Introduction
Pancreatic cancer is a devastating disease with an
extremely poor prognosis, and patients often do not undergo
curative surgery (1). Additionally,
effective chemotherapy and radiotherapy treatments for pancreatic
cancer are limited (1). The 5-year
survival of patients with pancreatic cancer is 0.4–4.0%, and has
not significantly improved over the past three decades (2). Elucidating the molecular mechanism of
pancreatic cancer may contribute to the early diagnosis and
effective therapies for pancreatic cancer. Smad4, also referred to
as deleted in pancreatic cancer locus 4, is localized to chromosome
18q21, and was originally identified as a signaling mediator of the
transforming growth factor-β (TGF-β) signaling pathway (3). It has been reported that tumor
development is induced by a decrease in Smad4 in pancreatic cancer,
and mutations of the Smad4 gene predict a poorer prognosis of
patients with pancreatic ductal adenocarcinoma and pancreatic
cancer (4–6). Furthermore, verbal evidence supports the
role of Smad4 as a tumor suppressor gene in pancreatic
tumorigenesis (7).
c-Jun N-terminal kinase (JNK) is a member of the
mitogen-activated protein kinase (MAPK) family (8). JNK has two ubiquitously expressed
isoforms, JNK1 and JNK2, and a tissue-specific isoform JNK3. Each
isoform has two different splicing forms, p54 and p46 (9). The activation of JNK is mediated by
sequential protein phosphorylation through MAPK kinase (MKK)4 and
MKK7, which primarily function as two upstream kinases for JNK
activation (10). The inactivation of
JNK primarily depends on the dephosphorylation effect of
phosphatases, including MAPK phosphatase-1 (MKP-1) (11). Numerous studies have revealed that JNK
is pivotal in tumorigenesis by enhancing cell proliferation and
migration, and antagonizing apoptosis in digestive system tumors,
including hepatocellular carcinoma and pancreatic cancer (12–14). A
previous study has demonstrated that JNK is a potential therapeutic
target for pancreatic cancer (15).
In addition, knocking down JNK or introducing a JNK inhibitor
factor results in growth inhibition of human pancreatic carcinoma
cells. In a previous study, a mouse model with JNK inhibitor factor
inhibits tumor growth and prolongs the survival time of the mice
(16).
Various signaling pathways in cells constitute a
complex network that interact with each other, which is referred to
as cross-talk (17–19). Previous studies have revealed that the
Smad signaling pathway downstream of TGF-β has complicated
interactions with MAPK members, including p38, JNK and
extracellular signal-regulated kinases (ERKs) (19,20).
Previous studies conducted over the past decade have revealed that
the Smad2/3 complex is phosphorylated by JNK and p38 through direct
or indirect ways; and this complex subsequently binds to Smad4,
thus regulating downstream gene transcription (20). However, little is known regarding
whether Smad4 regulates JNK and p38, and whether it affects the
occurrence, development and metastasis of tumors. The present study
reports that Smad4 suppresses JNK activity, and also inhibits the
migration of human pancreatic epithelioid carcinoma PANC-1 cells by
upregulating the expression of MKP-1.
Materials and methods
Cell culture and transfection
Human embryonic kidney (HEK)-293T, human cervix
adenocarcinoma epithelial HeLa and human pancreatic epithelioid
carcinoma PANC-1 cells were purchased from the American Type
Culture Collection (Manassas, VA, USA). Human pancreatic
adenocarcinoma AsPC-1, BxPC-3 and SW850 cells were obtained from
Professor Hongyang Wang (National Center for Liver Cancer,
Secondary Military Medical University, Shanghai, China). The cells
were cultured in Dulbecco's modified Eagle medium (DMEM; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) supplemented with 10%
fetal bovine serum (FBS; Lanzhou Bailing Biotechnology Co., Ltd.,
Lanzhou, China), 100 U/ml penicillin (North China Pharmaceutical
Group Co., Ltd., Shijiazhuang, China) and 100 U/ml streptomycin
(North China Pharmaceutical Group Co., Ltd.), and were maintained
at 37°C with 5% CO2. Cell transfection was performed
using Lipofectamine® 2000 (Thermo Fisher Scientific,
Inc.), according to the manufacturer's protocol. Stable clones were
selected using 800 µg/ml puromycin (Thermo Fisher Scientific, Inc.)
for ~2 months.
Mice
Female BALB/c mice (n=3), 6–8 weeks-old, were
purchased from Institute of Experimental Animals, Military Medical
Sciences Institution (Beijing, China). All mice were maintained at
room temperature under specific pathogen-free conditions and
exposed to 12 h light/dark cycles. The care, use and treatment of
mice in the present study was in strict agreement with guidelines
in the care and use of Laboratory Animal Manual set out by the
Institute of Basic Medical Sciences (Beijing, China). The protocol
was approved by the Institute of Basic Medical Sciences. All
surgery was performed under sodium pentobarbital anesthesia, and
all efforts were made to minimize suffering. Following anesthesia,
the mouse thymus was removed, lyzed and subjected to western blot
analysis.
Plasmid and small interfering (si)
RNA
The HA-Smad4 expression vector was generated by
cloning a polymerase chain reaction (PCR) product into the
pCDNA3.1+ vector (Thermo Fisher Scientific, Inc.), which
was confirmed by DNA sequencing. The PCR were performed using a
MasterCycler® Personal (Eppendorf, Hamburg, Germany).
