Introduction
In previous years, the association between ion
channels and tumors has drawn particular attention. Increasing
evidence has demonstrated that ion channels are involved in the
regulation of tumor progression, including potassium (1–3),
calcium (4) and sodium channels
(5,6). Therefore, understanding the
underlying molecular mechanisms of ion channels in tumorigenesis,
and tumor progression and migration provides novel insights into
tumor pathogenesis, and also identifies potential targets for tumor
prevention and treatment.
Chloride channels are expressed ubiquitously and are
important in various cellular processes, including the cell cycle
and proliferation, cell volume modulation, epithelial secretion and
membrane excitability (7). To
date, chloride channel family members, including CLC proteins,
ionotropic receptors for γ-aminobutyric acid and glycine, and the
cystic fibrosis transmembrane conductance regulator (CFTR), have
been described (8).
Calcium-activated chloride channels (CaCCs) are reported to be
major modulators of cell volume and epithelial secretion (9). However, the composition of CaCCs
remains unknown. More recently, transmembrane protein with unknown
function 16 (TMEM16) A, also known as anoctamin 1, has been
hypothesized to be a candidate CaCC (10–12).
TMEM16A belongs to the TMEM16 family, which consists of nine other
members, including TMEM16B-K, characterized by eight transmembrane
segments and a highly conserved domain of unknown function. TMEM16A
has been demonstrated to be involved in the regulation of
gastrointestinal tract motility, epithelial fluid transport and
saliva production (13–16). CaCCs have been found to be
proapoptotic, acting as tumor suppressor genes in mammary
epithelium (17). However,
bestrophin, a putative CaCC that promotes cell proliferation, has
been shown to be overexpressed in colon cancer (18). Notably, TMEM16A is localized on
chromosome 11q13, a region that is frequently amplified in human
cancers, including those of the head and neck, esophagus, bladder
and breast (19–21). Therefore, TMEM16A dysregulation may
be associated with tumor progression. As hypothesized, several
studies have revealed that TMEM16A is overexpressed in various
tumor types, including gastrointestinal stromal tumors, esophageal
cancers, and head and neck cancers (22–24).
However, the molecular mechanisms of TMEM16A in the regulation of
tumors remains poorly understood.
Several studies have hypothesized that chloride
channels are associated with the nuclear factor-κB (NF-κB)
signaling pathway (25–29). NF-κB, which is tightly controlled,
regulates a wide range of cellular processes and is extensively
involved in cancer progression (30). Normally, the binding of NF-κB to
inhibitory inhibitor of κB (IκB) proteins inactivates NF-κB in the
cytoplasm (31). Degradation of
IκB proteins releases NF-κB and enables it to translocate to the
nucleus and activate target genes. Dysregulation of this process is
involved in a number of diseases (32,33).
NF-κB has also been demonstrated to be a critical regulator of
tumorigenesis involved in cell survival, metastasis and
angiogenesis (31). However,
although TMEM16A has been hypothesized to be a transmembrane
protein regulating cellular ion exchanges, its role in the
regulation of the NF-κB signal pathway remains unclear.
To date, no studies have determined the exact role
of TMEM16A in gliomas. The aim of the present study was to
determine whether TMEM16A is involved in gliomas and the potential
underlying mechanisms.
Materials and methods
Cell lines and cell culture
One normal human astrocyte line (SVGp12) and four
glioma cell lines (U87MG, U118, U251 and SHG44) were obtained from
the American Type Culture Collection (Manassas, VA, USA). All cells
were maintained according to standard protocols. Briefly, cells
were cultured in Dulbecco’s modified Eagle’s medium (DMEM)
supplemented with 10% fetal bovine serum to which 100 U/ml
penicillin, 100 μg/ml streptomycin and 2 mM L-glutamate were added.
All cells were cultured at 37°C with 5% CO2 in an
incubator (Invitrogen Life Technologies, Carlsbad, CA, USA).
Tumor tissue preparation
All tumor tissues were obtained from Tangdu Hospital
(Xi’an, China) according to institutional guidelines for consent
with the approval of patients and the hospital. Brain tissue was
obtained from 20 patients with gliomas (eight females and 12 males;
median age, 43.6 years; range, 16–78 years; five each of grade I,
II, III and IV). Normal brain tissue samples were obtained from
five patients with brain trauma (median age, 42 years; range, 21–68
years). All samples were obtained from the initial surgery, prior
to chemotherapy and radiotherapy. The malignant grades of the tumor
tissues were analyzed as per the standard classification
established by the World Health Organization (34).
Quantitative polymerase chain reaction
(qPCR) analysis
Total RNA was extracted from cultured cells using
TRIzol reagent (Invitrogen Life Technologies), following the
manufacturer’s instructions. In total, ≤5 μg RNA was
reverse-transcribed into cDNA using Maloney-murine leukemia virus
reverse transcriptase (Clontech Laboratories, Inc., Palo Alto, CA,
USA). The cDNA molecules were used as templates for qPCR. Genes
were amplified using primers for TMEM16A, cyclin D1, cyclin E and
c-myc. The primers were as follows: TMEM16A forward,
5′-CACAAGAGAGCCTCGGGTAG-3′ and reverse, 5′-ATCTTCACAAACCCGACACC-3′;
cyclin D1 forward, 5′-GCCAACCTCCTCAACGACCGG-3′ and reverse,
5′-GTCCATGTTCTGCTGGGCCTG-3′; cyclin E forward,
5′-GTCCTGGCTGAATGATACATC-3′ and reverse,
5′-CCCTATTTTGTTCAGACAACATGGC-3′; c-myc forward
5′-ACACATCAGCACAACTACGC-3′ and reverse,
5′-CCTCTTGACATTCTCCTCGGT-3′; GAPDH forward,
5′-CGGAGTCAACGGATTTGGTCGTAT-3′ and reverse
5′-AGCCTTCTCCATGGTGGTGAA-3′. The qPCR mixture system contained 5 μl
SsoFast™ EvaGreen Supermix (Bio-Rad, Hercules, CA, USA), 1 μl cDNA
(diluted at 1:50) and 2 μl each of the forward and reverse primers
(1 μM), to a final volume of 10 μl. The PCR procedure was as
follows: 94°C for 4 min; 94°C for 20 sec, 55°C for 30 sec and 72°C
for 20 sec; 2 sec for plate reading for 35 cycles; melting curve,
65–95°C. GAPDH was used as a control for normalizing the gene
expression. Three independent experiments were performed. The data
obtained were analyzed by the 2−ΔΔCt method and
statistically analyzed as described previously (35), followed by Student’s unpaired
sample t-test.
