Introduction
Bee venom (BV) has been used as a non-steroidal
anti-inflammatory drug for the treatment of inflammatory diseases
for a number of years (1,2). Hong et al (3) demonstrated that BV was able to induce
apoptosis through caspase-3 activation in synovial fibroblasts.
Jang et al (4) reported
that BV also triggered apoptosis through inhibiting cyclooxygenase
(Cox)-2 expression in human lung cancer cells. Moon et al
(5) identified that the key
regulators in BV-induced apoptosis were B-cell lymphoma 2 (Bcl-2)
and caspase-3 in human leukemic cells through the downregulation of
mitogen-activated signal pathways. There are several biologically
active peptides in BV extracts, including melittin (MEL), a major
component, apamin, phospholipase A2 (PLA2), adolapin and mast cell
degranulating peptide (6).
In recent years, a number of studies have reported
that the MEL may also induce apoptosis, and with even higher
activities. Son et al (7)
demonstrated that MEL induces apoptosis in vascular smooth muscle
cells through the suppression of NF-κB and Akt activation. However,
Shaposhnikova et al (8)
reported that MEL may activate PLA2 in tymocytes and cause
necrosis, but not apoptosis. Kim et al (9) proved that MEL may regulate
mitochondrial apoptosis-associated gene expression to induce
apoptosis in rheumatoid arthritis. Although a number of studies
have illustrated that MEL has significant anti-proliferative and
pro-apoptotic effects, the specific mechanisms of MEL remain
elusive in human osteosarcoma cells.
A novel pathway of apoptosis due to endoplasmic
reticulum (ER) stress has been identified recently (10). In endoplasmic reticulum stress, the
adaptive responses of cells are referred to as the unfolded protein
response (UPR). Phosphorylated-protein kinase R (PKR)-like ER
kinase (p-Perk), inositol-requiring protein-1 and activating
transcription factor 6a (ATF6a) are key transmembrane signaling
proteins involved in UPR (11).
In the present study, an attempt is made to explore
the pro-apoptotic effect and the specific mechanism of MEL in human
osteosarcoma cells.
Materials and methods
Plasmid construction
Amplification of the MEL gene was performed by
polymerase chain reaction (PCR) using the cDNA of BV extracts, with
the following primers: Forward, 5′-CGTGGATCC GGAATTGGAGCAGTTCTC-3′
and reverse, 5′-AGTCTC GAGCGCCTTTGAGTGAGCT-3′. The PCR product was
ligated to vector pMD18-T, and subcloned into vector pcDNA3.1(+),
yielding a recombinant plasmid, pcDNA3.1-LPT. Amplification was
performed in a programmable thermal controller (Thermo-Hybaid,
Hybaid Limited, Cambridge, UK) with one cycle of 94°C for 3 min
followed by 30 cycles of denaturation at 94°C for 90 sec, annealing
at 63°C for 60 sec, followed by extension at 94°C for 60 sec and a
final step of 72°C for 10 min.
Cell lines, culture and transfection
The human osteosarcoma cell line MG63 and the human
fetal-osteoblast cell line hFOB 1.19 were obtained from the
American Type Culture Collection (CRL-6253; Manassas, VA, USA) and
cultured in Dulbecco’s modified Eagle medium supplemented with 10%
heat-inactivated fetal bovine serum, 100 U/ml penicillin and 100
μg/ml streptomycin. All the cells were cultured at 37°C with 5%
CO2. The MG63 and hFOB 1.19 cells were plated into 6 or
96-well plates (Falcon, Osaka, Japan) 24 h prior to transfection.
The plasmids were transfected into MG63 and hFOB 1.19 monolayer
cells with Lipofectamine™ 2000 transfection reagent (Invitrogen
Life Technologies, Carlsbad, CA, USA; employed as the MEL-exp
group). The MG63 and hFOB 1.19 cells were harvested by trypsin/EDTA
in PBS 24 h following transfection. The cells were then pelleted by
a short centrifugation (5,000 × g), suspended in the lysis buffer
as previously described by Wang et al (12) and supplemented with a complete
proteasomal inhibitor mixture (Merck KGaA, Darmstadt, Germany).
Western blot analysis
The cell lysates were separated by 15% SDS-PAGE and
electro-transferred onto nitrocellulose membranes. Following
blocking with 5% skimmed milk in phosphate-buffered saline
overnight at 4°C, the membranes were then incubated with 1:1,000
MEL-specific monoclonal antibody (mAb; Santa Cruz Biotechnology,
Inc., Santa Cruz, CA, USA), 1:1,000 goat anti-human CHOP polyclonal
antibody (pAb), 1:3,000 mouse anti-human p-Perk mAb, 1:3,000 mouse
anti-human inositol-requiring protein-1α (IRE-α) mAb, 1:600
anti-human β-actin mAb (Santa Cruz Biotechnology, Inc.), 1:1,000
anti-human caspase 3 pAb (Santa Cruz Biotechnology, Inc.), 1:1,000
anti-full length and spliced XBP1 mAb (Stressgen, New York, NY,
USA), 1:1,000 anti-full length and cleaved ATF6 mAb (Santa Cruz
Biotechnology, Inc.) and 1:1,000 anti-eIF2-α (Santa Cruz
Biotechnology, Inc.) for 2 h at room temperature, and then
incubated with 1:4,000 horseradish peroxidase-conjugated anti-mouse
and 1:1,000 anti-rabbit or anti-goat immunoglobulin G (Santa Cruz
Biotechnology, Inc.). The reactive signals were visualized by an
enhanced chemiluminescence kit (PE Applied Biosystems, Waltham, MA,
USA).