The synthesis of the primers for PCR and the DNA sequencing were
performed by Beijing Sino Geno Max Co., Ltd. (Beijing, China). PCR
was performed using Taq 2X PCR Mastermix [Tiangen Biotech (Beijing)
Co., Ltd., Beijing, China]. The primers for the PCR were as
follows: HA-Smad-4, forward
5′-CCCAAGCTTGCCACCATGTACGATGTTCCAGATTACGCTATGGACAATATGTCTATTACG-3′
and reverse 5′-GCTCTAGATACGTCTAAAGGTTGTGGG-3′. Smad4 siRNA
(Smad4-1, CGAAUACACCAACAAGUAATT; Smad4-2, AGAUGAAUUGGAUUCUUUATT),
MKP-1 siRNA-1 (GCAUAACUGCCUUGAUCAA), MKP-1 siRNA-2
(CCAAUUGUCCCAACCAUUU) and non-targeting control siRNA (scramble,
UUCUCCGAACGUGUCACGUTT) were purchased from Shanghai GenePharma Co.,
Ltd. (Shanghai, China). siRNAs that target human JNK1 and JNK2
messenger RNA were designed based on the 1,013-1,031 nt (JNK1) and
461–479 nt (JNK2) sequences, relative to the translation start
sites, and were purchased from GE Dharmacon (Lafayette, CO, USA)
(21). The JNK siRNA sequences were
as follows: 5′-CUGACAAGCAGUUAGAUGA-3′ for JNK1;
5′-CUAGCAACAUUGUUGUGAA-3′ for JNK2. PCR was performed under the
following conditions: Denaturation at 98°C for 30 sec, followed by
30 cycles of 58°C for 1 min, and a final extension step at 72°C for
2 min. The PCR products were analyzed by 1.2% agarose gel
(Sigma-Aldrich) electrophoresis and observed under ultra-violet
light (UV/White TMW-20 Transilluminator; UVP LLC, Upland, CA,
USA).
Western blot analysis
Western blot analysis was performed as previously
described (22). Briefly, the cells
were washed twice with ice-cold phosphate-buffered saline and were
lysed using 20 mM Tris/HCl (pH7.6; Amresco, Inc., Solon, OH, USA),
250 mM NaCl, 3 mM EDTA (Sigma-Aldrich, St. Louis, MO, USA), 3 mM
ethylene glycol tetraacetic acid (Sigma-Aldrich), 0.5% NP-40 lysis
buffer (Amresco, Inc.), 1 mM dithiothreitol (Sigma-Aldrich), 1 mM
p-nitrophenyl phosphate (Sigma-Aldrich), 2 mM
Na3VO4 (Sigma-Aldrich) and 10 µg/ml aprotinin
(Sigma-Aldrich). The whole cell extract was centifuged (Fresco 21;
Thermo Fisher Scientific, Inc.) at 13,600 × g for 15 min at 4°C,
and the supernatants were subjected to 12% sodium
dodecy1sulfate-polyacrylamide gel electrophoresis (2 h).
Subsequently, the proteins were transferred to Hybond-P
polyvinylidene difluoride membranes (GE Healthcare Life Sciences,
Little Chalfont, UK). The membranes were initially incubated with
primary antibody at 4°C overnight, and then with horseradish
peroxidase (HRP)-conjugated secondary antibodies for 1 h at room
temprature. Bound antibody was detected using Amersham ECL Western
Blotting Detection kit (catalog no. RPN2106; GE Healthcare Life
Sciences) and X-ray film (Kodak, Rochester, NY, USA). The following
primary antibodies were used in a dilution of 1:1,000: Rabbit
polyclonal anti-human phosphorylated (p-)JNK (catalog no. 9251;
Cell Signaling Technology, Inc., Danvers, MA, USA), rabbit
monoclonal anti-human p-p38 MAPK (catalog no. 9215; Cell Signaling
Technology, Inc.), mouse monoclonal anti-human JNK1 (catalog no.
51–1570GR; BD Pharmingen™; BD Biosciences, Franklin Lakes, NJ,
USA), rabbit monoclonal anti-human JNK2 (catalog no., 2037–1;
Epitomics, Burlingame, CA, USA), rabbit polyclonal anti-human p38
(catalog no. sc-535; Santa Cruz Biotechnology, Inc., Dallas, TX,
USA), rabbit polyclonal anti-hemagglutinin (HA)-probe (catalog no.
sc-805; Santa Cruz Biotechnology, Inc.), rabbit polyclonal
anti-human Smad4 (catalog no. 9515; Cell Signaling Technology,
Inc.,), rabbit polyclonal anti-human MKP-1 (catalog no. sc-1199;
Santa Cruz Biotechnology, Inc.), monoclonal mouse anti-human
vascular endothelial growth factor (VEGF; catalog no. MAB293;
R&D Systems, Inc., Minneapolis, MN, USA) and mouse monoclonal
anti-human β-actin (catalog no. sc-47778; Santa Cruz Biotechnology,
Inc.). A polyclonal goat anti-rabbit or goat anti-mouse
HRP-conjugated secondary antibody (catalog no. ZB-2301 and ZB-2305,
respectively; both dilution 1:5000; Zhongshan Golden Bridge
Biotechnology Co., Ltd., Beijing, China) were used at a dilution of
1:5,000. Densitometric analysis was performed using Gel-Pro
Analyzer 4.0 (Media Cybernetics, Inc., Rockville, MD, USA).
