. 10:702019.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhang HN, Xu QQ, Thakur A, Alfred MO,
Chakraborty M, Ghosh A and Yu XB: Endothelial dysfunction in
diabetes and hypertension: Role of microRNAs and long non-coding
RNAs. Life Sci. 213:258–268. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cersosimo E and DeFronzo RA: Insulin
resistance and endothelial dysfunction: The road map to
cardiovascular diseases. Diabetes Metab Res Rev. 22:423–436. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Potenza MA, Gagliardi S, Nacci C, Carratu
MR and Montagnani M: Endothelial dysfunction in diabetes: From
mechanisms to therapeutic targets. Curr Med Chem. 16:94–112. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Muniyappa R, Iantorno M and Quon MJ: An
integrated view of insulin resistance and endothelial dysfunction.
Endocrinol Metab Clin North Am. 37:685–711. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bakke J and Haj FG: Protein-tyrosine
phosphatase 1B substrates and metabolic regulation. Semin Cell Dev
Biol. 37:58–65. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tonks NK, Diltz CD and Fischer EH:
Purification of the major protein-tyrosine-phosphatases of human
placenta. J Biol Chem. 263:6722–6730. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Galic S, Klingler-Hoffmann M,
Fodero-Tavoletti MT, Puryer MA, Meng TC, Tonks NK and Tiganis T:
Regulation of insulin receptor signaling by the protein tyrosine
phosphatase TCPTP. Mol Cell Biol. 23:2096–2108. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Abdelsalam SS, Korashy HM, Zeidan A and
Agouni A: The role of protein tyrosine phosphatase (PTP)-1B in
cardiovascular disease and its interplay with insulin resistance.
Biomolecules. 9:2862019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Vercauteren M, Remy E, Devaux C, Dautreaux
B, Henry JP, Bauer F, Mulder P, van Huijsduijnen RH, Bombrun A,
Thuillez C and Richard V: Improvement of peripheral endothelial
dysfunction by protein tyrosine phosphatase inhibitors in heart
failure. Circulation. 114:2498–2507. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gomez E, Vercauteren M, Kurtz B,
Ouvrard-Pascaud A, Mulder P, Henry JP, Besnier M, Waget A, Van
Huijsduijnen RH, Tremblay ML, et al: Reduction of heart failure by
pharmacological inhibition or gene deletion of protein tyrosine
phosphatase 1B. J Mol Cell Cardiol. 52:1257–1264. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Agouni A, Tual-Chalot S, Chalopin M, Duluc
L, Mody N, Martinez MC, Andriantsitohaina R and Delibegović M:
Hepatic protein tyrosine phosphatase 1B (PTP1B) deficiency protects
against obesity-induced endothelial dysfunction. Biochem Pharmacol.
92:607–617. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gogiraju R, Schroeter MR, Bochenek ML,
Hubert A, Münzel T, Hasenfuss G and Schäfer K: Endothelial deletion
of protein tyrosine phosphatase-1B protects against pressure
overload-induced heart failure in mice. Cardiovasc Res.
111:204–216. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Agouni A, Mody N, Owen C, Czopek A, Zimmer
D, Bentires-Alj M, Bence KK and Delibegović M: Liver-specific
deletion of protein tyrosine phosphatase (PTP) 1B improves
obesity-and pharmacologically-induced endoplasmic reticulum stress.
Biochem J. 438:369–378. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Owen C, Lees EK, Grant L, Zimmer DJ, Mody
N, Bence KK and Delibegović M: Inducible liver-specific knockdown
of protein tyrosine phosphatase 1B improves glucose and lipid
homeostasis in adult mice. Diabetologia. 56:2286–2296. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Villalobos-Labra R, Subiabre M, Toledo F,
Pardo F and Sobrevia L: Endoplasmic reticulum stress and
development of insulin resistance in adipose, skeletal, liver, and
foetoplacental tissue in diabesity. Mol Aspects Med. 66:49–61.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Maamoun H, Abdelsalam SS, Zeidan A,
Korashy HM and Agouni A: Endoplasmic reticulum stress: A critical
molecular driver of endothelial dysfunction and cardiovascular
disturbances associated with diabetes. Int J Mol Sci. 20:16582019.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Flamment M, Hajduch E, Ferré P and
Foufelle F: New insights into ER stress-induced insulin resistance.
Trends Endocrinol Metab. 23:381–390. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Özcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi
NN, Ozdelen E, Tuncman G, Görgün C, Glimcher LH and Hotamisligil
GS: Endoplasmic reticulum stress links obesity, insulin action, and
type 2 diabetes. Science. 306:457–461. 2004. View Article : Google Scholar
|
|
20
|
Özcan U, Yilmaz E, Özcan L, Furuhashi M,
Vaillancourt E, Smith RO, Görgün CZ and Hotamisligil GS: Chemical
chaperones reduce ER stress and restore glucose homeostasis in a
mouse model of type 2 diabetes. Science. 313:1137–1140. 2006.