Assessment of proliferation and
apoptosis
A
2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide
(XTT) assay was employed to measure the cell proliferation using a
cytotoxicity detection kit (Cayman Chemical, Ann Arbor, MI, USA).
The detailed processes were performed according to the
manufacturer’s instructions. The multi-well plates were read at 490
nm on an ELISA plate reader (Thermo Scientific, Waltham, MA, USA).
Cell apoptosis was detected by flow cytometric analysis, which
monitored annexin V fluorescein isothiocyanate binding and
propidium iodide uptake simultaneously, according to the
manufacturer’s instructions (Sigma-Aldrich, St. Louis, MO, USA).
The samples were analyzed by fluorescence on a FACScan flow
cytometer (Beckman Coulter, Miami, FL, USA). Every analysis was
performed in at least six wells and in duplicate.
MEL peptide treatments
The human MEL peptide (sequence,
GIGAVLKVLTTGLPALISWIKRKRQQ-CONH2; Invitrogen Life
Technologies) was synthesized by the solid-phase method using
9-fluorenyl-methoxycarbonyl-chemistry, according to a study by Park
and Lee (13). The crude peptide
was repeatedly washed with diethylether, dried under vacuum and
purified using reverse-phase preparative high-performance liquid
chromatography on a Waters 15-μm Deltapak C18 column (Waters,
Milford, MA, USA). The MG63 and hFOB 1.19 cells were incubated with
the MEL peptide (100 nm) for 24 h (employed as the MEL-pep
group).
Statistical analysis
A quantitative analysis of the immunoblot images was
performed using computer-assisted software (Image Total Tech;
Pharmacia, New York, NY, USA). Briefly, the image of the immunoblot
was scanned with Typhoon (Pharmacia), digitalized and saved in TIF
format. The values of each target blot were evaluated. All the data
are presented as the mean ± standard deviation. A statistical
analysis was performed using the t-test. P<0.05 was considered
to indicate a statistically significant difference.
Results
MEL inhibits cell viability and triggers
apoptosis in MG63 and hFOB 1.19 cells
In order to explore the potential effects of MEL on
the viability of human MG63 and hFOB 1.19 cells, the cells were
treated with the MEL gene or MEL peptide for 24 h. In the
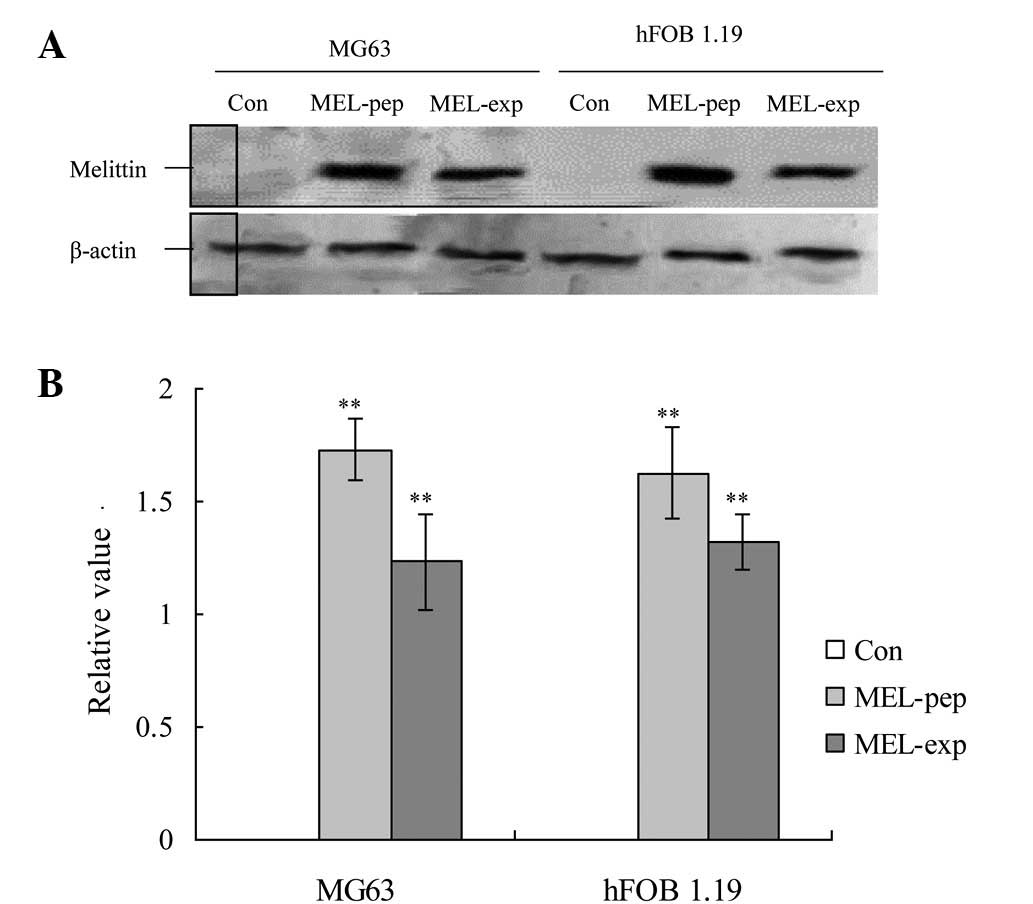
preparations of MG63 cells, high levels of MEL were expressed 24 h
following culturing in the MEL-exp and MEL-pep groups (Fig. 1). MEL expression or treatment also
activated high levels of MEL in the hFOB 1.19 cells in the two
groups (Fig. 1).