Cell proliferation and migration
assay
Cells were seeded into a 24-well plate at a density
of 2×104cells/well for 12 h. Cells were then incubated
with 20 µM SP600125 (JNK inhibitor; Sigma-Aldrich) or 20 µM
dimethyl sulfoxide. The viable cells were counted daily with a
trypan blue stain (Yeasen Corporation, Shanghai, China). A
migration assay was performed using a Transwell chamber (diameter,
6.5 mM; pore size, 8 µM; polycarbonate membrane; Corning
Incorporated, Corning, NY, USA). In total, 3×104 PANC-1
cells in 0.1 ml serum-free DMEM were placed in the upper chamber,
while the lower chamber was loaded with 0.5 ml 10% FBS-DMEM as a
chemoattractant. Following incubation at 37°C and 5% CO2
for 6 h, the non-invading cells in the inserts were removed with
cotton swabs. The migrated cells on the lower surface were stained
with sulforhodamine B (Sigma-Aldrich) and counted under a
microscope (ECLIPSE TS100; Nikon Corporation, Tokyo, Japan) in five
randomly selected fields at a magnification of ×400.
Statistical analysis
Data are expressed as the mean ± standard deviation.
The Student's t-test was used to compare the differences between
groups. All statistical analysis was performed using SPSS 13.0
software (SPSS, Inc., Chicago, IL, USA). P<0.05 was considered
to indicate a statistically significant difference.
Results
Smad4 suppressed JNK activity in
PANC-1 cells and HEK-293T cells
To investigate whether Smad4 affects JNK activity in
human pancreatic cancer cells, the present study first analyzed the
effects of Smad4 using a gain-of-function analysis in PANC-1 cells
(Fig. 1A). In addition, single clone
PANC-1 cells that express stable levels of exogenous Smad4 were
established. Clones 7, 13, 14, 15 16, 20 and 22 were screened as
exogenous Smad4 stably expressing clones, while clone 1 was
referred to as a control clone transfected with a pCDNA3.1(+) empty
vector. Western blot analysis revealed that these single clone
cells exhibited a reduced JNK phosphorylation compared to the
control clone, which was associated with Smad4 expression,
particularly in clone no. 20 (Fig.
1B). Additionally, the present study investigated whether JNK
phosphorylation was suppressed by Smad4 in non-pancreatic cells. A
transient knockdown of Smad4 using siRNA partially elevated basal
and tumor necrosis factor (TNF)-α (10 ng/mL; R&D Systems, Inc.)
or 302 nM UV light (CL-1000M Midrange Ultraviolet Crosslinker; UVP,
Inc., Upland, CA, USA)-induced JNK and p38 phosphorylation compared
to control siRNA in HEK-293T cells (Fig.
1C). Similarly, overexpression of Smad4 in HEK-293T cells
