Introduction
Stroke is a leading global cause of morbidity and
mortality (1). Ischemic stroke
occurs when the blood supply to the brain is obstructed, and the
majority of ischemic strokes result from acute thrombosis.
Currently, tissue plasminogen activator (tPA) is the only approved
agent by the Food and Drug Administration for ischemic stroke
treatment; however, tPA must be administered within 4.5 h of stroke
onset for it to exert therapeutic effects (2). Thus, tPA has limited applicability
and is currently used in <5% of stroke victims (3). Therefore, studies are required to
identify therapies with an increased efficacy and extended
treatment window for stroke patients.
The N-methyl-D-aspartate (NMDA) receptor (NMDAR)
complex is a tetrameric or pentameric structure composed of at
least two NR1 subunits and two or three subunits from the NR2
family (NR2A-D) (4,5). An additional NMDA receptor subunit,
NR3A, has been identified in mammalian brains (6,7).
However, unlike the conventional NR1/NR2 receptors, those
containing NR3 subunits exhibit decreased single-channel
conductance, insensitivity to magnesium blockade and reduced
calcium (Ca2+) permeability (7–10).
NR3 subunits act in a novel, dominant-negative manner to suppress
NMDAR activity (6,11).
Calcitriol is the biologically active metabolite of
vitamin D and the predominant Ca2+-regulatory steroid
hormone in peripheral tissues (12,13).
Previous studies have demonstrated that the chronic peripheral
treatment of rats with calcitriol retarded the age-related decrease
in neuronal density observed in the rodent hippocampus (14) and protected against damage in a
rodent model of stroke (15). In
addition, a previous study also demonstrated that calcitriol
exhibits a direct consistent neuroprotective action against
excitotoxic insults (16).
The aim of this study was to determine whether
calcitriol protected the brain from ischemic injury through a
signaling mechanism involving elevated levels of NR3A and
Ca2+-response element binding protein (p-CREB), and to
determine whether mitogen-activated protein kinase kinase
(MEK)/extracellular signal-regulated kinase (ERK) is involved in
the regulatory mechanism of NR3A-mediated p-CREB expression.
Materials and methods
Animals and treatment
Healthy male Sprague-Dawley rats (weight, 200–250 g)
were purchased from Hunan Weasleyg Scene of Experimental Animals
Co., Ltd. (Changsha, China). Experimental protools were approved by
the Ethics Committee of Tongji Medical College, Huazhong University
of Science and Technology (Wuhan, China), and conformed to
internationally accepted ethical standards (Guide For the Care and
Use of Laboratory Animals; NIH Publication 85-23, revised 1985).
The rats were allowed access to food and water ad libitum.
Rats were randomly divided into four groups (n=12): Sham-operated
rats (group S); rats with middle cerebral artery occlusion (MCAO)
(group I); rats with MCAO followed by calcitriol treatment (group
C); and rats with MCAO followed by calcitriol plus PD98059
treatment (group P) (Fig. 1).
Calcitriol (Cayman Chemical Company, Ann Arbor, MI,
USA) was dissolved in ethanol and diluted with 0.9% NaCl solution
immediately prior to intraperitoneal (i.p.) administration. The
drug was applied either acutely (a single dose of 2 μg/kg,
immediately following ischemia) and subchronically (2 μg/kg on six
consecutive days). Control animals (groups I and S) received 0.9%
NaCl supplemented with the required volume of ethanol. On day six,
the final dose was administered 1 h prior to surgery. An MEK
inhibitor, PD98059 (dissolved in 1% dimethylsulfoxide; 0.75
mg/rat), was administered alone or in combination with
calcitriol.
Focal cerebral ischemia
Stroke was induced using the intraluminal filament
MCAO model (17). Throughout the
surgical procedure, rectal temperature was monitored and maintained
at 37°C using a circulating heating pad. Briefly, the animals were
anesthetized with 10% chloral hydrate (400 mg/kg, i.p.), and the
right common carotid artery (CCA) and its proximal branches were
isolated. The CCA and external carotid artery were ligated, and the
internal carotid artery (ICA) was temporarily occluded using a
metal microvessel clip. A nylon monofilament (Beijing Sunbio
Biotech Co., Ltd., Beijing, China) with a rounded tip was inserted
and advanced through the CCA and ICA until resistance was felt. The
filament was left in place for 2 h and then withdrawn. Rats in the
sham-operated group were subjected to the same surgical procedure,
however, they did not undergo MCAO. All animals were placed in a
warm environment until they had fully recovered from the
anesthesia.