MEL expression inhibits the proliferation
of MG63 and triggers apoptosis
To observe the effect of MEL expression or
incubation on the proliferation viability in the MG63 and hFOB 1.19
cells, the proliferation viabilities were measured by XTT analysis
at 24 h post-transfection (or incubation). The XTT analysis showed
no difference in the proliferation viabilities among the control
(Con), MEL-pep and MEL-exp groups in the hFOB 1.19 cells. However,
in the MG63 cells, the proliferation viability of the MEL-pep and
MEL-exp groups was significantly lower than in the Con group
(Fig. 2A).
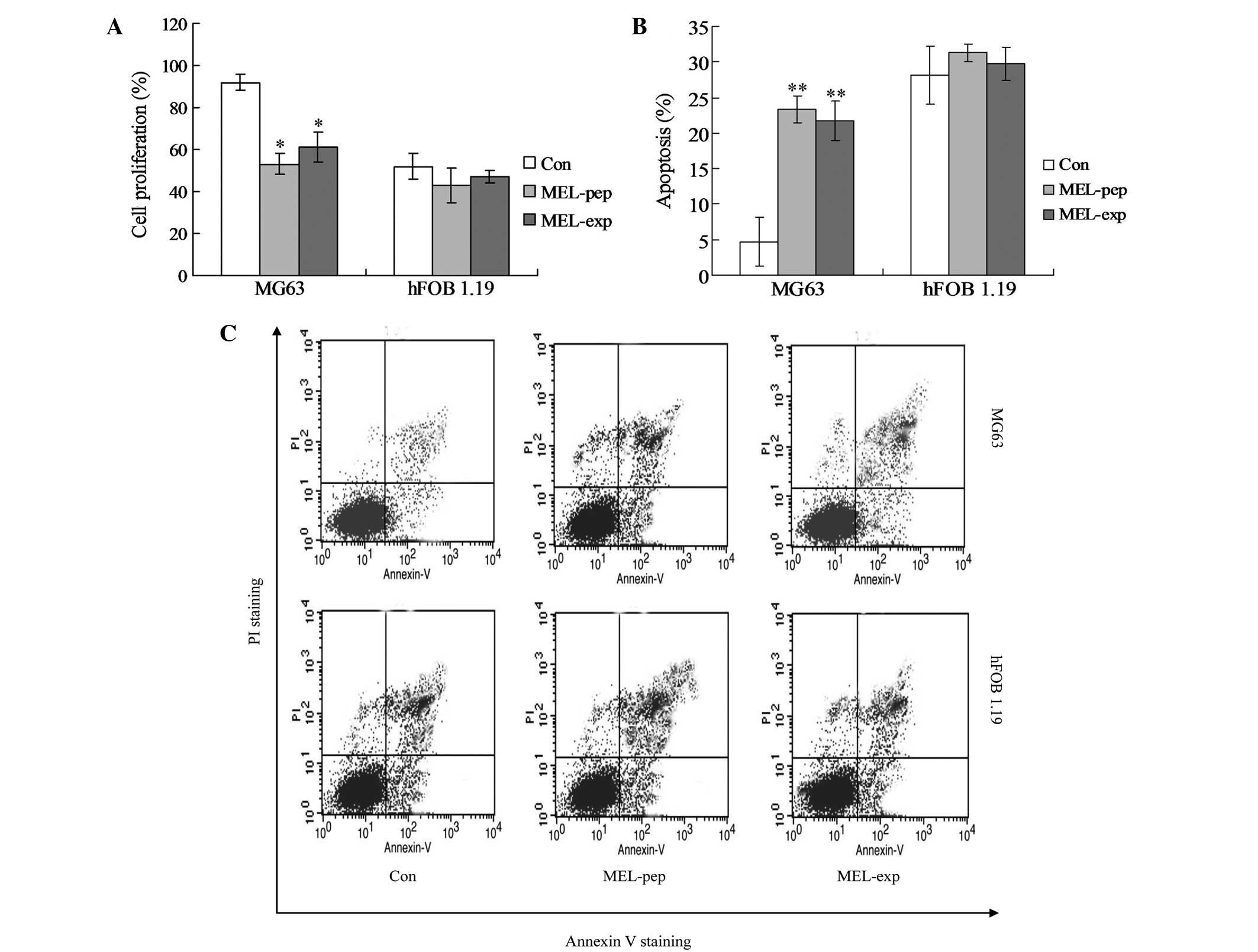 | Figure 2Cell proliferation or apoptosis
effects of MEL on the MEL-pep, MEL-exp and Con groups. (A) Cell
proliferation of the MEL-pep, MEL-exp and Con groups. The cell
proliferation was measured by the XTT method. (B) Annexin V/PI
double staining assays of the MEL-pep, MEL-exp and Con groups. (C)
Statistical analysis of apoptotic cells. The y-axis indicates the
numbers of PI-stained cells, and the x-axis indicates the numbers
of Annexin V-fluorescein isothiocyanate-stained cells. Results for
three independent experiments were taken. The average data of each
preparation were evaluated from three independent blots and