attenuated the phosphorylation of JNK and p38 (Fig. 1D). These results suggest that Smad4
suppressed JNK activity in various cell types.
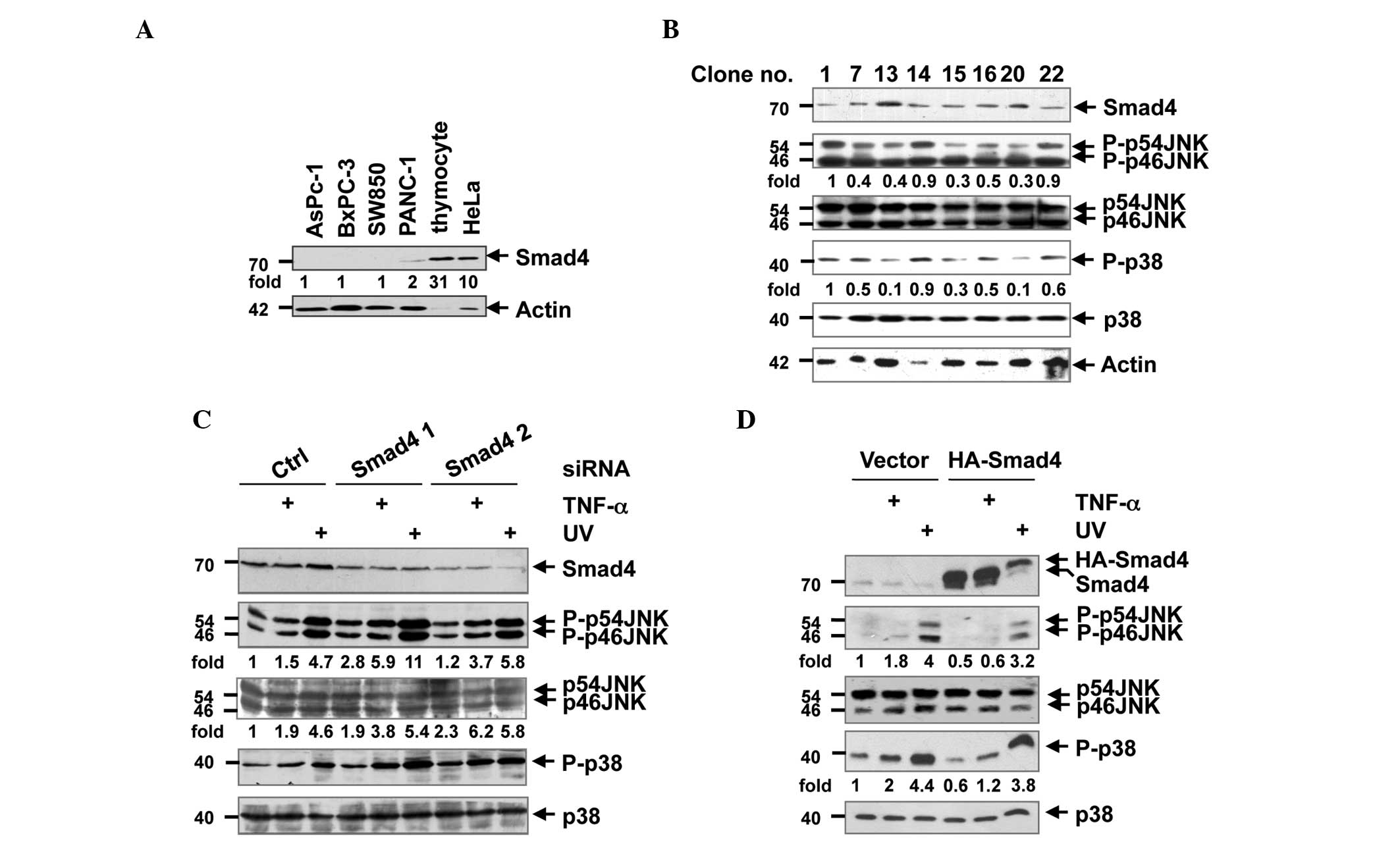 | Figure 1.Smad4 suppressed JNK activity in human
pancreatic epithelioid carcinoma PANC-1 and HEK-293T cells. (A)
Human pancreatic adenocarcinoma AsPC-1, BxPC-3, SW850 and PANC-1
cells, human epithelial carcinoma HeLa cells and mouse thymocyte
tissue were lysed with a lysis buffer, and the lysates were
subjected to western blot analysis for the detection of Smad4 and
actin. DR are shown for Smad4 and normalized with actin. (B) PANC-1
single clones that stably expressed Smad4 were lysed and subjected
to western blot analysis with antibodies against Smad4, p-JNK, JNK,
p-p38, p38 and actin. (C) HEK-293T cells were transfected with
control or Smad4 siRNA. Following incubation for 72 h, the cells
were stimulated with TNF-α or UV light for 15 min, and lysed on
ice. The lysates were subjected to western blot analysis with
antibodies against Smad4, p-JNK, JNK, p-p38 and p38. (D) HEK-293T
cells were transfected with control vector or and
hemagglutinin-Smad4 plasmid. Following TNF-α or UV light
stimulation 24 h later, the cells were lysed, and the cell lysates
were subjected to western blot analysis with antibodies against
Smad4, p-JNK, JNK, p-p38 and p38. (B-D) DR are shown for p-JNK and
normalized with JNK; DR are shown for p-p38 and normalized with
p38. HEK, human embryonic kidney; p-, phosphorylated; JNK, c-Jun
N-terminal kinase; siRNA; small interfering RNA; TNF, tumor
necrosis factor; UV, ultraviolet; HA, hemagglutinin probe; Ctrl,
control; DR, densitometric readings. |
Smad4 suppressed JNK activity by
upregulating MKP-1
MKP-1 is a nuclear-localized phosphatase that
dephosphorylates MAPK ERK, p38 and JNK (23). To investigate the potential role of
MKP-1 in the association between Smad4 and JNK, HA-Smad4 and Smad4
siRNA were transfected into PANC-1 cells. Western blot analysis
demonstrated that MKP-1 protein was upregulated with Smad4
overexpression, but downregulated in Smad4 knockdown cells
(Fig. 2A and B). To additionally
examine the role of MKP-1 in this biological process, PANC-1 cells
were transfected with the HA-Smad4 vector, followed by silencing of
MKP-1 using siRNA, and stimulation with TNF-α. As expected, the
suppression of JNK phosphorylation by Smad4 was partially reversed
under the conditions of MKP-1 knockdown (Fig. 2C). Therefore, MKP-1 upregulation was
pivotal in Smad4-regulated suppression of JNK phosphorylation.