Nuclear protein extraction
Nuclear proteins were extracted using a Nuclear
Extraction kit (Sangon Biotech, Shanghai, China) according to the
manufacturer’s instructions. Briefly, cells were lysed in
cytoplasmic buffer containing protease inhibitors, mixed and
incubated for 15 min at 4°C. Next, cells were centrifuged at 12,000
rpm for 20 min at 4°C. Supernatants containing cytoplasmic
extraction products were collected. Cell sediments were collected
and resuspended in nucleus buffer for 10 min at 4°C. Next, the
sample was centrifuged at 12,000 rpm for 10 min at 4°C and the
supernatant was collected for analysis.
Western blotting
A total of 20–30 μg protein was fractionated by 12%
SDS-PAGE electrophoresis and transferred to nitrocellulose
membranes (Amersham, Little Chalfont, UK). The membrane was treated
under agitation and blocking at room temperature, with 2% non-fat
dry milk in Tris-buffered saline (TBS) for 1 h, followed by
incubation with primary rabbit anti-human TMEM16A, IκB, p65, MMP-2,
MMP-9, GAPDH and Histone polyclonal antibodies. (Santa Cruz
Biotechnology, CA, USA). Antibodies were diluted in blocking buffer
(1:2,000) and incubated with the membrane at 4°C overnight. Next,
membranes were washed three times with TBS and Tween 20 [TBST; 10
mM Tris-HCl (pH 7.5), 150 mM NaCl and 0.05% Tween-20] for 10 min at
room temperature. Subsequently, the membrane was incubated in
horseradish peroxidase (HRP)-conjugated secondary antibody goat
anti-rabbit Ig G (diluted 1:5,000 in the blocking buffer; Boster
Biological Technology Ltd., Wuhan, China) for 1 h. Following
washing three times with TBST and once with TBS each for 10 min, 1
ml 4-chloro-1-naphthol was used as the HRP substrate, with 9 ml TBS
and 6 μl H2O2, to visualize the target
protein in the dark for 5–30 min.
Cell transfection and luciferase
assay
The full length cDNA-encoding sequence of TMEM16A
was subcloned into pcDNA3.1 vectors according to standard
protocols. Small interfering RNAs (siRNAs) targeting TMEM16A and
control siRNA were purchased from Santa Cruz Biotechnology. Cells
were transfected with vectors or siRNA according to the
manufacturer’s instructions. Briefly, plasmid DNA (1 μg) or siRNA
(30 pmol) were diluted in 500 μl DMEM with 5 μl Lipofectamine
(Invitrogen Life Technologies), mixed and incubated at room
temperature for 15 min. Next, the mixtures were added to the cells
at a final volume of 3 ml medium and incubated for 24 h. Cells were
co-transfected with the promoter luciferase reporter plasmid and
harvested after 24 h. Luciferase activity was analyzed by the Dual
Luciferase Reporter Assay system (Promega Corporation, Madison, WA,
USA).
Cell proliferation assay
For the MTT assay, cells were plated in 96-well
plates and cultured under standard conditions until they reached
80% confluency. Plasmids or siRNA were transfected, according to
standard protocols, and incubated with cells for the indicated
times. Next, the initial culture medium was replaced with fresh
medium containing MTT (5 mg/ml in PBS; 200 μl/well; Sigma, St.
Louis, MO, USA) and incubated for an additional 4 h. The formazan
was dissolved in dimethylsulfoxide (150 μl/well; Sigma) for 10 min
and the absorbance at 490 nm was determined with an ELISA reader
(Bio-Tek Instruments, Winooski, VA, USA). Each cell viability assay
was performed in quadruplicate and repeated three times. For the
5-bromo-2-deoxyuridine (BrdU) assay, a BrdU Cell Proliferation
Assay kit (Millipore, Billerica, MA, USA) was used. A total of 10
μl BrdU solution was added per well and incubated with cells for 2
h. Next, the medium was discarded and 100 μl/well fixing/denaturing
solution was added for 15 min. Following this, the solution was
removed and 100 μl/well antibody detection solution was added and
incubated for 1 h at room temperature. Thereafter, plates were
washed three times with wash buffer, followed by the addition of
100 μl/well prepared HRP-conjugated secondary antibody solution,
and incubated for 30 min at room temperature. Plates were washed
three times with wash buffer and 100 μl
3,3′,5,5′-tetramethylbenzidine substrate was added prior to
incubation for 30 min at room temperature. The volume of BrdU
incorporated into the cells was determined at 450 nm by a
microplate reader (Bio-Rad). Data are expressed as the mean ±
standard error of the mean and differences were analyzed by
Student’s-t test.
Cell invasion and migration assays
Cells were suspended in a volume of 50 μl serum-free
medium, which was then added to the upper chamber of chemotaxis
chambers (Neuro Probe, Inc., Gaithersburg, MD, USA). Complete
medium was added to the lower chamber. Polycarbonate membrane was
placed between the two chambers and culture medium, supplemented
with 20 μl Matrigel (BD Biosciences, Franklin Lakes, NJ, USA), was
applied. Cells were incubated for 36 h at 37°C. Next, the membrane
was fixed and stained with methanol and Giemsa, respectively. In
total, ~10 visual fields were randomly selected and quantified per
membrane. Each sample was assayed in triplicate. A similar system
was performed for the migration assay.
Statistical analysis
All experiments were performed independently at
least three times. Differences between groups were analyzed by
Student’s t-test and P<0.01 was considered to indicate a
statistically significant difference.