Measurement of the infarct volume
For 2,3,5-triphenyltetrazolium chloride (TTC)
staining, brain tissues were sectioned into 2-mm thick coronal
slices seven days following reperfusion. These tissues were stained
for 20 min in a 2% TTC solution (Sigma-Aldrich, St. Louis, MO, USA)
and fixed in 4% paraformaldehyde. The stained tissues were
photographed by a digital camera (COOLPIX P500; Nikon, Tokyo,
Japan) and measured for ischemic lesions by Image J software
(National Institutes of Health, Bethesda, MD, USA). The ischemic
lesion percentage of each slice was calculated by the ratio of the
infarction area to the whole slice area.
Western blot analysis
Subsequent to seven days of reperfusion, the rats
were euthanized by decapitation and the hippocampal tissues were
harvested. Total protein extraction was performed using the total
protein extraction kit (Nanjing Keygen Biotech Co., Ltd., Nanjing,
China). Total protein extracts were prepared for protein
determination and analyzed by western blot analysis for NR3A,
phosphorylated MEK (p-MEK) and MEK. Nuclear protein extraction was
performed according to the manufacturer’s instructions (Fermentas
International, Glen Burnie, MD, USA). Nuclear protein extracts were
prepared to determine the expression of p-CREB. Protein
concentration was analyzed by a bicinchoninic acid assay kit
(Nanjing Keygen Biotech Co., Ltd.). Equal quantities of protein
were loaded and separated by sodium dodecyl sulfate-polyacrylamide
gel electrophoresis and transferred onto a polyvinylidene
difluoride membrane. The membrane was blocked and incubated
overnight at 4°C with the following antibodies: Anti-NR3A (1:1,000;
Millipore, Billerica, MA, USA), anti-phospho-MEK, anti-MEK,
anti-p-CREB (dilution, 1:1,000; Cell Signaling Technology Inc.,
Beverly, MA, USA), anti-Lamin B1 (dilution, 1:500; Bioworld
Merchandising, Inc., Minneapolis, MN, USA) and anti-GAPDH
(dilution, 1:1,000; Proteintech Group, Inc, Chicago, IL, USA).
Following three washes with Tris-buffered saline and Tween 20 for
15 min, the membrane was incubated with the appropriate horseradish
peroxidase-conjugated secondary antibodies (dilution, 1:5,000) for
1 h at room temperature. Labeled proteins were detected with the
ChemiDoc XRS chemiluminescence imaging system (Bio-Rad, Hercules,
CA, USA). Protein bands were quantified by Image Lab™ image
acquisition and analysis software (Bio-Rad). The experiments were
repeated in triplicate.
Quantum dot-based immunofluorescence
Subsequent to seven days of reperfusion, the animals
were anesthetized with chloral hydrate (400 mg/kg, i.p.) and
perfused transcardially with 0.9 % sodium chloride at 4°C, followed
by 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4). The
brains were then rapidly removed, blocked and embedded in paraffin.
Paraffin-embedded brains were cut into 4-μm-thick sections
according to standard procedures. The paraffin sections (n=3) were
incubated overnight with antibodies against NR3A (dilution, 1:100;
Millipore) at 4°C, following blocking with bovine serum albumin
(BSA). The samples were then incubated with a biotinylated
secondary antibody at 37°C for 30 min. Following blocking with BSA,
the paraffin sections were incubated with streptavidin-conjugated
QDs605 (dilution, 1:100; Wuhan Jiayuan Quantum Dot Technological
Development, Co, Ltd., Wuhan, Hubei, China). NR3A-positive cells
were measured at ×200 magnification per visual field in the cortex;
three visual fields per section and three brain sections per rat
were analyzed. Fluorescent signals were detected with a
fluorescence microscope (BX51; Olympus, Tokyo, Japan) and signal
intensities were collected for statistical analysis. Images were
captured with a Doppler imaging system (CRi Nuance Fx; Caliper Life
Sciences, Hopkinton, MA, USA).
Statistical analysis
All values are presented as the mean ± standard
error of the mean. One-way analysis of variance followed by a post
hoc Newman-Keuls test was performed for statistical comparison of
several groups. The unpaired t-test was used for the comparison of
two groups. P<0.05 was considered to indicate a statistically
significant difference. GraphPad Prism for Windows (version 5;
GraphPad Software Inc., San Diego, CA, USA) was used for all
statistical analyses.