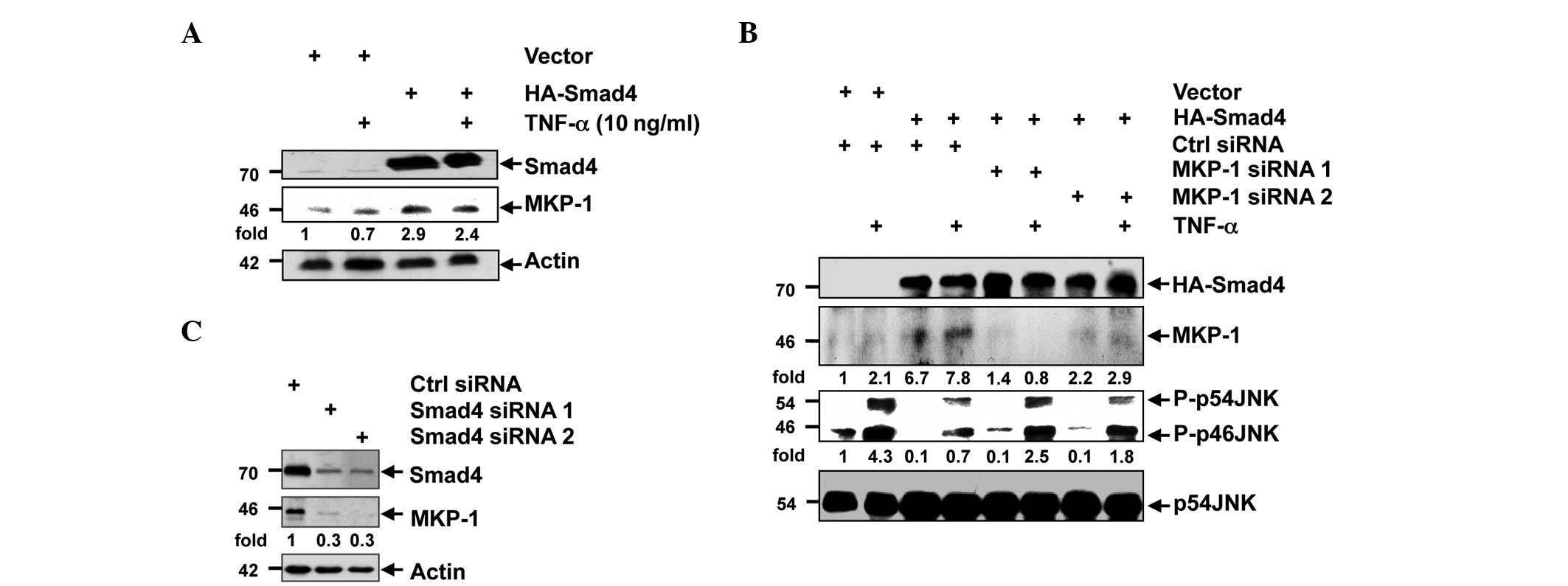 | Figure 2.Smad4 upregulated the expression of
MKP-1. (A) HEK-293T cells were transfected with control vector or
HA-Smad4 plasmid. Following 24 h, the cells were stimulated with
TNF-α, and the protein levels of Smad4, MKP-1 and actin were
analyzed using western blot analysis. (B) HEK-293T cells were
transfected with control or Smad4 siRNA, and 72 h later, the
expression levels of Smad4, MKP-1 and actin were analyzed using
western blot analysis. DR are shown for MKP-1 and normalized with
actin. (C) HEK-293T cells were co-transfected with control vector
or HA-Smad4 plasmid and control or MKP-1 siRNA. Following 15 min of
TNF-α stimulation, the cells were lysed, and the expression levels
of Smad4, MKP-1, JNK and phosphorylated JNK were analyzed using
western blot analysis. DR are shown for p-JNK and normalized with
JNK; densitometric readings are shown for MKP-1 and normalized with
JNK. MKP-1, mitogen-activated protein kinase phosphatase-1; HEK,
human epithelial kidney; TNF, tumor necrosis factor; p-,
phosphorylated; JNK, c-Jun N-terminal kinase; siRNA; small
interfering RNA; HA, hemagglutinin; Ctrl, control; DR,
densitometric readings. |
Smad4 suppressed migration, but not
proliferation, via JNK activity in PANC-1 cells
JNK activity has been reported to contribute to
oncogenicity by promoting tumor cell proliferation and migration
(24). Therefore, it is important to
investigate whether Smad4 affects the oncogenicity of PANC-1 cells
via JNK. Notably, a PANC-1 single clone that stably expressed
HA-Smad4 exhibited no differences, compared with the control vector
(Fig. 3A). The JNK inhibitor also
exhibited no effects on PANC-1 cell proliferation (Fig. 3B). However, in the migration analysis,
the PANC-1 cells that stably overexpressed Smad4 exhibited a
weakened ability for migration. Knockdown of JNK and the use of the
JNK inhibitor demonstrated a similar ability to attenuate the
migration of the PANC-1 cells (Fig.
3C; P<0.01). VEGF is a key factor in tumor cell migration
(25). Therefore, JNK siRNAs were
used in the present study to examine the association between JNK
activity and migration in PANC-1 cells. Silencing of JNK1 and JNK2
using siRNA inhibited VEGF expression in PANC-1 cells (Fig. 3D). These results collectively suggest
that Smad4 regulates cell migration by suppressing JNK activity and
JNK-dependent VEGF expression.