View Article : Google Scholar
|
|
21
|
Kassan M, Galán M, Partyka M, Saifudeen Z,
Henrion D, Trebak M and Matrougui K: Endoplasmic reticulum stress
is involved in cardiac damage and vascular endothelial dysfunction
in hypertensive mice. Arterioscler Thromb Vasc Biol. 32:1652–1661.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Galán M, Kassan M, Choi SK, Partyka M,
Trebak M, Henrion D and Matrougui K: A novel role for epidermal
growth factor receptor tyrosine kinase and its downstream
endoplasmic reticulum stress in cardiac damage and microvascular
dysfunction in type 1 diabetes mellitus. Hypertension. 60:71–80.
2012. View Article : Google Scholar
|
|
23
|
Ghemrawi R, Battaglia-Hsu SF and Arnold C:
Endoplasmic reticulum stress in metabolic disorders. Cells.
7:632018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Schwarz DS and Blower MD: The endoplasmic
reticulum: Structure, function and response to cellular signaling.
Cell Mol Life Sci. 73:79–94. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Pandey VK, Mathur A and Kakkar P: Emerging
role of unfolded protein response (UPR) mediated proteotoxic
apoptosis in diabetes. Life Sci. 216:246–258. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Battson ML, Lee DM and Gentile CL:
Endoplasmic reticulum stress and the development of endothelial
dysfunction. Am J Physiol Heart Circul Physiol. 312:H355–H367.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Minamino T, Komuro I and Kitakaze M:
Endoplasmic reticulum stress as a therapeutic target in
cardiovascular disease. Circ Res. 107:1071–1082. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bilekova S, Sachs S and Lickert H:
Pharmacological targeting of endoplasmic reticulum stress in
pancreatic beta cells. Trends Pharmacol Sci. 42:85–95. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zeng L, Zampetaki A, Margariti A, Pepe AE,
Alam S, Martin D, Xiao Q, Wang W, Jin ZG, Cockerill G, et al:
Sustained activation of XBP1 splicing leads to endothelial
apoptosis and atherosclerosis development in response to disturbed
flow. Proc Natl Acad Sci USA. 106:8326–8331. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Maamoun H, Zachariah M, McVey JH, Green FR
and Agouni A: Heme oxygenase (HO)-1 induction prevents Endoplasmic
Reticulum stress-mediated endothelial cell death and impaired
angiogenic capacity. Biochem Pharmacol. 127:46–59. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Huang J, Wan L, Lu H and Li X: High
expression of active ATF6 aggravates endoplasmic reticulum
stress-induced vascular endothelial cell apoptosis through the
mitochondrial apoptotic pathway. Mol Med Rep. 17:6483–6489.
2018.PubMed/NCBI
|
|
32
|
Choy JC, Granville DJ, Hunt DW and McManus
BM: Endothelial cell apoptosis: Biochemical characteristics and
potential implications for atherosclerosis. J Mol Cell Cardiol.
33:1673–1690. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Osman A, El-Gamal H, Pasha M, Zeidan A,
Korashy HM, Abdelsalam SS, Hasan M, Benameur T and Agouni A:
Endoplasmic reticulum (ER) stress-generated extracellular vesicles
(Microparticles) self-perpetuate ER stress and mediate endothelial
cell dysfunction independently of cell survival. Front Cardiovasc
Med. 7:5847912020. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Abdullahi A, Stanojcic M, Parousis A,
Patsouris D and Jeschke MG: Modeling acute ER stress in vivo and in
vitro. Shock. 47:506–513. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Andersen HS, Olsen OH, Iversen LF,
Sørensen AL, Mortensen SB, Christensen MS, Branner S, Hansen TK,
Lau JF, Jeppesen L, et al: Discovery and SAR of a novel selective
and orally bioavailable nonpeptide classical competitive inhibitor
class of protein-tyrosine phosphatase 1B. J Med Chem. 45:4443–4459.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Agouni A, Mostefai HA, Porro C, Carusio N,
Favre J, Richard V, Henrion D, Martínez MC and Andriantsitohaina R:
Sonic hedgehog carried by microparticles corrects endothelial
injury through nitric oxide release. FASEB J. 21:2735–2741. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Coquerel D, Neviere R, Delile E, Mulder P,
Marechal X, Montaigne D, Renet S, Remy-Jouet I, Gomez E, Henry JP,
et al: Gene deletion of protein tyrosine phosphatase 1B protects
against sepsis-induced cardiovascular dysfunction and mortality.
Arterioscler Thromb Vasc Biol. 34:1032–1044. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang Y, Fan Y, Song Y, Han X, Fu M, Wang
J, Cui X, Cao J, Chen L, Hu K, et al: Angiotensin II induces
apoptosis of cardiac microvascular endothelial cells via regulating
PTP1B/PI3K/Akt pathway. In vitro Cell Dev Biol Animal. 55:801–811.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lavandero S, Chiong M, Rothermel BA and
Hill JA: Autophagy in cardiovascular biology. J Clin Invest.