Results
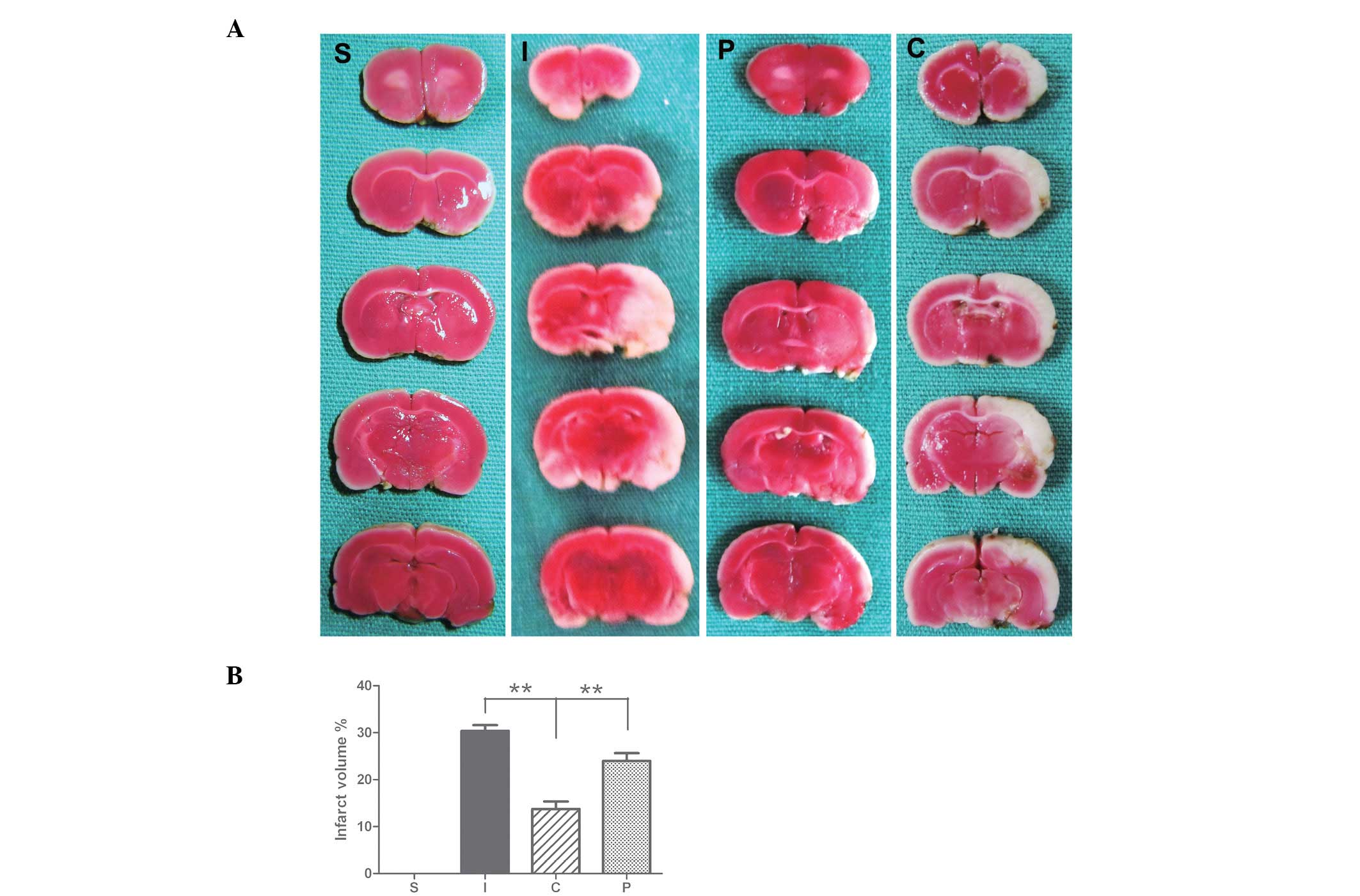
Effect of calcitriol on infarct area and
volume following focal cerebral ischemia
Seven days following ischemia/reperfusion (I/R),
rats developed infarcts affecting the cortex and striatum (Fig. 2). The calcitriol treatment group
had a significantly smaller infarct area and volume of total
hemisphere infarction seven days following MCAO compared with those
of the control (P<0.01; Fig.
2).