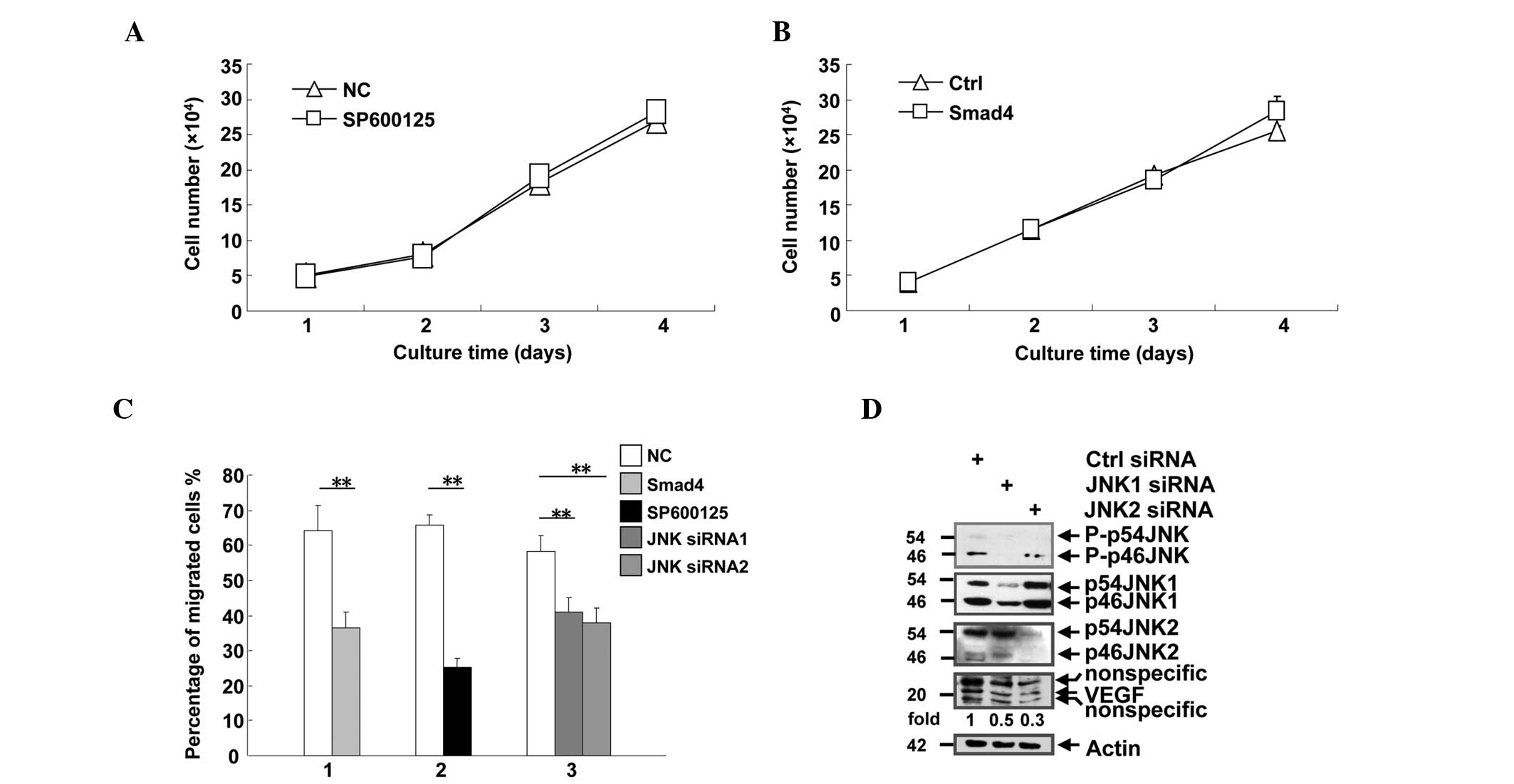 | Figure 3.Smad4 suppressed cell migration, but
not proliferation. (A) Human pancreatic epithelioid carcinoma
PANC-1 cells were seeded into 24-well plates at a density of
2×104cells/well, and treated with DMSO or SP600125, a
JNK inhibitor. The cells were counted daily between days 1 and 4.
(B) PANC-1 single clones that stably expressed the control vector
or Smad4 were seeded into 24-well plates at a density of
2×104cells/well, and counted daily between days 1 and 4.
(C) PANC-1 cells subjected to various treatments were analyzed by
Transwell assay. The migrated cells on the lower surface of the
membrane were stained with sulforhodamine B, and counted under a
microscope in five randomly selected fields at a magnification of
×400. Group 1, cells transfected with control vector or
hemagglutinin-Smad4 plasmid; group 2, cells treated with DMSO or
the JNK inhibitor SP600125; group 3, cells transfected with control
or JNK siRNA. (D) PANC-1 cells were transfected with control, JNK1
or JNK2 siRNA. The cells were lysed following 72 h, and the lysates
were subjected to western blot analysis with antibodies against
phosphorylated JNK, JNK1, JNK2, vascular endothelial growth factor
and actin. DR are shown for VEGF and normalized with actin.
**P<0.01 vs. NC. JNK, c-Jun N-terminal kinase; siRNA; small
interfering RNA; DMSO, dimethyl sulfoxide; p-, phosphorylated;
VEGF, vascular endothelial growth factor; NC, negative control;
Ctrl, control; DR, densitometric readings. |
Discussion
Pancreatic cancer is a devastating disease with an
extremely poor prognosis and a 0.4–4% survival rate (2). Elucidating the molecular mechanisms of
pancreatic cancer may contribute to the early diagnosis and
effective therapies for patients with pancreatic cancer. It has
been demonstrated that Smad4 deletions are associated with
pancreatic cancer metastasis, and Smad4 gene mutations are
associated with poor prognosis in pancreatic cancer (1,2). Previous
studies have established that Smad4 may interact with other
signaling pathways, including Notch and MAPK, and function
independently of TGF-β (20). Despite
clear evidence of the association between Smad4 and pancreatic
cancer (1,4–6), the
mechanism of the cross-talk between Smad4 and other signaling
pathways that affect the development of pancreatic cancer remains
unclear.
JNK activity is reported to be pivotal in tumor
development, particularly in gastrointestinal tumors, including
hepatocellular carcinoma (12), colon
cancer (26) and pancreatic cancer
(27). In human colorectal cancer,
JNK phosphorylates Smad2/3, which results in normal colorectal
epithelial cells transforming to invasive adenocarcinoma (28). However, whether and how Smad4
regulates JNK activity in pancreatic cancer is unknown. The present
study demonstrated that Smad4 suppresses JNK activity by elevating
MKP1 expression, resulting in reduced VEGF expression and inhibited
cell migration. For additional confirmation of these results, the
JNK inhibitor SP600125 and JNK siRNA were used in the present
study. The present results revealed that the inhibition of JNK by
siRNA or SP600125 impaired cell migration and caused overexpression
of Smad4 in PANC-1 cells. Overall, the present results suggest that
Smad4 acts as a tumor suppressor not only through the TGF-β
pathway, but also through the JNK/MAPK pathway.