presented as the mean ± standard deviation. Statistical differences
in the data for MEL-exp or MEL-pep compared with that of the Con
group are illustrated as *P<0.05 and
**P<0.01, respectively. MEL, melittin; XTT,
2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide;
MEL-exp, MEL-expressing group; MEL-pep, MEL peptide treated group;
Con, control; PI, propidium iodide. |
In order to investigate the mechanism of cell death
(or inhibition of cell proliferation) caused by MEL, the apoptosis
(the early and late apoptosis) of every group was detected by flow
cytometric analysis. The hFOB 1.19 cells showed no significant
differences in apoptosis among the Con, MEL-pep and MEL-exp groups
(Fig. 2B and C; P>0.05). In
MG63 cells, the apoptosis rates of the MEL-pep and MEL-exp groups
were significantly decreased compared with the Con group, but no
difference was observed between the MEL-pep and MEL-exp groups
(Fig. 2B and C; P<0.01). This
indicates that MEL expression only triggers apoptosis in the MG63
cells.
IRE-α UPR pathway is involved in MG63
cell proliferation inhibition
In order to identify the mechanism of the inhibition
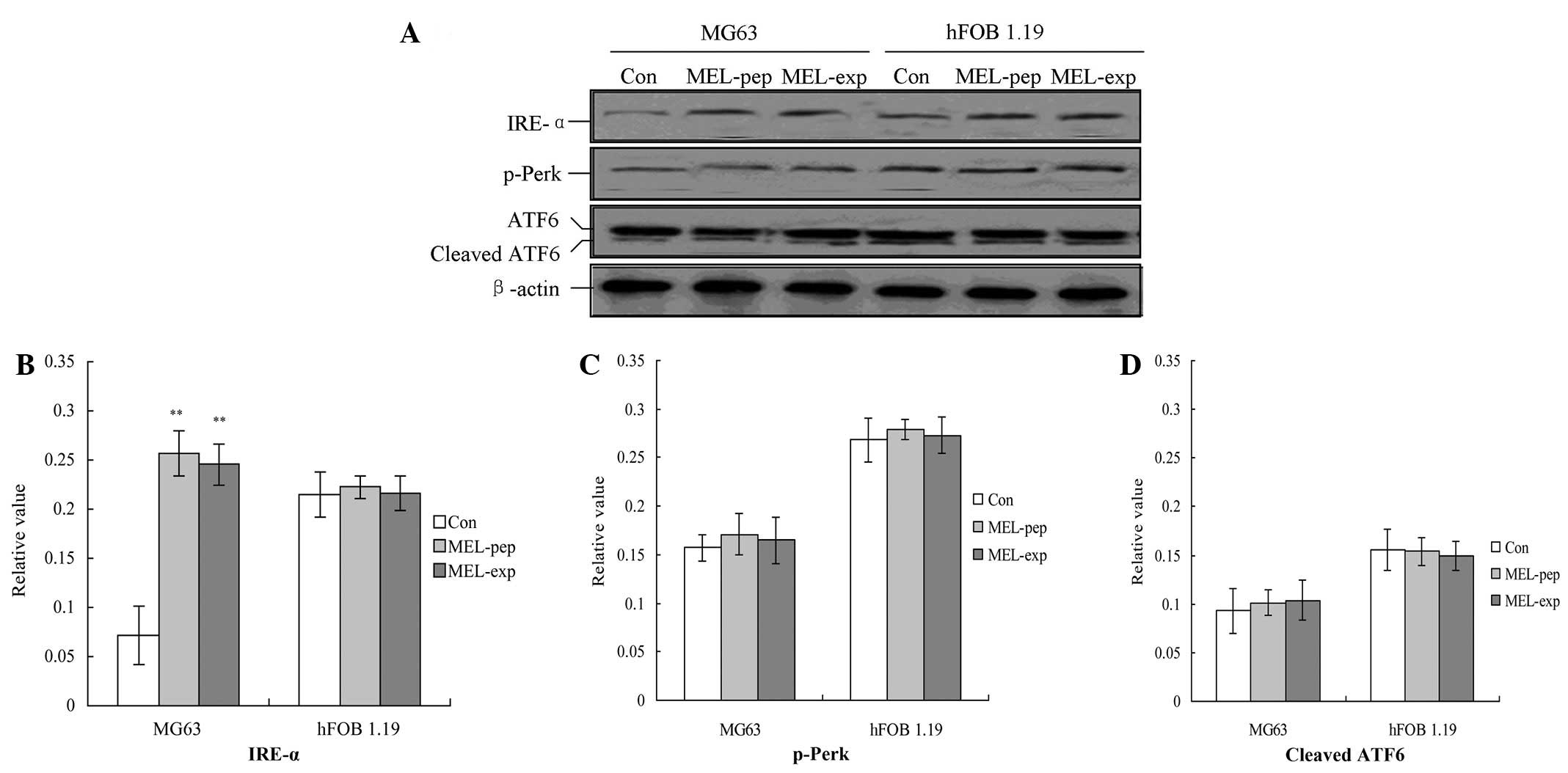
of the proliferation of MEL on MG63 cells, three UPR factors,
p-Perk, IRE-1 and ATF6, were detected using western blot analysis.