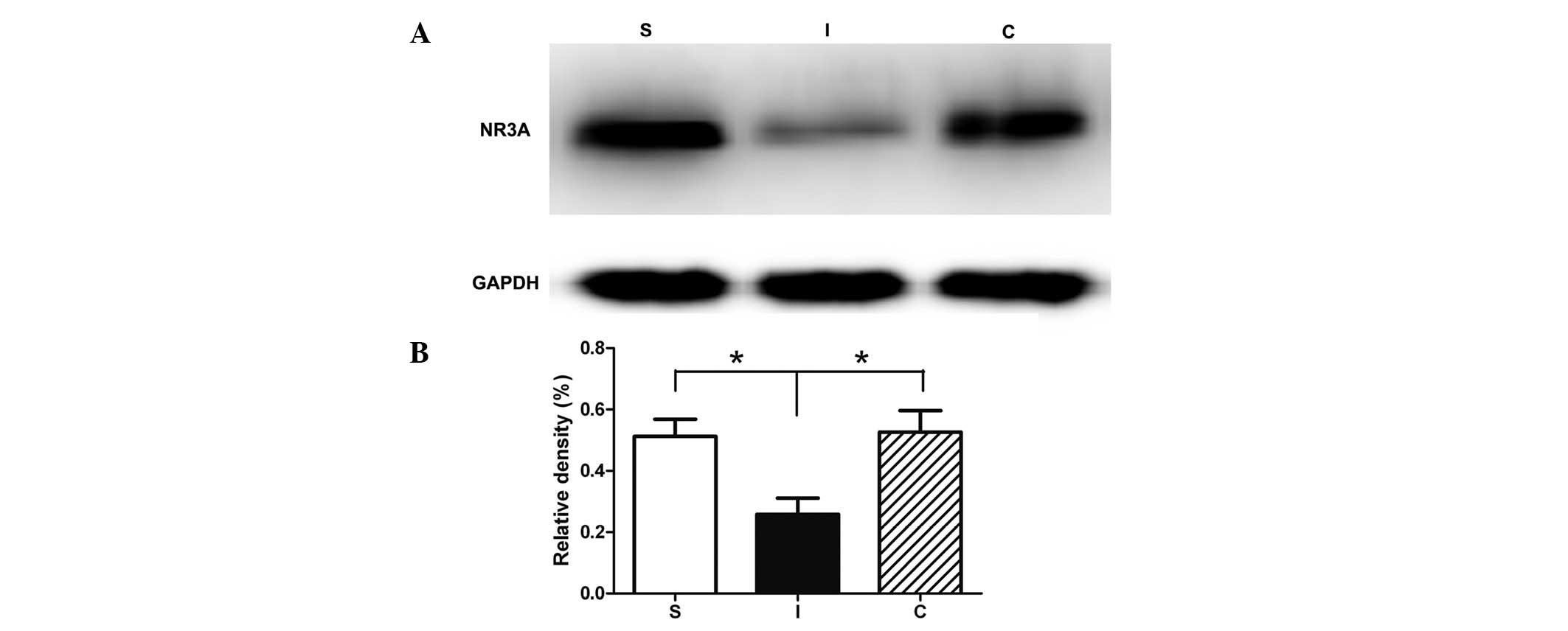
Calcitriol reduces the degradation of
NR3A in I/R injury
To determine whether calcitriol exerted a
neuroprotective effect through the regulation of the NR3A level,
the NR3A levels in the rat hippocampus were investigated by western
blot analysis (Fig. 3). In the
hippocampal tissues collected seven days following ischemia, the
NR3A expression level was significantly reduced in group I compared
with that of the sham-operated animals (Fig. 3). Treatment with calcitriol
significantly restored the level of NR3A to that observed in the
uninjured rats.
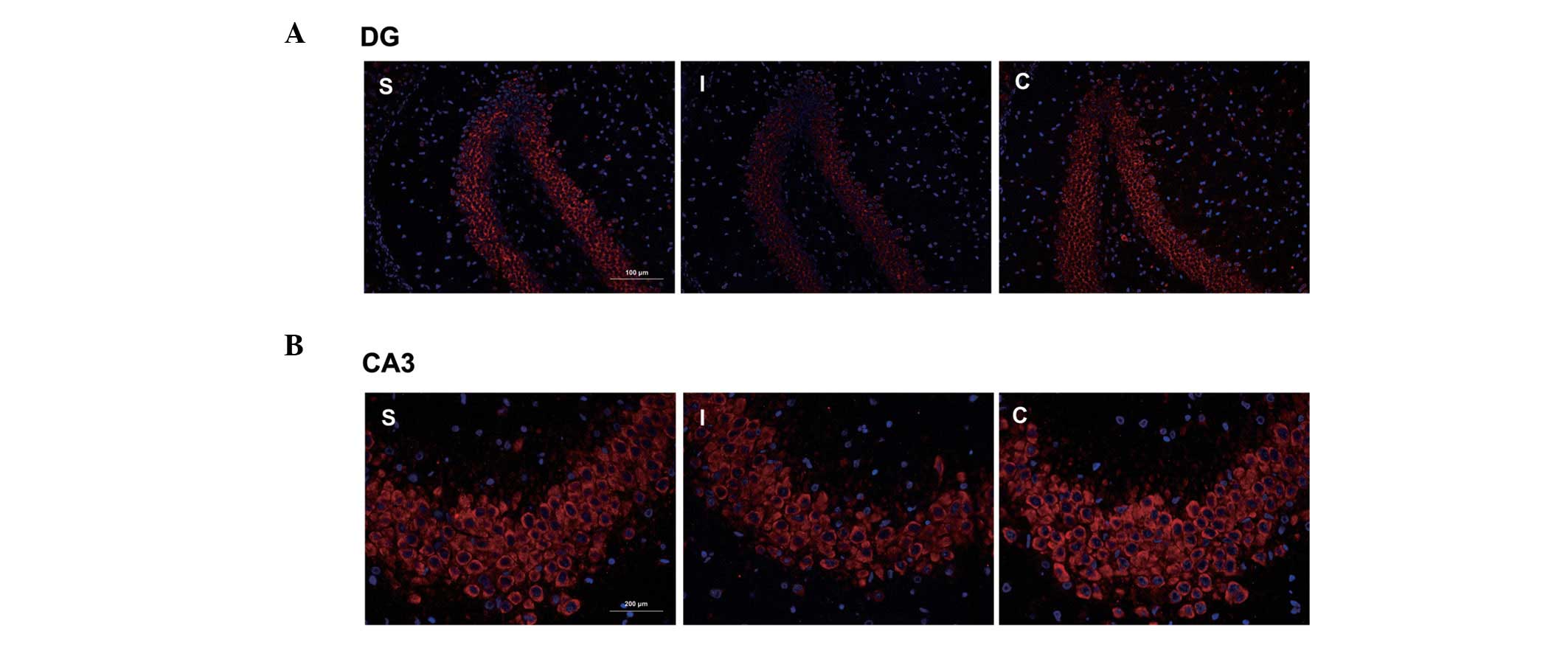
Immunofluorescence results showed a cytomembrane
staining pattern of NR3A protein in neurons of the hippocampal
dentate gyrus and CA3 areas, and these findings were corroborated
by the results of the western blot analysis (Fig. 4).
PD98059 specifically inhibits MEK
phosphorylation
To determine the effectiveness and specificity of
the MEK inhibitor PD98059, p-MEK levels in the rat hippocampus were
determined by western blot analysis (Fig. 5). Subsequent to the application of
PD98059, the protein levels of p-MEK in the rat hippocampus were
significantly decreased seven days following reperfusion compared
with those of the calcitriol-treated group (P<0.01; Fig. 5).
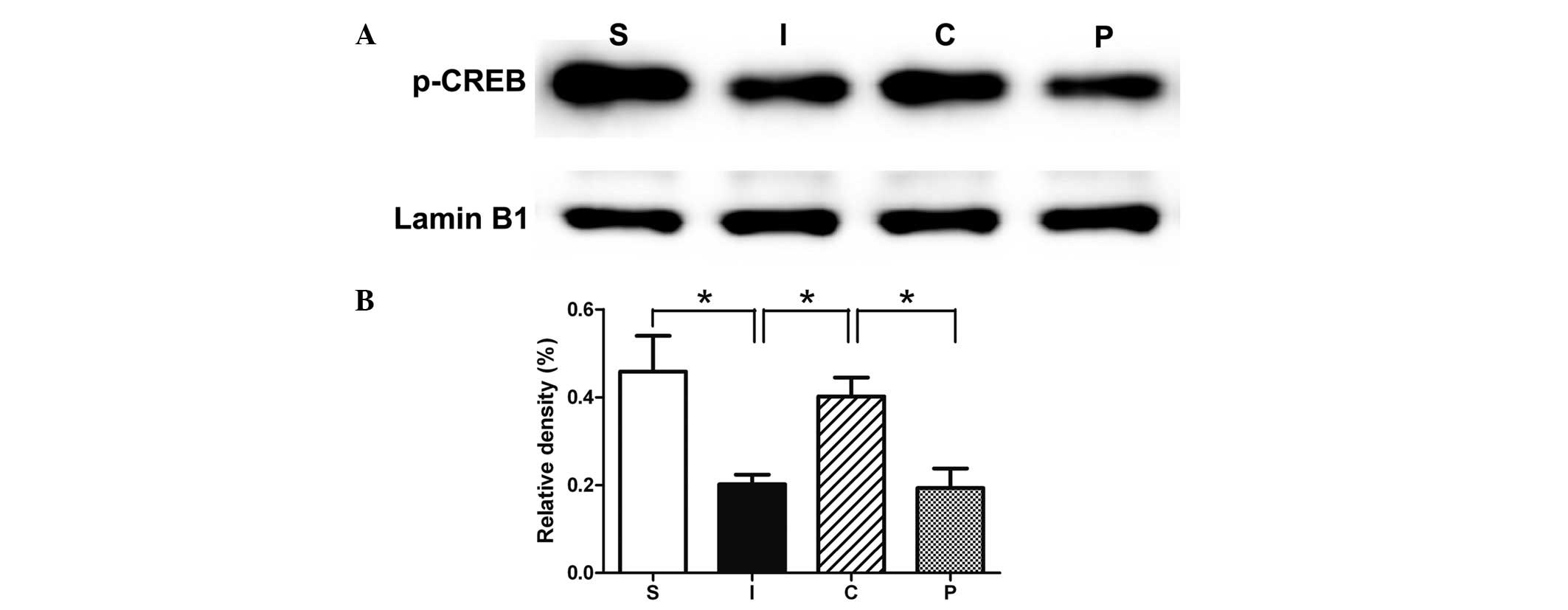
Calcitriol maintains the level of p-CREB
via the NR3A-MEK/ERK pathway
In the hippocampal tissues collected seven days
following ischemia, the p-CREB expression levels in the rat
hippocampus were significantly reduced compared with those of the
sham-operated animals. The treatment with calcitriol significantly
restored the levels of p-CREB to those of the uninjured rats. This
upregulation was prevented by the inhibition of the ERK (PD98059)