Results
TMEM16A expression is high in glioma
tissue
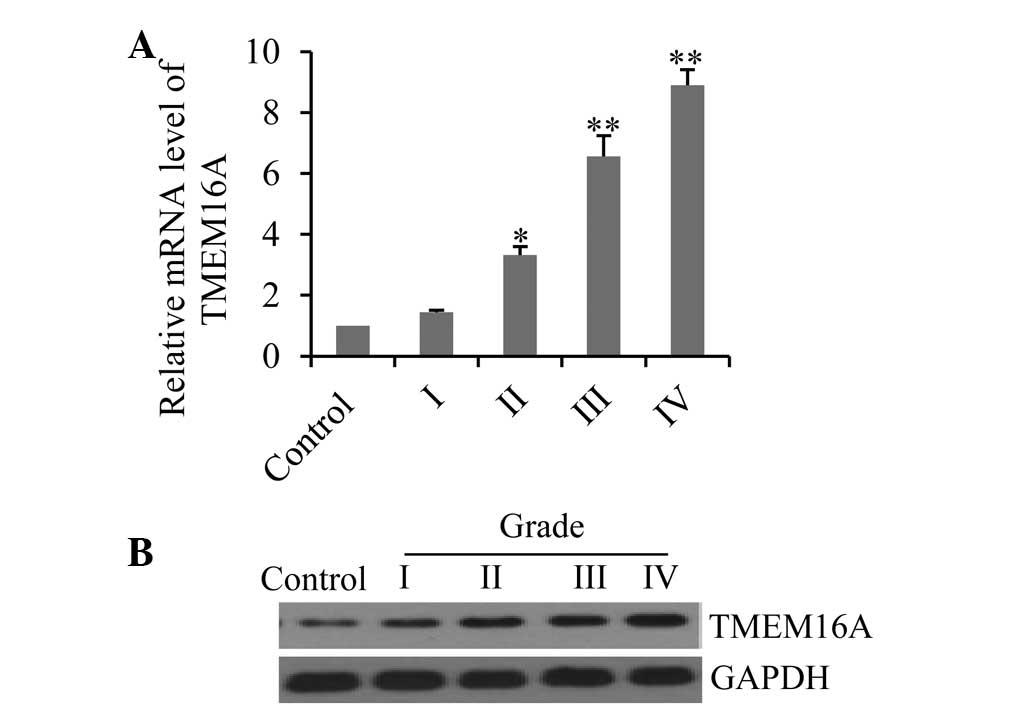
To determine whether TMEM16A is involved in glioma
formation, the expression profiles of TMEM16A in the tissue sample
of patients with various pathological grade gliomas were analyzed
by qPCR and western blotting. Results demonstrated that mRNA
expression levels increased with increasing pathological glioma
grades (Fig. 1A), with
particularly high levels seen in grade III and IV gliomas. The data
were further confirmed by western blotting, which demonstrated that
TMEM16A proteins were also overexpressed in glioma tissues and
correlated with high pathological grade (Fig. 1B).
TMEM16A expression is high in glioma cell
lines
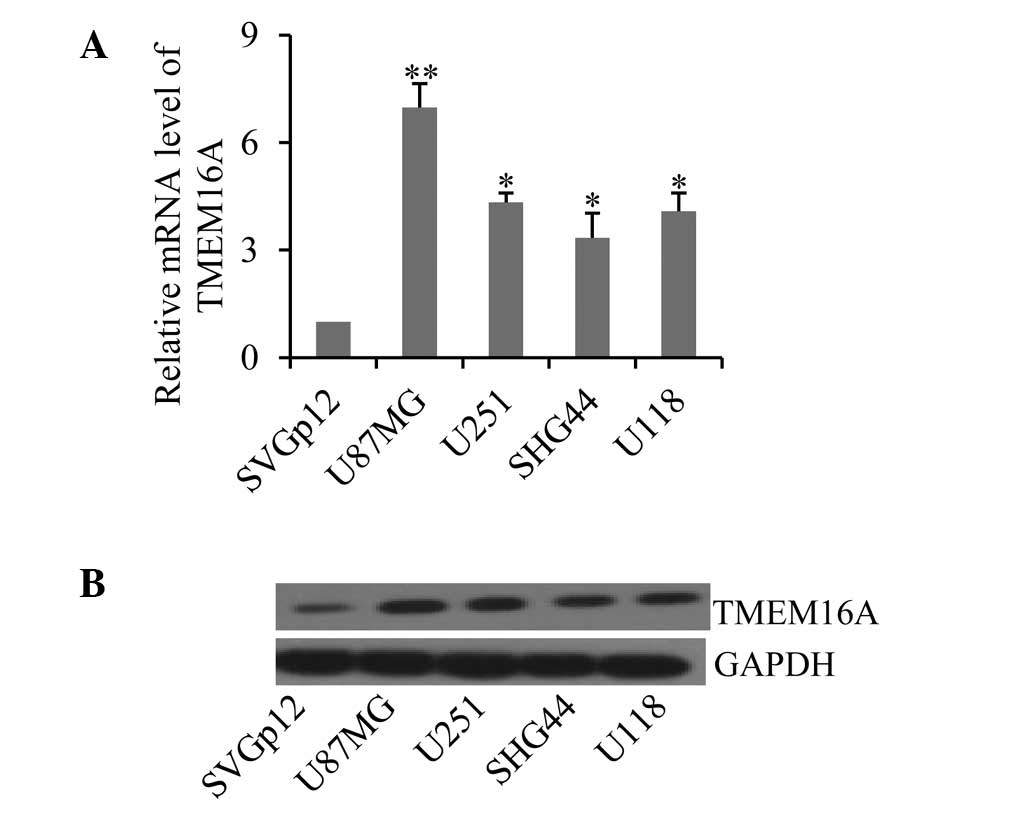
Next, the expression profiles of TMEM16A were
measured in four cultured glioma cell lines and one normal
astrocyte line. Consistent with results from glioma tissues,
TMEM16A demonstrated high mRNA expression levels in the four
cultured glioma cell lines, U87MG, U251, SHG44 and U118, compared
with the normal astrocyte cell line, SVGp12 (Fig. 2A; P<0.05). Additionally, TMEM16A
levels were more abundant in U87MG cells (P<0.01). These results
were further confirmed by western blotting of the protein levels of
TMEM16A in these cell lines (Fig.