The results indicated that, for the MG63 and hFOB 1.19 cells, the
expression or incubation of MEL protein triggered the activation of
IRE-α (Fig. 3A), but not for the
Con group. Furthermore the amounts of IRE-α for the MEL-pep and
MEL-exp groups were significantly enhanced compared with the Con
group (Fig. 4A; P<0.05).
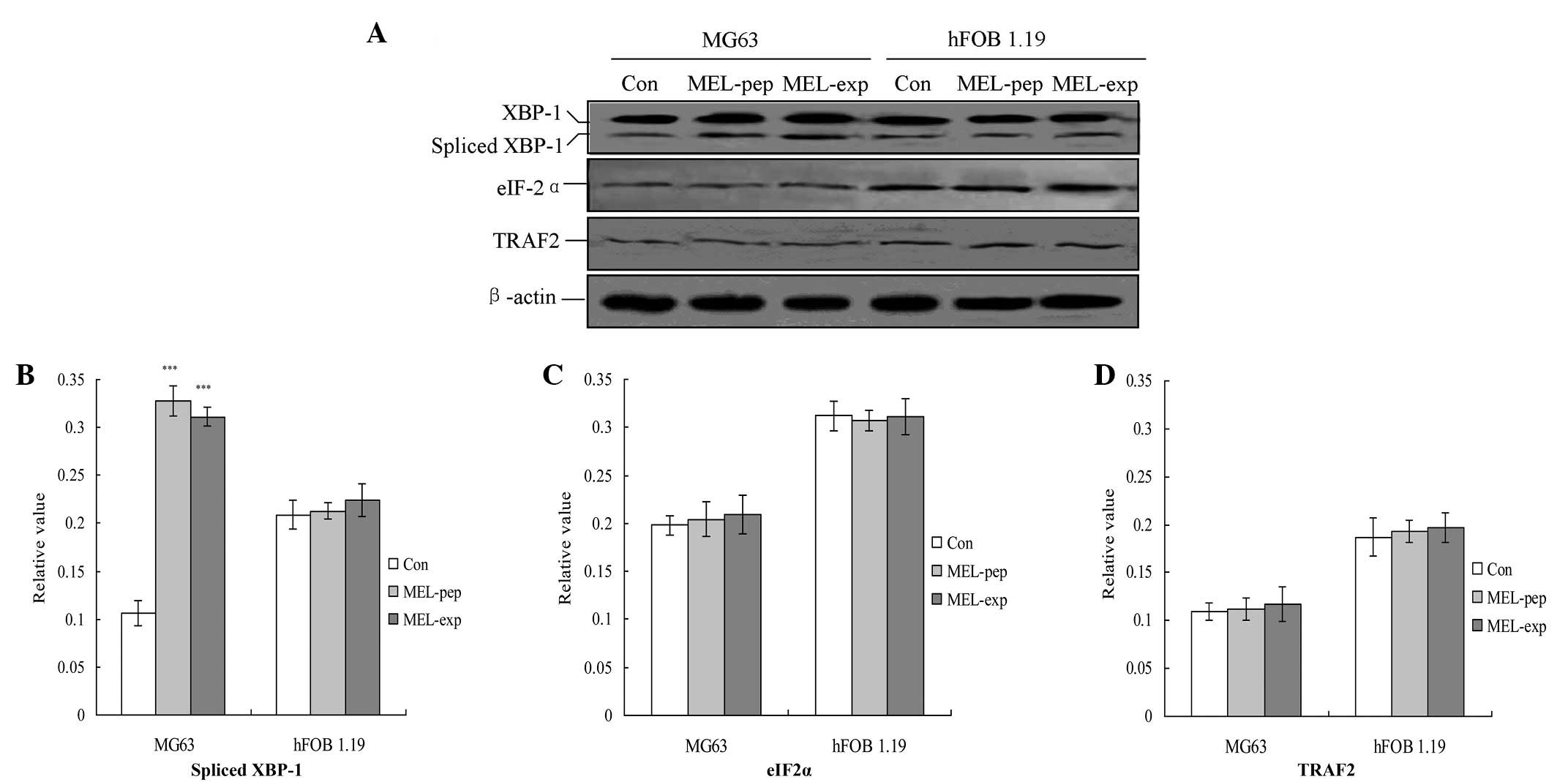 | Figure 4Observation of endoplasmic reticulum
stress (unfolding protein response) downstream proteins. (A)
Observation of the spliced XBP1, eIF-2α and TRAF2 proteins.