pathway (Fig. 6).
Discussion
The present results demonstrated that the NR3A
subunit was effective in protecting the brain from ischemic injury.
In this study, treatment with calcitriol for seven days
significantly decreased the infarct volumes, and was correlated
with elevated NR3A and p-CREB activities, following cerebral I/R
injury. This neuroprotective effect was attenuated by cotreatment
with PD98059, an MEK (the upstream kinase of ERK) inhibitor.
Therefore, the results clearly demonstrated that calcitriol exerted
neuroprotective effects against ischemic injury through the
NR3A-MEK/ERK-CREB pathways.
In stroke, excessive extracellular glutamate
overstimulates glutamate receptors, initiating excessive calcium
entry mainly through the NMDARs, which is the predominant
contributory factor to neuronal excitotoxicity injury during the
process of ischemic stroke (18,19).
NMDARs are molecularly organized as heteromeric complexes
incorporating different subunits of three subtypes: NR1, NR2 and
NR3, the latter of which has two subunits (NR3a and NR3b) (20). In vitro and in vivo
studies have suggested that NMDAR antagonists are effective in
ischemic neuronal death, and pharmacological agents that block
glutamate release or glutamate-mediated postsynaptic excitability
are able to reduce neural degeneration in rat stroke models
(21,22). However, studies concerning the
discovery of neuroprotective agents in the last few decades focused
on NMDAR antagonists, which although promising in preclinical
studies, failed during clinical trials (23,24).
Among numerous possible reasons for this failure, it is suggested
that the NR2A-containing NMDARs mediate neuronal survival while the
NR2B-containing NMDARs are coupled to neuronal apoptosis. Blockade