It is of interest that Smad4 inhibits JNK activity
not only in malignant cells, but also in normal cells (29,30).
Western blot analysis performed in the present study indicated that
Smad4 also suppressed JNK activity in HEK-293T cells, suggesting
that the Smad4-mediated suppression of JNK may be a universal
event. Furthermore, in isolated murine T cells, JNK phosphorylation
was inversely associated with Smad4 protein expression (data not
shown). Previous studies have indicated that constitutive JNK
activity may promote the malignancy of B and T cells (21,31), while
mice with a Smad4 deletion were observed to spontaneously develop
gastrointestinal cancer (32,33). The possibility that a Smad4 deletion
may initiate pancreatic cancer in humans is unclear. The data
presented in the current study associated a decrease in Smad4
expression with increased migration of PANC-1 cells, which was
accompanied by enhanced JNK activity due to the lack of Smad4.
Therefore, the present results suggest that a Smad4 deletion may
cause the development of pancreatic cancer, which may be
accelerated due to increased JNK activity.
Acknowledgements
This study was supported by the Basic Research for
Application Foundation of General Logistic Department (grant no.
13QNP149).
References
|
1
|
Lepage C, Capocaccia R, Hackl M, Lemmens
V, Molina E, Pierannunzio D, Sant M, Trama A and Faivre J: Survival
in patients with primary liver cancer, gallbladder and extrahepatic
biliary tract cancer and pancreatic cancer in Europe 1999–2007:
Results of EUROCARE-5. Eur J Cancer pii. S0959-8049(15)00714-5.
2015. View Article : Google Scholar
|
|
2
|
Hruban RH and Adsay NV: Molecular
classification of neoplasms of the pancreas. Hum Pathol.
40:612–623. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hahn SA, Schutte M, Hoque AT, Moskaluk CA,
da Costa LT, Rozenblum E, Weinstein CL, Fischer A, Yeo CJ, Hruban
RH and Kern SE: DPC4, a candidate tumor suppressor gene at human
chromosome 18q21.1. Science. 271:350–353. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Oshima M, Okano K, Muraki S, Haba R, Maeba
T, Suzuki Y and Yachida S: Immunohistochemically detected
expression of 3 major genes (CDKN2A/p16, TP53, and SMAD4/DPC4)
strongly predicts survival in patients with resectable pancreatic
cancer. Ann Surg. 258:336–346. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Singh P, Srinivasan R and Wig JD: SMAD4
genetic alterations predict a worse prognosis in patients with
pancreatic ductal adenocarcinoma. Pancreas. 41:541–546. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Blackford A, Serrano OK, Wolfgang CL,
Parmigiani G, Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ,
Eshleman JR, et al: SMAD4 gene mutations are associated with poor
prognosis in pancreatic cancer. Clin Cancer Res. 15:4674–4679.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
McCarthy DM, Hruban RH, Argani P, Howe JR,
Conlon KC, Brennan MF, Zahurak M, Wilentz RE, Cameron JL, Yeo CJ,
et al: Role of the DPC4 tumor suppressor gene in adenocarcinoma of
the ampulla of Vater: analysis of 140 cases. Mod Pathol.
16:272–278. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Johnson GL and Lapadat R:
Mitogen-activated protein kinase pathways mediated by ERK, JNK, and
p38 protein kinases. Science. 298:1911–1912. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang J, Tang R, Lv M, Wang Q, Zhang X, Guo
Y, Chang H, Qiao C, Xiao H, Li X, et al: Defective anchoring of
JNK1 in the cytoplasm by MKK7 in Jurkat cells is associated with
resistance to Fas-mediated apoptosis. Mol Biol Cell. 22:117–127.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Takekawa M, Tatebayashi K and Saito H:
Conserved docking site is essential for activation of mammalian MAP
kinase kinases by specific MAP kinase kinase kinases. Mol Cell.
18:295–306. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liu Y, Gorospe M, Yang C and Holbrook NJ:
Role of mitogen-activated protein kinase phosphatase during the
cellular response to genotoxic stress. Inhibition of c-Jun
N-terminal kinase activity and AP-1-dependent gene activation. J
Biol Chem. 270:8377–8380. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Guo Y, Wang W, Wang J, Feng J, Wang Q, Jin
J, Lv M, Li X, Li Y, Ma Y, et al: Receptor for activated C kinase 1
promotes hepatocellular carcinoma growth by enhancing
mitogen-activated protein kinase kinase 7 activity. Hepatology.
57:140–151. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sakurai T, Maeda S, Chang L and Karin M:
Loss of hepatic NF-kappa B activity enhances chemical
hepatocarcinogenesis through sustained c-Jun N-terminal kinase 1
activation. Proc Natl Acad Sci USA. 103:10544–10551. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kuntzen C, Sonuc N, De Toni EN, Opelz C,
Mucha SR, Gerbes AL and Eichhorst ST: Inhibition of
c-Jun-N-terminal-kinase sensitizes tumor cells to CD95-induced
apoptosis and induces G2/M cell cycle arrest. Cancer Res.