2B). Collectively, these results indicate that dysregulation of
TMEM16A is associated with gliomas.
Alteration of TMEM16A expression impairs
cell proliferation, migration and invasion
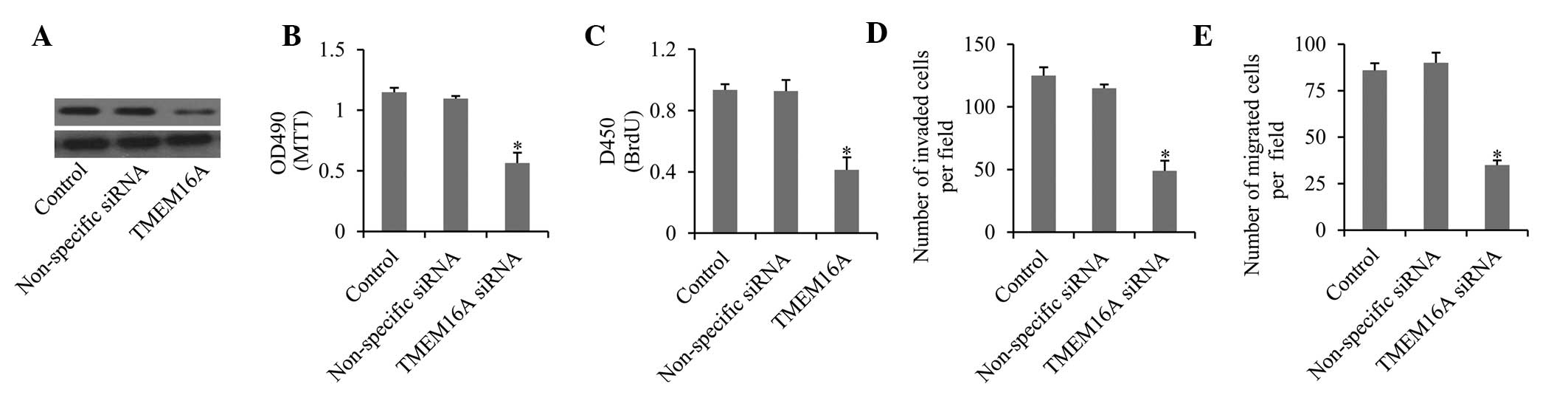
To investigate the role of TMEM16A in gliomas, the
expression of TMEM16A was silenced in U87MG cells by specific
siRNAs (Fig. 3A). First, the
effects of TMEM16A knockdown on cell proliferation were detected.
The MTT assay revealed that cell growth and viability were
significantly inhibited compared with controls (Fig. 3B). This was further confirmed by
BrdU assay, which demonstrated that cell proliferation was markedly
decreased upon knockdown of TMEM16A (Fig. 3C). Furthermore, the migration
(Fig. 3D) and invasion (Fig. 3E) ability were also significantly
impaired upon TMEM16A knockdown. These results indicate that
TMEM16A is important in cell proliferation and in the migration and
invasion of glioma cells. By contrast, SVGp12 cells transfected
with TMEM16A overexpression vectors exhibited high levels of cell
proliferation, migration and invasion (data not shown).
TMEM16A is involved in activation of
NF-κB
Various studies have demonstrated that chloride
channels are involved in the activation of NF-κB (25,26).
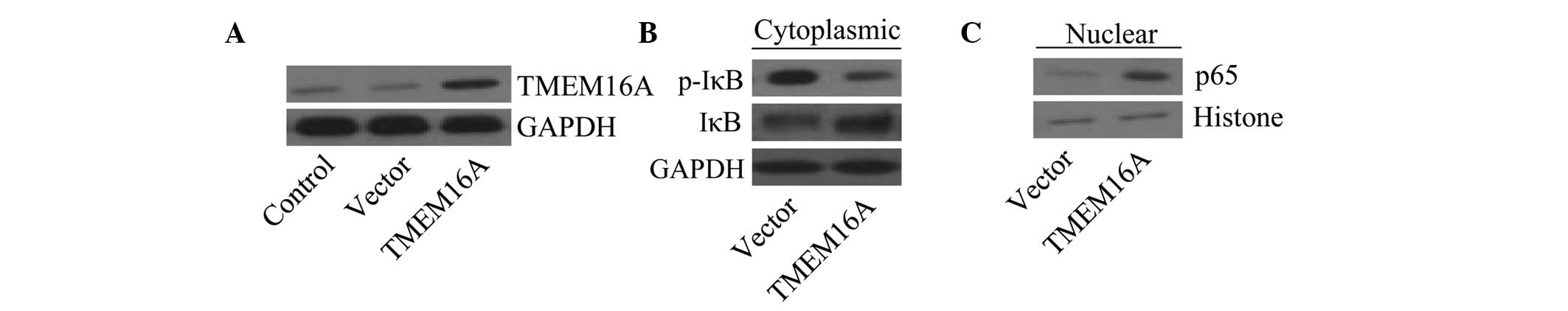
In order to assess whether TMEM16A is associated with the
activation of NF-κB, SVGp12 cells were used to generate stable
TMEM16A overexpression cell lines. Cells transfected with TMEM16A
overexpression vectors exhibited an increase in TMEM16A expression
levels compared with the vector control (Fig. 4A). Cytoplasmic and nuclear extracts
were prepared from various cell groups. Results revealed that IκBα
phosphorylation was decreased in TMEM16A overexpressing cells,
while unphosphorylated IκBα was increased in the cytoplasm
(Fig. 4B), compared with control
vector-transfected cells. Furthermore, the NF-κB subunit p65
accumulated in the nucleus in TMEM16A-overexpressing cells
(Fig. 4C). The data suggest that
TMEM16A is associated with activation of the NF-κB signaling
pathway.