Statistical analyzes of the (B) spliced XBP1, (C) eIF-2α and (D)
TRAF2 proteins are also shown. The relative values of the spliced
XBP1, TRAF2 and eIF-2α proteins were calculated by the gray
numerical value of each specific product versus that of β-actin.
The mean data of each preparation are evaluated based on three
independent reactions and presented as the mean ± standard
deviation. Statistical differences in the data for expressed or
incubated MEL compared with that of the Con group are illustrated
as ***P<0.001. eIF-2α, eukaryotic translation
initiation factor-2α; TRAF2, tumor necrosis factor
receptor-associated factor 2; XBP1, spliced X-box transcription
factor-1; MEL, melittin; MEL-exp, MEL-expressing group; MEL-pep,
MEL peptide treated group; Con, control. |
UPR downstream proteins are highly
inhibited in MEL-expressing cells
The downstream proteins of the UPR pathway
associated with ER stress, including spliced XBP1, tumor necrosis
factor receptor-associated factor 2 (TRAF2) and eIF-2α, were
analyzed with semi-quantitative PCR 24 h following transfection.
The results indicated that transfection or incubation with MEL
significantly increased the levels of TRAF2 compared with the Con
group in MG63 cells (Fig. 4;
P<0.01). However, MEL was not able to affect the levels of the
three factors in the hFOB 1.19 cells (Fig. 4).
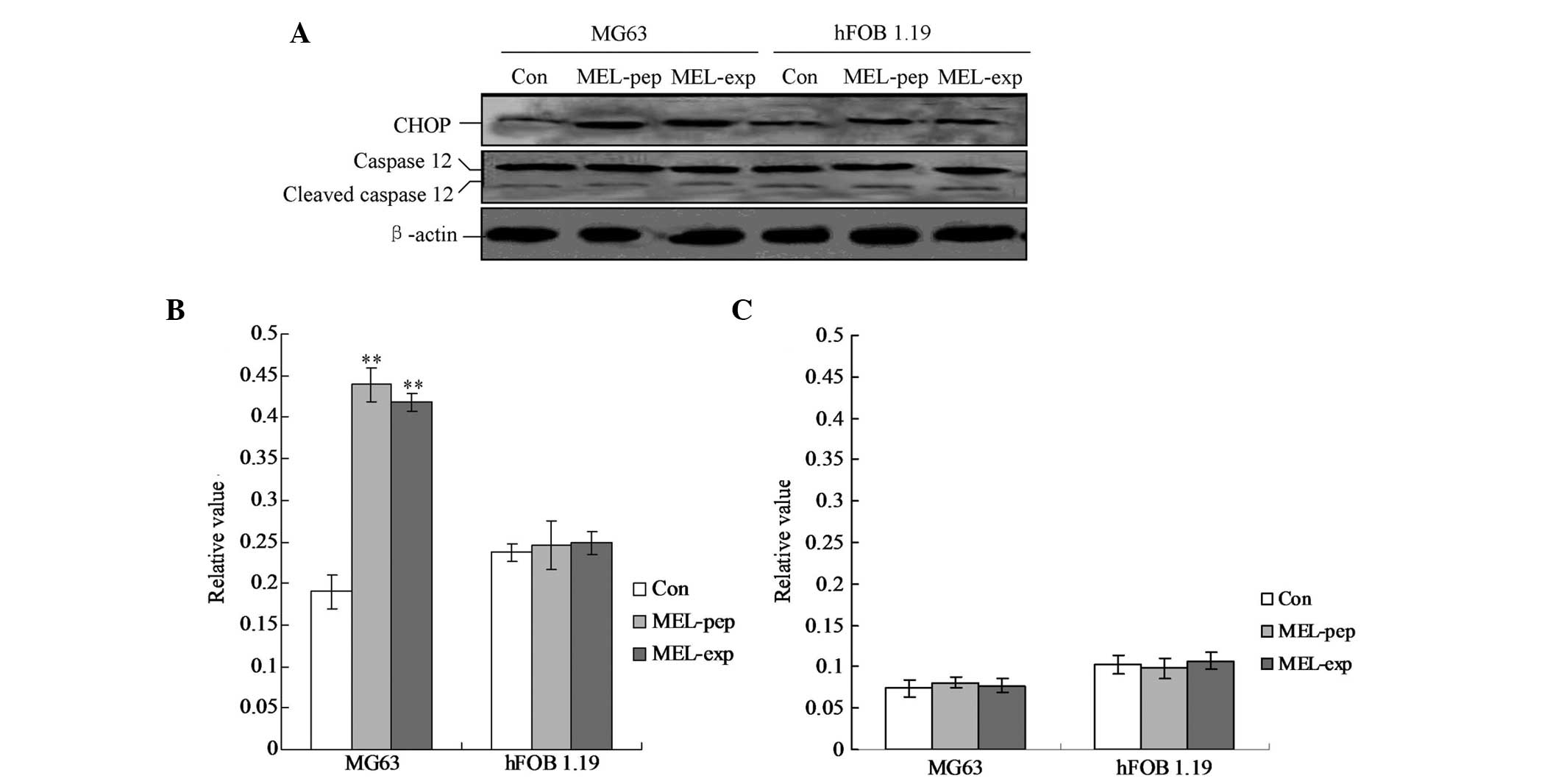
MEL expression activates CHOP-triggered
apoptosis
To clarify the pro-apoptotic factors of the cells
following expression or incubation with MEL, the cellular levels of
cleaved caspase-12 and CHOP protein were evaluated by individual
western blot analysis (Fig. 5).