65:6780–6788. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pearson G, Robinson F, Beers Gibson T, Xu
BE, Karandikar M, Berman K and Cobb MH: Mitogen-activated protein
(MAP) kinase pathways: Regulation and physiological functions.
Endocr Rev. 22:153–183. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Takahashi R, Hirata Y, Sakitani K, Nakata
W, Kinoshita H, Hayakawa Y, Nakagawa H, Sakamoto K, Hikiba Y,
Ijichi H, et al: Therapeutic effect of c-Jun N-terminal kinase
inhibition on pancreatic cancer. Cancer Sci. 104:337–344. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Thornley JA, Trask HW, Ringelberg CS,
Ridley CJ, Wang S, Sal-Lari RC, Moore JH, Korc M and Tomlinson CR:
SMAD4-dependent polysome RNA recruitment in human pancreatic cancer
cells. Mol Carcinog. 51:771–782. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Derynck R and Zhang YE: Smad-dependent and
Smad-independent pathways in TGF-beta family signalling. Nature.
425:577–584. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hayashida T, Decaestecker M and Schnaper
HW: Cross-talk between ERK MAP kinase and Smad signaling pathways
enhances TGF-beta-dependent responses in human mesangial cells.
FASEB J. 17:1576–1578. 2003.PubMed/NCBI
|
|
20
|
He S, Liu X, Yang Y, Huang W, Xu S, Yang
S, Zhang X and Roberts MS: Mechanisms of transforming growth factor
beta(1)/Smad mediated by mitogen-activated protein kinase pathways
in keloid fibroblasts. Br J Dermatol. 162:538–546. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cui J, Wang Q, Wang J, Lv M, Zhu N, Li Y,
Feng J, Shen B and Zhang J: Basal c-Jun NH2-terminal protein kinase
activity is essential for survival and proliferation of T-cell
acute lymphoblastic leukemia cells. Mol Cancer Ther. 8:3214–3222.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang J, Wang Q, Zhu N, Yu M, Shen B,
Xiang J and Lin A: Cyclic AMP inhibits JNK activation by
CREB-mediated induction of c-FLIP(L) and MKP-1, thereby
antagonizing UV-induced apoptosis. Cell Death Differ. 15:1654–1662.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chi H, Barry SP, Roth RJ, Wu JJ, Jones EA,
Bennett AM and Flavell RA: Dynamic regulation of pro- and
anti-inflammatory cytokines by MAPK phosphatase 1 (MKP-1) in innate
immune responses. Proc Natl Acad Sci USA. 103:2274–2279. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wagner EF and Nebreda AR: Signal
integration by JNK and p38 MAPK pathways in cancer development. Nat
Rev Cancer. 9:537–549. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kinney CM, Chandrasekharan UM, Mavrakis L
and DiCorleto PE: VEGF and thrombin induce MKP-1 through distinct
signaling pathways: Role for MKP-1 in endothelial cell migration.
Am J Physiol Cell Physiol. 294:C241–C250. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kwon GT, Cho HJ, Chung WY, Park KK, Moon A
and Park JH: Isoliquiritigenin inhibits migration and invasion of
prostate cancer cells: Possible mediation by decreased JNK/AP-1
signaling. J Nutr Biochem. 20:663–676. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li M, Feurino LW, Li F, Wang H, Zhai Q,
Fisher WE, Chen C and Yao Q: Thymosinalpha1 stimulates cell
proliferation by activating ERK1/2, JNK, and increasing cytokine
secretion in human pancreatic cancer cells. Cancer Lett. 248:58–67.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yamagata H, Matsuzaki K, Mori S, Yoshida
K, Tahashi Y, Furukawa F, Sekimoto G, Watanabe T, Uemura Y, Sakaida
N, et al: Acceleration of Smad2 and Smad3 phosphorylation via c-Jun
NH(2)-terminal kinase during human colorectal carcinogenesis.
Cancer Res. 65:157–165. 2005.PubMed/NCBI
|
|
29
|
Itatani Y, Kawada K, Fujishita T, Kakizaki
F, Hirai H, Matsumoto T, Iwamoto M, Inamoto S, Hatano E, Hasegawa
S, et al: Loss of SMAD4 from colorectal cancer cells promotes CCL15
expression to recruit CCR1+ myeloid cells and facilitate
liver metastasis. Gastroenterology. 145:1064–1075. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Aitchison AA, Veerakumarasivam A, Vias M,
Kumar R, Hamdy FC, Neal DE and Mills IG: Promoter methylation
correlates with reduced Smad4 expression in advanced prostate
cancer. Prostate. 68:661–674. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gururajan M, Chui R, Karuppannan AK, Ke J,
Jennings CD and Bondada S: c-Jun N-terminal kinase (JNK) is
required for survival and proliferation of B-lymphoma cells. Blood.
106:1382–1391. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hahn JN, Falck VG and Jirik FR: Smad4
deficiency in T cells leads to the Th17-associated development of
premalignant gastroduodenal lesions in mice. J Clin Invest.
121:4030–4042. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chen YW, Hsiao PJ, Weng CC, Kuo KK, Kuo
TL, Wu DC, Hung WC and Cheng KH: SMAD4 loss triggers the phenotypic
changes of pancreatic ductal adenocarcinoma cells. BMC Cancer.
14:1812014. View Article : Google Scholar : PubMed/NCBI
|