TMEM16A regulates NF-κB-mediated gene
transcription
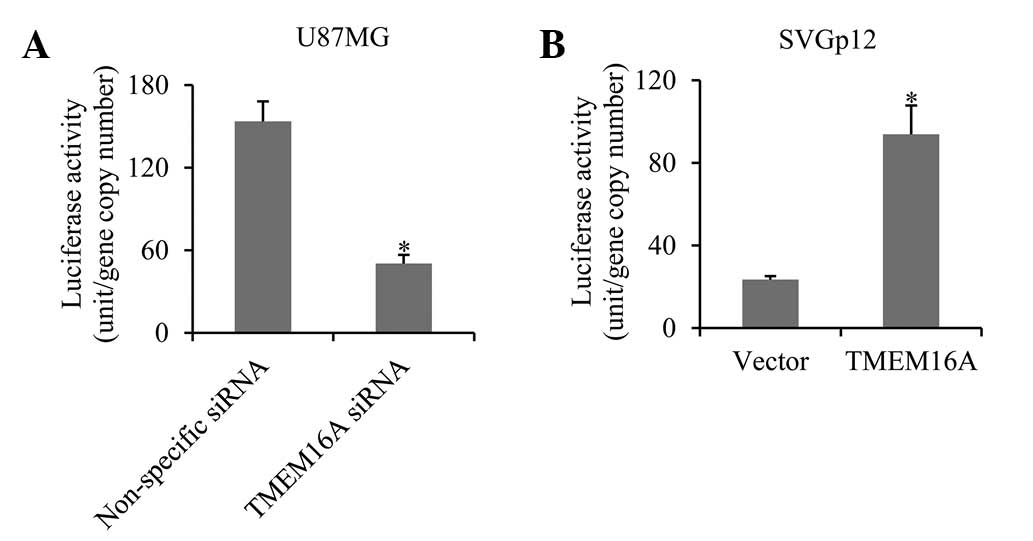
In order to further determine whether TMEM16A
regulates NF-κB-mediated gene transcription in glioma cells, a
luciferase reporter vector containing the NF-κB response element
promoter and TMEM16A siRNA were co-transfected into U87MG cells.
Results demonstrated that the luciferase activity was decreased
three-fold in TMEM16A-silenced cells compared with the control
group (Fig. 5A). In
TMEM16A-overexpressing SVGp12 cells, luciferase activity increased
four-fold (Fig. 5B). The results
indicate that TMEM16A promotes NF-κB-mediated gene
transcription.
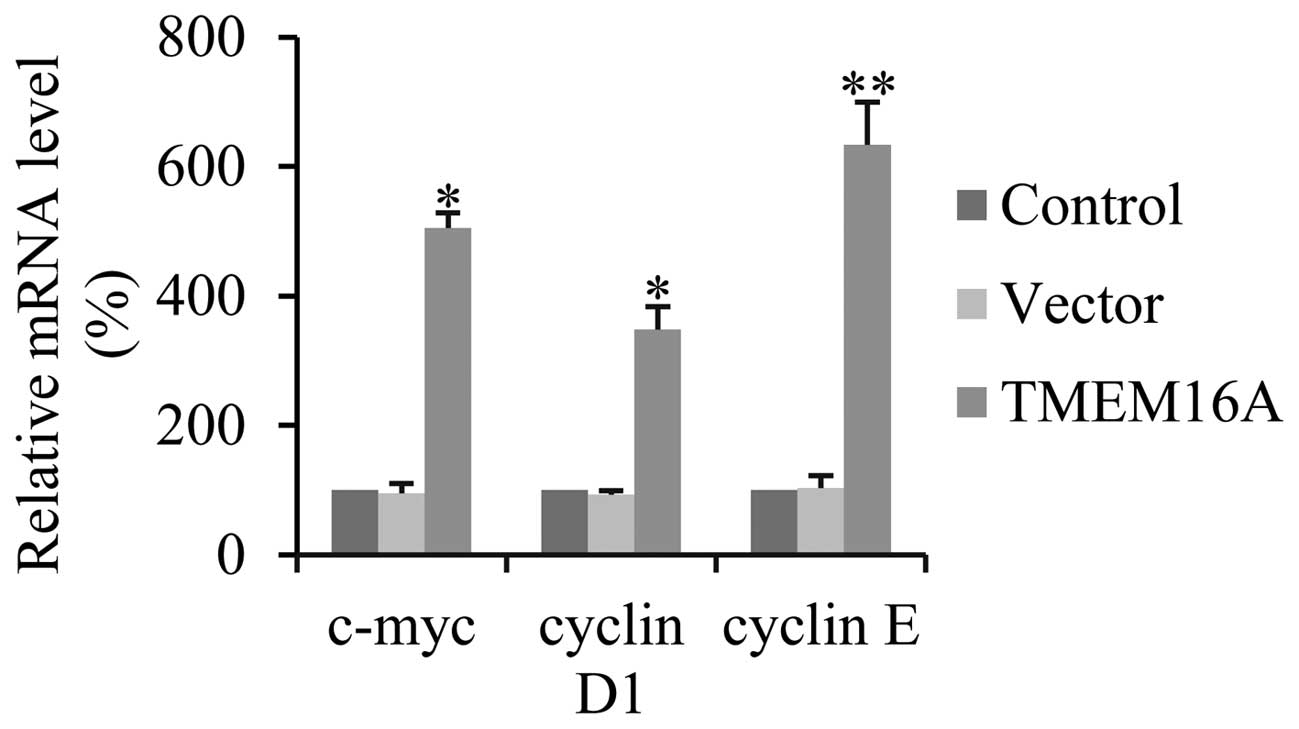
TMEM16A increases cell proliferation
through cyclin D1, cyclin E and c-myc activated by NF-κB
Various studies have demonstrated that NF-κB is
involved in the regulation of cell proliferation through cyclin D1,
cyclin E and c-myc (36–38). In order to define whether
overexpression of TMEM16A activates NF-κB-mediated cell
proliferation, the transcription levels of cyclin D1, cyclin E and
c-myc were determined by qPCR. TMEM16A overexpression, compared
with controls, significantly increased the expression of cyclin D1,
cyclin E and c-myc in SVGp12 cells (Fig. 6). These results indicate that
TMEM16A promotes cell proliferation in glioma cells via the
NF-κB-target gene.