The hFOB 1.19 cells showed no changes in caspase-12 and CHOP in any
group (Fig. 5). The MG63 cells
exhibited significant differences in cleaved caspase-12 levels in
the MEL-pep and MEL-exp groups compared with the Con group
(Fig. 5; both P<0.01). However,
no differences were identified between the MEL-pep and MEL-exp
groups. The aforementioned results indicated that in the MG63 cells
of the MEL-pep and MEL-exp groups CHOP was activated and triggered
apoptosis.
Discussion
To the best of our knowledge, the present study was
the first to explore the inhibition of MEL-triggered proliferation
or the activation of apoptosis in the human osteosarcoma cell line
MG63. However, there was no such effect of MEL on the cell
proliferation in the human fetal-osteoblast hFOB 1.19 cell line.
The present study indicates that MEL triggers apoptosis through the
IRE-α pathway, activated by inducing CHOP protein expression.
The MEL protein was detected by western blot
analysis in MG63 and hFOB 1.19 cells. The results indicated that
the MEL-pep and MEL-exp groups contain MEL protein. It is known
that the malignant transformations leading to cancer require the
cells to keep proliferating and evade apoptosis of the tumor cells.
MEL expression in MG63 cells may activate apoptosis and induce cell
death to block the proliferation of the tumor cells. The cell
proliferation of the MEL-pep, MEL-exp and Con groups was detected
by the XTT assay. The results indicated that the cell proliferation
of the MG63 cells in the MEL-pep and MEL-exp groups was
significantly decreased compared with the Con group. Notably, there
were no significant differences among all three groups in the hFOB
1.19 cells. The lower proliferation in the MG63 cells is likely to
be due to the induction of apoptosis. Therefore, apoptosis was
observed in all three groups. When MG63 cells expressed or were
incubated with MEL, the percentage of apoptotic cells was
significantly increased in the MEL-pep and MEL-exp groups compared
with the Con group (P<0.01). However, the expression of or
incubation with MEL had no significant effects on the rate of
apoptosis in the hFOB 1.19 cells. These differences indicated that
the expression of MEL triggered apoptosis only in the osteosarcoma
cells, but not in the normal osteoblast cells.
In order to explore the mechanism of apoptosis in
MG63 cells, the levels of ER stress (UPR pathway)-associated
proteins, including p-Perk, IRE-α and cleaved ATF6, were detected
in cells following expression of or incubation with MEL. The
results revealed that, in the MG63 cells, only the IRE-α protein
was activated in the MEL-pep and MEL-exp groups. However, no
significant changes were identified in all three ER
stress-associated proteins in hFOB 1.19 cells. Therefore, the
IRE-α-mediated UPR pathway may be involved in MEL-triggered
apoptosis. MEL-induced apoptosis may aid in the further
illumination of the therapeutic role of MEL in the progression of
osteosarcomas. Furthermore, downstream factors, including spliced
XBP1, TRAF2 and eIF-2α, were also detected. The results indicated
that spliced XPB1 was significantly activated in the MEL-pep and
MEL-exp groups in MG63 cells compared with the Con group
(P<0.01). These data strongly indicated the emergence of an ER
stress (or UPR pathway) following the expression of or incubation
with MEL in MG63 cells. Therefore, it is thought that, in the human
osteosarcoma cell line, MEL may be involved in the pathogenic
process of osteosarcomas.
In the present study, the pro-apoptotic factors,
cleaved caspase-3 and -12, and CHOP protein, were also detected in
the MG63 and hFOB 1.19 cells. Studies have reported that cleaved
caspase-12 may trigger caspase-mediated apoptosis, and that CHOP is
able to directly induce ER stress-associated apoptosis (14). In all three groups of the MG63 and
hFOB 1.19 cells, cleaved (activated) caspase-12 levels were not
significantly increased. Notably, when the MG63 cells were treated
with MEL, the CHOP levels in the MEL-pep and MEL-exp groups were
significantly increased compared with the Con group (P<0.05);
however there were no changes in the hFOB 1.19 cells. Therefore, it
may be concluded that the expression of or incubation with MEL in
osteosarcoma cells may indirectly activate CHOP protein-mediated
apoptosis. The induction of the transcription factor CHOP/GADD153
may kill cells by an apoptotic mechanism (15,16).