of the NR2A-containing NMDARs does not confer neuroprotection, and,
by contrast leads to the exacerbation of neuronal death. However,
blocking NR2B-mediated cell death was effective in reducing infarct
volume only when the receptor antagonist was administered prior to
the onset of stroke and not 4.5 h subsequent to stroke (25,26).
Therefore, the common conception concerning treatment of ischemic
brain damage with NMDAR antagonists may have to be reconsidered.
Conventional NMDARs are composed of NR1 and NR2 subunits, while the
incorporation of NR3A gives the NMDAR unconventional properties,
such as low Ca2+ permeability and decreased sensitivity
to Mg2+ blocking (27).
NR3A subunits modulate the susceptibility of oligodendroglial
lineage NMDARs to glycine or D-serine activation (28,29).
In situ hybridization and immunohistochemistry analyses have
demonstrated that the NR3A subunit is widely distributed in the rat
brain with predominant expression of the novel NR3B subunit by
motor neurons (30). The
co-expression of NR3A and NR3B subunits prevents Ca2+
mobilization into the mitochondria following the activation of
NMDAR channels composed of NR1/NR2A and NR1/NR2B subunits on the
cell surface, in association with the rescue from cell death
(31). Cultured neurons expressing
transgenic (TG) NR3A exhibited greater resistance to NMDA-mediated
neurotoxicity than wild type (WT) neurons. Similarly, in
vivo, adult NR3A TG mice subjected to focal cerebral ischemia
exhibited less damage than WT mice (32). A previous study has demonstrated
that calcitriol provides neuroprotection against I/R injury
(15). In addition, calcitriol has
a direct and highly consistent neuroprotective action against
excitotoxic insults (16).