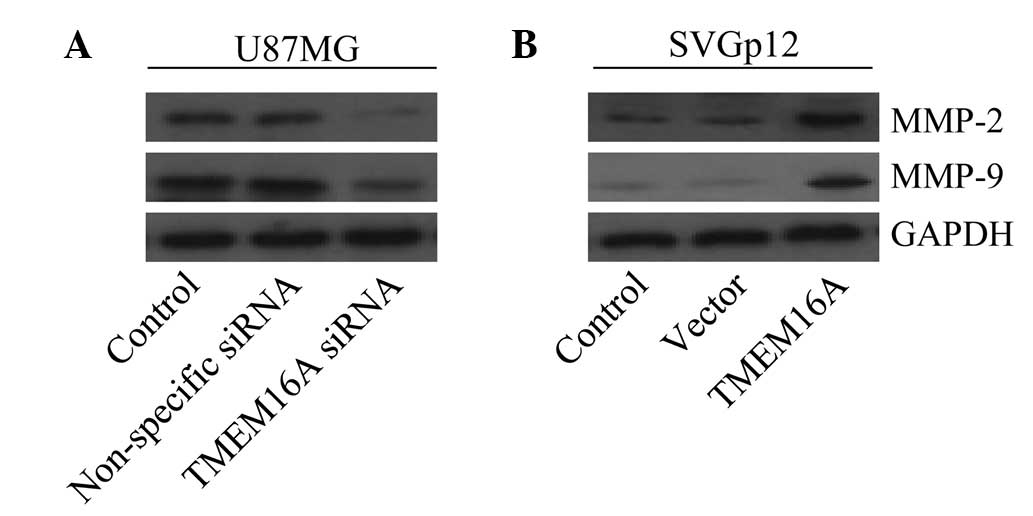
TMEM16A promotes cell migration and
invasion by MMP-2 and -9 mediated by NF-κB
Previous studies have demonstrated that MMPs,
particularly MMP-2 and -9, are markedly associated with cell
migration and invasion of gliomas and are the target genes of NF-κB
(39–41). The present study aimed to determine
whether TMEM16A impaired cell migration and invasion by MMP-2 and
-9, mediated by NF-κB. Results revealed that knockdown of TMEM16A
in U87MG cells effectively downregulated MMP-2 and -9 protein
expression levels (Fig. 7A).
Additionally, it was found that overexpression of TMEM16A in SVGp12
cells markedly upregulated protein expression levels of MMP-2 and
-9 (Fig. 7B). These results
indicate that TMEM16A is involved in the regulation of MMP-2 and
-9.
Discussion
In previous years, increasing evidence has
demonstrated that chloride channels regulate tumor growth and
progression (42,43). In addition, contradictory functions
of CaCCs, including upregulation and downregulation of tumor
growth, have been reported (17,18).
High levels of TMEM16A have been reported in gastrointestinal
stromal tumors, squamous cell carcinomas of the head and neck, and
esophageal cancer (22–24). In the present study, TMEM16A was
demonstrated to be overexpressed in gliomas. Notably, a higher
abundance was found in grade III and IV gliomas, indicating that
high TMEM16A expression may be correlate with pathological grade.
However, low levels of TMEM16A were also detected in normal brain
tissues and normal human astrocytes, indicating that TMEM16A may be
essential for maintaining basic cellular activities. In addition,
TMEM16A has been shown to be expressed in a broad spectrum of mouse
and human tissues (44). However,
the precise mechanism by which TMEM16A regulates tumor development
and progression remains poorly understood.
Chloride channels have previously been hypothesized
to be involved in the regulation of cellular proliferation. The
present study found that knockdown of TMEM16A significantly
suppressed the proliferation of glioma cells. In addition,
silencing of TMEM16A downregulated the migration and invasion of
glioma cells. It has been reported that TMEM16A overexpression
contributes to tumor metastasis by promoting cell motility
(45). Recently, TMEM16A was
suggested to influence cellular proliferation through activation of
the Ras-Raf-mitogen-activated protein kinase-extracellular
signal-related kinase 1/2 pathway (46). Blockade of TMEM16A has also been
shown to markedly increase the expression of insulin-like growth
factor-binding protein 5, a potent anti-angiogenic factor,
therefore implicating TMEM16A in the modulation of the tumor
microenvironment through the signaling pathway (47). However, it was also demonstrated
that suppression of TMEM16A did not affect cell growth in
vitro but significantly delayed the growth of xenografts in
vivo. Mazzone et al (48) reported that inhibition of TMEM16A,
by its selective inhibitor T16A(inh)-A01, reduced the proliferation
of cultured pancreatic cancer cells and interstitial cells. Liu
et al (49) provided data
demonstrating that TMEM16A was involved in tumorigenesis and the
development of metastatic prostate cancer. Increasing evidence has
indicated that TMEM16A is involved in the development and
progression of tumors. Thus, further study into the exact role of
TMEM16A in tumors is of extreme importance.
Chloride channels have been hypothesized to be
involved in the regulation of NF-κB. A decrease in intracellular
chloride concentration activates the NF-κB signaling pathway
through the chloride channel CLC-3 (25). Inhibition of the CFTR impairs tight
junctions through activation of the NF-κB signaling pathway
(26). However, whether TMEM16A is
associated with the activation of NF-κB remains unclear. NF-κB is
important in the regulation of various cellular functions that are
frequently overactivated in tumor cells. In order to determine
whether TMEM16A overexpression is involved in the activation of
NF-κB, in the present study, the key components of the activation
of NF-κB were examined in TMEM16A-expressing SVGp12 cells.
Phosphorylation levels of the inhibitor of NF-κB, IκBα, were high
following TMEM16A overexpression, which was further confirmed by
the accumulation of the NF-κB subunit p65 protein in the nucleus.
Furthermore, a luciferase reporter assay also demonstrated that
overexpression of TMEM16A significantly upregulated NF-κB-mediated
gene transcription activity. These results suggest that the
increased activity of NF-κB is associated with TMEM16A
overexpression. Thus, the present study may present a mechanism for
the role of TMEM16A in tumors.
NF-κB has been implicated in the regulation of
glioma cell proliferation through the upregulation of oncogenes,
including cyclin D1, cyclin E, and c-myc (50–54).
The present study revealed that overexpression of TMEM16A
significantly increased transcription levels of these genes, which
may explain why TMEM16A promotes cell proliferation in gliomas.