The present study therefore produced a novel result stating that
MEL may trigger CHOP-induced ER stress. Notably and significantly,
MEL protein was not capable of triggering ER-stress-associated
apoptosis. Therefore, MEL may be clinically significant in the
antitumor mechanisms in osteosarcomas. However, the specific
mechanism of this distinctive function of MEL for normal or
osteosarcoma cells should be addressed in further experiments.
In conclusion, MEL may be employed as a therapeutic
factor that inhibits the proliferation of MG63 cells through
activation of the ER stress-mediated pathway. This activation is
triggered by the IRE-α pathway through the induction of CHOP
protein expression.
References
|
1
|
Alvarez-Fischer D, Noelker C, Vulinović F,
Grünewald A, Chevarin C, Klein C, Oertel WH, Hirsch EC, Michel PP
and Hartmann A: Bee Venom and its component apamin as
neuroprotective agents in a Parkinson disease mouse model. PLoS
One. 8:e617002013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Park HJ, Lee SH, Son DJ, Oh KW, Kim KH,
Song HS, Kim GJ, Oh GT, Yoon DY and Hong JT: Antiarthritic effect
of bee venom: inhibition of inflammation mediator generation by
suppression of NF-kappaB through interaction with the p50 subunit.
Arthritis Rheum. 50:3504–3515. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hong SJ, Rim GS, Yang HI, Yin CS, Koh HG,
Jang MH, Kim CJ, Choe BK and Chung JH: Bee venom induces apoptosis
through caspase-3 activation in synovial fibroblasts of patients
with rheumatoid arthritis. Toxicon. 46:39–45. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jang MH, Shin MC, Lim S, Han SM, Park HJ,
Shin I, Lee JS, Kim KA, Kim EH and Kim CJ: Bee venom induces
apoptosis and inhibits expression of cyclooxygenase-2 mRNA in human
lung cancer line NCI-H1299. J Pharmacol Sci. 91:95–104. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Moon DO, Park SY, Heo MS, Kim KC, Park C,
Ko WS, Choi YH and Kim GY: Key regulators in bee venom-induced
apoptosis are Bcl-2 and caspase-3 in human leukemic U937 cells
through downregulation of ERK and Akt. Int Immunopharmacol.
6:1796–1807. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lariviere WR and Melzack R: The bee venom
test: a new tonic-pain test. Pain. 66:271–277. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Son DJ, Ha SJ, Song HS, Lim Y, Yun YP,
Moon DC, Park YH, Park BS, Song MJ and Hong JT: Melittin inhibits
vascular smooth muscle cell proliferation through induction of
apoptosis via suppression of NF-kappaB and Akt activation and
enhancement of apoptotic protein expression. J Pharmacol Exp Ther.
317:627–634. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shaposhnikova VV, Egorova MV, Kudryavtsev
AA, Levitman MK and Korystov YuN: The effect of melittin on
proliferation and death of thymocytes. FEBS Lett. 410:285–288.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kim SK, Park KY, Yoon WC, Park SH, Park
KK, Yoo DH and Choe JY: Melittin enhances apoptosis through
suppression of IL-6/sIL-6R complex-induced NF-κB and STAT3
activation and Bcl-2 expression for human fibroblast-like
synoviocytes in rheumatoid arthritis. Joint Bone Spine. 78:471–477.
2011.PubMed/NCBI
|
|
10
|
Rasheva VI and Domingos PM: Cellular
responses to endoplasmic reticulum stress and apoptosis. Apoptosis.
14:996–1007. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ron D and Walter P: Signal integration in
the endoplasmic reticulum unfolded protein response. Nat Rev Mol
Cell Biol. 8:519–529. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang X, Dong CF, Shi Q, Shi S, Wang GR,
Lei YJ, Xu K, An R, Chen JM, Jiang HY, Tian C, Gao C, Zhao YJ, Han
T and Dong XP: Cytosolic prion protein induces apoptosis in human
neuronal cell SH-SY5Y via mitochondrial disruption pathway. BMB
Rep. 42:444–449. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Park C and Lee DG: Melittin induces
apoptotic features in Candida albicans. Biochem Biophys Res
Comm. 394:170–172. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang X, Shi Q, Xu K, Gao C, Chen C, Li XL,
Wang GR, Tian C, Han J and Dong XP: Familial CJD associated PrP
mutants within transmembrane region induced Ctm-PrP retention in ER
and triggered apoptosis by ER stress in SH-SY5Y cells. PLoS One.
6:e146022011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kim KM, Kim HC, Jeon KN, Kim HG, Kang JH,
Hahm JR and Lee GW: Rituximab-CHOP induced interstitial pneumonitis
in patients with disseminated extranodal marginal zone B cell
lymphoma. Yonsei Med J. 49:155–158. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang SC, Lu MC, Chen HL, Tseng HI, Ke YY,
Wu YC and Yang PY: Cytotoxicity of calotropin is through caspase
activation and downregulation of anti-apoptotic proteins in K562
cells. Cell Biol Int. 33:1230–1236. 2009. View Article : Google Scholar : PubMed/NCBI
|