Therefore, the results suggest that calcitriol may protect the
brain from ischemic injury through a signaling mechanism involving
elevated levels of NR3A. In the present study, the variation of
NR3A in the MCAO group suggested that brain ischemic injury induced
downregulation of NR3A in the hippocampal CA1 region; however,
calcitriol treatment reversed this tendency and significantly
increased the NR3A levels.
The nuclear transcription factor CREB, active form
p-CREB, exhibits numerous functions. The phosphorylation of
serine-133 in CREB allows it to interact with the co-activator,
CREB-binding protein/p300, and is required for its activation. A
previous study demonstrated that p-CREB stimulated neurogenesis and
prevented infarct expansion in the penumbra region of cerebral
ischemia (33). The CREB
activation was a critical event in neuroprotection against ischemic
injury (34,35) suggesting that the NR3A subunit may
rescue neurons from glutamate excitotoxicity mediated by NMDAR
through activation of CREB. In the present study, calcitriol
treatment markedly reduced the brain infarct area and enhanced the
expression levels of NR3A and p-CREB. In addition, PD98059 was used
to investigate the pathway by which calcitriol protected rats from
cerebral ischemia. It was demonstrated that when PD98059 was
co-administered with calcitriol, the p-CREB protein levels were
significantly decreased seven days following reperfusion compared
with the levels measured in calcitriol-treated rats. These results
demonstrated that the activation of CREB through the MEK/ERK
pathway is a pivotal downstream effector for the protective effect
of the NR3A subunit in neurons.
In conclusion, MCAO rats receiving calcitriol
treatment exhibited a markedly reduced brain infarct area and
enhanced expression levels of NR3A and p-CREB. Furthermore, MEK/ERK
is involved in the regulatory mechanism of NR3A-mediated p-CREB
expression. The results may provide insights into the pleiotropic
role of calcitriol and the functional modulation of NR3A, and
provide a basis for the protective effect of calcitriol on brain
ischemia.
References
|
1
|
Feigin VL: Stroke epidemiology in the
developing world. Lancet. 365:2160–2161. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fisher M, Feuerstein G, Howells DW, et al:
Update of the stroke therapy academic industry roundtable
preclinical recommendations. Stroke. 40:2244–2250. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Grotta JC, Burgin WS, El-Mitwalli A, et
al: Intravenous tissue-type plasminogen activator therapy for
ischemic stroke: Houston experience 1996 to 2000. Arch Neurol.
58:2009–2013. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
McBain CJ and Mayer ML:
N-methyl-D-aspartic acid receptor structure and function. Physiol
Rev. 74:723–760. 1994.PubMed/NCBI
|
|
5
|
Dingledine R, Borges K, Bowie D and
Traynelis SF: The glutamate receptor ion channels. Pharmacol Rev.
51:7–61. 1999.
|
|
6
|
Sucher NJ, Akbarian S, Chi CL, et al:
Developmental and regional expression pattern of a novel NMDA
receptor-like subunit (NMDAR-L) in the rodent brain. J Neurosci.
15:6509–6520. 1995.PubMed/NCBI
|
|
7
|
Chatterton JE, Awobuluyi M, Premkumar LS,
et al: Excitatory glycine receptors containing the NR3 family of
NMDA receptor subunits. Nature. 415:793–798. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Das S, Sasaki YF, Rothe T, et al:
Increased NMDA current and spine density in mice lacking the NMDA
receptor subunit NR3A. Nature. 393:377–381. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Perez-Otano I, Schulteis CT, Contractor A,
et al: Assembly with the NR1 subunit is required for surface
expression of NR3A-containing NMDA receptors. J Neurosci.
21:1228–1237. 2001.PubMed/NCBI
|
|
10
|
Sasaki YF, Rothe T, Premkumar LS, et al:
Characterization and comparison of the NR3A subunit of the NMDA
receptor in recombinant systems and primary cortical neurons. J
Neurophysiol. 87:2052–2063. 2002.PubMed/NCBI
|
|
11
|
Ciabarra AM, Sullivan JM, Gahn LG, Pecht
G, Heinemann S and Sevarino KA: Cloning and characterization of
chi-1: a developmentally regulated member of a novel class of the
ionotropic glutamate receptor family. J Neurosci. 15:6498–6508.