MMPs, the target genes of NF-κB, have long been implicated in tumor
progression processes, for example cell migration and invasion
(39). In particular, MMP-2 and -9
are heavily implicated in tumor progression and have been shown to
regulate cell migration and the invasion of gliomas (40,41).
In the present study, TMEM16A was demonstrated to be closely
associated with the expression of MMP-2 and -9.
The present study has shown that TMEM16A is
overexpressed in gliomas, particularly in high grade gliomas (III
and IV). Furthermore, it has been demonstrated that TMEM16A
overexpression promotes cell proliferation, migration and invasion
through activation of the NF-κB signaling pathway. In conclusion,
the present study provides novel insights into the role of TMEM16A
in the development and progression of gliomas, indicating that this
protein may act as a potential therapeutic target. However, the
specific role of TMEM16A in the regulation of tumors requires
further study.
Abbreviations:
|
CaCCs
|
calcium-activated chloride
channels
|
|
TMEM16A
|
transmembrane protein with unknown
function 16A
|
|
MMP
|
matrix metalloproteinase
|
|
NF-κB
|
nuclear factor-κB
|
|
CFTR
|
cystic fibrosis transmembrane
conductance regulator
|
|
BrdU
|
5-bromo-2-deoxyuridine
|
References
|
1
|
Stühmer W, Alves F, Hartung F, Zientkowska
M and Pardo LA: Potassium channels as tumour markers. FEBS Lett.
580:2850–2852. 2006.PubMed/NCBI
|
|
2
|
Lastraioli E, Taddei A, Messerini L, et
al: hERG1 channels in human esophagus: evidence for their aberrant
expression in the malignant progression of Barrett’s esophagus. J
Cell Physiol. 209:398–404. 2006.PubMed/NCBI
|
|
3
|
Masi A, Becchetti A, Restano-Cassulini R,
et al: hERG1 channels are overexpressed in glioblastoma multiforme
and modulate VEGF secretion in glioblastoma cell lines. Br J
Cancer. 93:781–792. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Panner A and Wurster RD: T-type calcium
channels and tumor proliferation. Cell Calcium. 40:253–259. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Diss JK, Stewart D, Pani F, et al: A
potential novel marker for human prostate cancer: voltage-gated
sodium channel expression in vivo. Prostate Cancer Prostatic Dis.
8:266–273. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fraser SP, Diss JK, Chioni AM, et al:
Voltage-gated sodium channel expression and potentiation of human
breast cancer metastasis. Clin Cancer Res. 11:5381–5389. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Furst J, Gschwentner M, Ritter M, et al:
Molecular and functional aspects of anionic channels activated
during regulatory volume decrease in mammalian cells. Pflugers
Arch. 444:1–25. 2002.PubMed/NCBI
|
|
8
|
Ferrera L, Caputo A and Galietta LJ:
TMEM16A protein: a new identity for Ca(2+)-dependent Cl-
channels. Physiology (Bethesda). 25:357–363. 2010.
|
|
9
|
Hartzell C, Putzier I and Arreola J:
Calcium-activated chloride channels. Annu Rev Physiol. 67:719–758.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Caputo A, Caci E, Ferrera L, et al:
TMEM16A, a membrane protein associated with calcium-dependent
chloride channel activity. Science. 322:590–594. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yang YD, Cho H, Koo JY, et al: TMEM16A
confers receptor-activated calcium-dependent chloride conductance.
Nature. 455:1210–1215. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Schroeder BC, Cheng T, Jan YN and Jan LY:
Expression cloning of TMEM16A as a calcium-activated chloride
channel subunit. Cell. 134:1019–1029. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rock JR, O’Neal WK, Gabriel SE, et al:
Transmembrane protein 16A (TMEM16A) is a Ca2+-regulated
Cl− secretory channel in mouse airways. J Biol Chem.
284:14875–14880. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Huang F, Rock JR, Harfe BD, et al: Studies
on expression and function of the TMEM16A calcium-activated
chloride channel. Proc Natl Acad Sci USA. 106:21413–21418. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hwang SJ, Blair PJ, Britton FC, et al:
Expression of anoctamin 1/TMEM16A by interstitial cells of Cajal is
fundamental for slow wave activity in gastrointestinal muscles. J
Physiol. 587:4887–4904. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Romanenko VG, Catalán MA, Brown DA, et al:
Tmem16A encodes the Ca2+-activated Cl−
channel in mouse submandibular salivary gland acinar cells. J Biol
Chem. 285:12990–13001. 2010.PubMed/NCBI
|
|
17
|
Elble RC and Pauli BU: Tumor suppression
by a proapoptotic calcium-activated chloride channel in mammary
epithelium. J Biol Chem. 276:40510–40517. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Spitzner M, Martins JR, Soria RB, et al:
Eag1 and Bestrophin 1 are up-regulated in fast-growing colonic
cancer cells. J Biol Chem. 283:7421–7428. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Katoh M and Katoh M: FLJ10261 gene,
located within the CCND1-EMS1 locus on human chromosome 11q13,
encodes the eight-transmembrane protein homologous to C12orf3,
C11orf25 and FLJ34272 gene products. Int J Oncol. 22:1375–1381.
2003.PubMed/NCBI
|
|
20
|
Akervall JA, Jin Y, Wennerberg JP, et al:
Chromosomal abnormalities involving 11q13 are associated with poor
prognosis in patients with squamous cell carcinoma of the head and
neck. Cancer. 76:853–859. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Schwab M: Amplification of oncogenes in
human cancer cells. Bioessays. 20:473–479. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Carles A, Millon R, Cromer A, et al: Head
and neck squamous cell carcinoma transcriptome analysis by
comprehensive validated differential display. Oncogene.
25:1821–1831. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Espinosa I, Lee CH, Kim MK, et al: A novel
monoclonal antibody against DOG1 is a sensitive and specific marker
for gastrointestinal stromal tumors. Am J Surg Pathol. 32:210–218.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kashyap MK, Marimuthu A, Kishore CJ, et
al: Genomewide mRNA profiling of esophageal squamous cell carcinoma
for identification of cancer biomarkers. Cancer Biol Ther. 8:36–46.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yang H, Huang LY, Zeng DY, et al: Decrease
of intracellular chloride concentration promotes endothelial cell
inflammation by activating nuclear factor-kappaB pathway.