125:55–64. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Thiebaut PA, Delile E, Coquerel D, Brunel
JM, Renet S, Tamion F and Richard V: Protein tyrosine phosphatase
1B regulates endothelial endoplasmic reticulum stress; role in
endothelial dysfunction. Vascul Pharmacol. 109:36–44. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Sun HJ, Wu ZY, Nie XW and Bian JS: Role of
endothelial dysfunction in cardiovascular diseases: The link
between inflammation and hydrogen sulfide. Front Pharmacol.
21:15682020. View Article : Google Scholar
|
|
43
|
Lubrano V and Balzan S: Roles of LOX-1 in
microvascular dysfunction. Microvasc Res. 105:132–140. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Grant L, Shearer KD, Czopek A, Lees EK,
Owen C, Agouni A, Workman J, Martin-Granados C, Forrester JV,
Wilson HM, et al: Myeloid-cell protein tyrosine phosphatase-1B
deficiency in mice protects against high-fat diet and
lipopolysaccharide-induced inflammation, hyperinsulinemia, and
endotoxemia through an IL-10 STAT3-dependent mechanism. Diabetes.
63:456–470. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Thompson D, Morrice N, Grant L, Sommer SL,
Ziegler K, Whitfield P, Mody N, Wilson HM and Delibegović M:
Myeloid protein tyrosine phosphatase 1B (PTP1B) deficiency protects
against atherosclerotic plaque formation in the
ApoE(−/−) mouse model of atherosclerosis with
alterations in IL10/AMPKα pathway. Mol Metab. 6:845–853. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Legeay S, Fautrat P, Norman JB, Antonova
G, Kennard S, Bruder-Nascimento T, Patel VS, Faure S and de
Chantemèle EJ: Selective deficiency in endothelial PTP1B protects
from diabetes and endoplasmic reticulum stress-associated
endothelial dysfunction via preventing endothelial cell apoptosis.
Biomed Pharmacother. 127:1102002020. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kim JA, Jang HJ and Hwang DH: Toll-like
receptor 4-induced endoplasmic reticulum stress contributes to
impairment of vasodilator action of insulin. Am J Physiol
Endocrinol Metab. 309:E767–E776. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Dixit M, Loot AE, Mohamed A, Fisslthaler
B, Boulanger CM, Ceacareanu B, Hassid A, Busse R and Fleming I:
Gab1, SHP2, and protein kinase A are crucial for the activation of
the endothelial NO synthase by fluid shear stress. Circ Res.
97:1236–1244. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Karbach S, Wenzel P, Waisman A, Munzel T
and Daiber A: eNOS uncoupling in cardiovascular diseases-the role
of oxidative stress and inflammation. Curr Pharm Des. 20:3579–3594.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Shukla N, Freeman N, Gadsdon P, Angelini
GD and Jeremy JY: Thapsigargin inhibits angiogenesis in the rat
isolated aorta: Studies on the role of intracellular calcium pools.
Cardiovasc Res. 49:681–689. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Zhang J, Li L, Li J, Liu Y, Zhang CY,
Zhang Y and Zen K: Protein tyrosine phosphatase 1B impairs diabetic
wound healing through vascular endothelial growth factor receptor 2
dephosphorylation. Arterioscler Thromb Vasc Biol. 35:163–174. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zhang Y, Li Q, Youn JY and Cai H: Protein
phosphotyrosine phosphatase 1B (PTP1B) in calpain-dependent
feedback regulation of vascular endothelial growth factor receptor
(VEGFR2) in endothelial cells: Implications in VEGF-dependent
angiogenesis and diabetic wound healing. J Biol Chem. 292:407–416.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Besnier M, Galaup A, Nicol L, Henry JP,
Coquerel D, Gueret A, Mulder P, Brakenhielm E, Thuillez C, Germain
S, et al: Enhanced angiogenesis and increased cardiac perfusion
after myocardial infarction in protein tyrosine phosphatase
1B-deficient mice. FASEB J. 28:3351–3361. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Jeon YM, Lee S, Kim S, Kwon Y, Kim K,
Chung CG, Lee S, Lee SB and Kim HJ: Neuroprotective effects of
protein tyrosine phosphatase 1B inhibition against ER
stress-induced toxicity. Mol Cells. 40:280–290. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Gu F, Nguyên DT, Stuible M, Dubé N,
Tremblay ML and Chevet E: Protein-tyrosine phosphatase 1B
potentiates IRE1 signaling during endoplasmic reticulum stress. J
Biol Chem. 279:49689–49693. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Hotamisligil GS and Davis RJ: Cell
signaling and stress responses. Cold Spring Harb Perspect Biol.
8:a0060722016. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Darling NJ and Cook SJ: The role of MAPK
signalling pathways in the response to endoplasmic reticulum
stress. Biochim Biophys Acta. 1843:2150–2163. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Wang S, Chen X, Nair S, Sun D, Wang X and
Ren J: Deletion of protein tyrosine phosphatase 1B obliterates
endoplasmic reticulum stress-induced myocardial dysfunction through
regulation of autophagy. Biochim Biophys Acta Mol Basis Dis.
1863:3060–3074. 2017. View Article : Google Scholar : PubMed/NCBI
|