1995.PubMed/NCBI
|
|
12
|
DeLuca HF and Zierold C: Mechanisms and
functions of vitamin D. Nutr Rev. 56:S4–S10, (discussion S54–S75).
1998. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Brown AJ, Dusso A and Slatopolsky E:
Vitamin D. Am J Physiol. 277:F157–F175. 1999.PubMed/NCBI
|
|
14
|
Landfield PW and Cadwallader-Neal L:
Long-term treatment with calcitriol (1,25(OH)2 vit D3) retards a
biomarker of hippocampal aging in rats. Neurobiol Aging.
19:469–477. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang Y, Chiang YH, Su TP, et al: Vitamin
D(3) attenuates cortical infarction induced by middle cerebral
arterial ligation in rats. Neuropharmacology. 39:873–880. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Brewer LD, Thibault V, Chen KC, Langub MC,
Landfield PW and Porter NM: Vitamin D hormone confers
neuroprotection in parallel with downregulation of L-type calcium
channel expression in hippocampal neurons. J Neurosci. 21:98–108.
2001.PubMed/NCBI
|
|
17
|
Longa EZ, Weinstein PR, Carlson S and
Cummins R: Reversible middle cerebral artery occlusion without
craniectomy in rats. Stroke. 20:84–91. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kostandy BB: The role of glutamate in
neuronal ischemic injury: the role of spark in fire. Neurol Sci.
33:223–237. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Szydlowska K and Tymianski M: Calcium,
ischemia and excitotoxicity. Cell Calcium. 47:122–129. 2010.
View Article : Google Scholar
|
|
20
|
Henson MA, Roberts AC, Pérez-Otaño I and
Philpot BD: Influence of the NR3A subunit on NMDA receptor
functions. Prog Neurobiol. 91:23–37. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Shen H, Chen GJ, Harvey BK, Bickford PC
and Wang Y: Inosine reduces ischemic brain injury in rats. Stroke.
36:654–659. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Shen H, Kuo CC, Chou J, et al: Astaxanthin
reduces ischemic brain injury in adult rats. FASEB J. 23:1958–1968.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hardingham GE, Fukunaga Y and Bading H:
Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB
shut-off and cell death pathways. Nat Neurosci. 5:405–414.
2002.PubMed/NCBI
|
|
24
|
Hoyte L, Barber PA, Buchan AM and Hill MD:
The rise and fall of NMDA antagonists for ischemic stroke. Curr Mol
Med. 4:131–136. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liu Y, Wong TP, Aarts M, et al: NMDA
receptor subunits have differential roles in mediating excitotoxic
neuronal death both in vitro and in vivo. J Neurosci. 27:2846–2857.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Terasaki Y, Sasaki T, Yagita Y, et al:
Activation of NR2A receptors induces ischemic tolerance through
CREB signaling. J Cereb Blood Flow Metab. 30:1441–1449. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cavara NA and Hollmann M: Shuffling the
deck anew: how NR3 tweaks NMDA receptor function. Mol Neurobiol.
38:16–26. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Stys PK and Lipton SA: White matter NMDA
receptors: an unexpected new therapeutic target? Trends Pharmacol
Sci. 28:561–566. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Piña-Crespo JC, Talantova M, Micu I, et
al: Excitatory glycine responses of CNS myelin mediated by NR1/NR3
‘NMDA’ receptor subunits. J Neurosci. 30:11501–11505.
2010.PubMed/NCBI
|
|
30
|
Chatterton JE, Awobuluyi M, Premkumar LS,
et al: Excitatory glycine receptors containing the NR3 family of
NMDA receptor subunits. Nature. 415:793–798. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Fukumori R, Takarada T, Nakamichi N, et
al: Requirement of both NR3A and NR3B subunits for dominant
negative properties on Ca2+ mobilization mediated by
acquired N-methyl-D-aspartate receptor channels into mitochondria.
Neurochem Int. 57:730–737. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Nakanishi N, Tu S, Shin Y, et al:
Neuroprotection by the NR3A subunit of the NMDA receptor. J
Neurosci. 29:5260–5265. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhu DY, Lau L, Liu SH, Wei JS and Lu YM:
Activation of cAMP-response-element-binding protein (CREB) after
focal cerebral ischemia stimulates neurogenesis in the adult
dentate gyrus. Proc Natl Acad Sci USA. 101:9453–9457. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Walton MR and Dragunow I: Is CREB a key to
neuronal survival? Trends Neurosci. 23:48–53. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Finkbeiner S: CREB couples neurotrophin
signals to survival messages. Neuron. 25:11–14. 2000. View Article : Google Scholar : PubMed/NCBI
|