Hypertension. 60:1287–1293. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen J, Fok KL, Chen H, et al:
Cryptorchidism-induced CFTR down-regulation results in disruption
of testicular tight junctions through up-regulation of
NF-κB/COX-2/PGE2. Hum Reprod. 27:2585–2597. 2012.PubMed/NCBI
|
|
27
|
He G, Ma Y, Chou SY, et al: Role of CLIC4
in the host innate responses to bacterial lipopolysaccharide. Eur J
Immunol. 41:1221–1230. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sheridan GK, Pickering M, Twomey C, et al:
NF-kappaB activity in distinct neural subtypes of the rat
hippocampus: Influence of time and GABA antagonism in acute slice
preparations. Learn Mem. 14:525–532. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Miller FJ Jr, Filali M, Huss GJ, et al:
Cytokine activation of nuclear factor kappa B in vascular smooth
muscle cells requires signaling endosomes containing Nox1 and
ClC-3. Circ Res. 101:663–671. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Laver T, Nozell S and Benveniste EN: The
NF-κB signaling pathway in GBMs: implications for apoptotic and
inflammatory responses and exploitation for therapy. CNS Cancer:
Models, Markers, Prognostic Factors, Targets and Therapeutic
Approaches. Van Meir EG: 1. Humana Press (Springer); New York, NY:
pp. 1011–1036. 2009
|
|
31
|
Gilmore TD: Introduction to NF-kappaB:
players, pathways, perspectives. Oncogene. 25:6680–6684. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Perkins ND: The Rel/NF-kappa B family:
friend and foe. Trends Biochem Sci. 25:434–440. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Perkins ND and Gilmore TD: Good cop, bad
cop: the different faces of NF-kappaB. Cell Death Differ.
13:759–772. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Louis DN, Ohgaki H, Wiestler OD, et al:
The 2007 WHO classification of tumours of the central nervous
system. Acta Neuropathol. 114:97–109. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
|
|
36
|
Lee CH, Jeon YT, Kim SH and Song YS:
NF-kappaB as a potential molecular target for cancer therapy.
Biofactors. 29:19–35. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sethi G, Sung B and Aggarwal BB: Nuclear
factor-kappaB activation: from bench to bedside. Experiment Biol
Med (Maywood). 233:21–31. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Van Waes C: Nuclear factor-kappaB in
development, prevention, and therapy of cancer. Clin Cancer Res.
13:1076–1082. 2007.PubMed/NCBI
|
|
39
|
Bauvois B: New facets of matrix
metalloproteinases MMP-2 and MMP-9 as cell surface transducers:
outside-in signaling and relationship to tumor progression. Biochim
Biophys Acta. 1825:29–36. 2012.PubMed/NCBI
|
|
40
|
Veeravalli KK and Rao JS: MMP-9 and uPAR
regulated glioma cell migration. Cell Adh Migr. 6:509–512. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kesanakurti D, Chetty C, Rajasekhar
Maddirela D, Gujrati M and Rao JS: Functional cooperativity by
direct interaction between PAK4 and MMP-2 in the regulation of
anoikis resistance, migration and invasion in glioma. Cell Death
Dis. 3:e4452012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Habela CW, Ernest NJ, Swindall AF and
Sontheimer H: Chloride accumulation drives volume dynamics
underlying cell proliferation and migration. J Neurophysiol.
101:750–757. 2009.PubMed/NCBI
|
|
43
|
Habela CW, Olsen ML and Sontheimer H: ClC3
is a critical regulator of the cell cycle in normal and malignant
glial cells. J Neurosci. 28:9205–9217. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kunzelmann K, Kongsuphol P, Aldehni F, et
al: Bestrophin and TMEM16-Ca(2+) activated Cl(−) channels with
different functions. Cell Calcium. 46:233–241. 2009.PubMed/NCBI
|
|
45
|
Ayoub C, Wasylyk C, Li Y, et al: ANO1
amplification and expression in HNSCC with a high propensity for
future distant metastasis and its functions in HNSCC cell lines. Br
J Cancer. 103:715–726. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Duvvuri U, Shiwarski DJ, Xiao D, et al:
TMEM16A induces MAPK and contributes directly to tumorigenesis and
cancer progression. Cancer Res. 72:3270–3281. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Simon S, Grabellus F, Ferrera L, et al:
DOG1 regulates growth and IGFBP5 in gastrointestinal stromal
tumors. Cancer Res. 73:3661–3670. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Mazzone A, Eisenman ST, Strege PR, et al:
Inhibition of cell proliferation by a selective inhibitor of the
Ca(2+)-activated Cl(−) channel, Ano1. Biochem Biophys Res Commun.
427:248–253. 2012.
|
|
49
|
Liu W, Lu M, Liu B, Huang Y and Wang K:
Inhibition of Ca(2+)-activated Cl(−) channel ANO1/TMEM16A
expression suppresses tumor growth and invasiveness in human
prostate carcinoma. Cancer Lett. 326:41–51. 2012.
|
|
50
|
Abdullah JM, Ahmad F, Ahmad KA, et al:
Molecular genetic analysis of BAX and Cyclin D1 genes in patients
with malignant glioma. Neurol Res. 29:239–242. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Arato-Ohshima T and Sawa H:
Over-expression of cyclin D1 induces glioma invasion by increasing
matrix metalloproteinase activity and cell motility. Int J Cancer.
83:387–392. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zhang X, Zhao M, Huang AY, et al: The
effect of cyclin D expression on cell proliferation in human
gliomas. J Clin Neurosci. 12:166–168. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Liao DJ, Thakur A, Wu J, Biliran H and
Sarkar FH: Perspectives on c-Myc, cyclin D1, and their interaction
in cancer formation, progression, and response to chemotherapy.
Crit Rev Oncogen. 13:93–158. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Robson S, Pelengaris S and Khan M: c-Myc
and downstream targets in the pathogenesis and treatment of cancer.
Recent Pat Anticancer Drug Discov. 1:305–326. 2006. View Article : Google Scholar : PubMed/NCBI
|